

Abstract
The hepatitis C Virus (HCV) presents a high degree of genetic variability which is explained by the combination of a lack of proof reading by the RNA dependant RNA polymerase and a high level of viral replication. The resulting genetic polymorphism defines a classification in clades, genotypes, subtypes, isolates and quasispecies. This diversity is known to reflect the range of responses to Interferon therapy. The genotype is one of the predictive parameters currently used to define the antiviral treatment strategy and the chance of therapeutic success. Studies have also reported the potential impact of the viral genetic polymorphism in the outcome of antiviral therapy in patients infected by the same HCV genotype. Both structural and non structural genomic regions of HCV have been suggested to be involved in the Interferon pathway and the resistance to antiviral therapy. In this review, we first detail the viral basis of HCV diversity. Then, the HCV genetic regions that may be implicated in resistance to therapy are described, with a focus on the structural region encoded by the E2 gene and the non-structural genes NS3, NS5A and NS5B. Both mechanisms of the Interferon resistance and of the new antiviral drugs are described in this review.
Keywords: Hepatitis C virus, Genetic diversity, Therapy resistance, E2, NS3, NS5A, NS5B
INTRODUCTION
The role of genetic variability in the natural history by hepatitis C Virus (HCV) infection and in the primary resistance to treatment remains unclear. In particular, the mechanisms underlying the resistance of HCV to Interferon treatment are very different from those by which the human immunodeficiency virus (HIV) becomes resistant to antiretroviral drugs. In this review, we will describe the impact of HCV genetic polymorphism in the treatment response.
HCV VARIABILITY: ORIGIN AND CLASSIFICATION
HCV has a single-strand positive RNA genome and displays a high genetic diversity. This diversity results from defects in the repair activity of the RNA-dependent RNA polymerase (resulting in nucleotide substitutions) and from the absence of 5' to 3' exonuclease activity (lack of error correction)[1]. The mean frequency of nucleotide mutations varies from 1.4 × 103 to 1.9 × 103 substitutions per nucleotide and per year. This estimate is based on comparisons of the major sequences of complete genomes obtained after eight years of evolution in a chimpanzee and 13 years in a human[2,3].
Some of the mutations accumulating during replication are silent or synonymous. These mutations have no impact on the amino-acid sequence of the viral protein, but may affect the secondary structure of the genomic RNA. Other so-called non-synonymous mutations lead to changes in protein sequence and the emergence of variants. Other mutations lead to the production of defective viral particles and are therefore lethal. The regions of the genome corresponding to essential viral functions (domains involved in translation or replication), or displaying major structural constraints (non-coding 5' and 3' ends) are the best conserved; indeed, the non coding 5' region is the most highly conserved region of the genome, with more than 90% identity between the sequences of distantly related strains[4]. The region encoding the capsid is also well conserved, with 81% to 88% sequence identity between isolates[5]. The most variable region of the genome is that encoding the envelope glycoproteins, E1 and E2. The sequences of hypervariable regions (HVR1 and HVR2) of E2 in strains isolated from different patients may differ by more than 50%. The polypeptides encoded by these hypervariable regions are therefore very tolerant to amino-acid substitutions[6,7].
The classification of HCV, redefined and simplified in 1998, is based on the topology of the trees obtained by phylogenetic relationships between viral variants. HCV variants can be classified into six clades, and then into subtypes corresponding to subgroups of the most closely related viruses within a clade[8]. The virus genotype is indicated by an Arabic number (from 1 to 6), associated with a lower-case letter to indicate the subtype. This new nomenclature has led to the reclassification of virus genotypes 7, 8, 9 and 11 as genotype 6 viruses, and the reclassification of genotype 10 viruses as genotype 3 viruses. Recently, the status of HCV-genotype nomenclature has been re-examined in an attempt to resolve conflicting subtype and genotype designations. A complete list of currently recognized genotypes and subtypes was published[9]. The euHCVDB (http://hepatitis.ibcp.fr), DDJB (http://www.ddbj.nig.ac.jp) and Los Alamos (http://hcv.lanl.gov) databases currently include a large number of HCV sequences, making it possible to compare an isolate with a large number of reference sequences[10]. Although currently restricted to specialist laboratories, the gold standard method for HCV genotyping is sequencing of the NS5B region followed by phylogenetic analysis including comparison to reference sequences[11,12].
A chimeric virus generated by an intergenotypic homologous recombination event (genotype 2k/genotype 1b) in the NS2 gene was first described in the St Petersburg area: this demonstrated the occurrence of recombination phenomena in HCV[13]. Another recombinant form (genotype 2i/genotype 6p) was recently described in Vietnam, with a point of recombination between NS2 and NS3[14]. Such recombination events have already been described for GBV-C and Dengue virus[15,16]. In HCV, homologous recombination may be favoured by the nature of HCV risk behaviour in which there may be frequent exposures, for example among Intra Venous Drug Users. In practice, genotyping is based solely on polymorphism in a single genomic region (5' NC or NS5B), so it is currently impossible to estimate the frequency of recombination events.
Studies of the global distribution of HCV genotypes and analyses of their phylogenetic relationships have provided insight into their emergence and diversification over the centuries. The worldwide distribution of genotype 2 began some 90 to 150 years ago; that of subtype 1b began 60 to 70 years ago and that of genotype 3 began about 40 years ago[17] Genotypes 4 and 6 emerged much earlier: 350 and 700 years ago, respectively[18]. The simultaneous presence of a large number of subtypes of a viral genotype in a limited geographic region is indicative of the long-standing endemic presence of the virus in the population studied. Genotype 2 is frequent in West Africa (59% to 100%, depending on the region studied), and many subtypes have been identified (2c to 2l)[19]. The same is true of genotype 4 in Central Africa (4b, 4c, 4e and 4m)[20]. Conversely, the limited number of subtypes in Europe (where genotype 1, 2, 3 are the more frequently found), North America and Japan is consistent with the more recent introduction of HCV into these populations. Another level of complexity is found within a given infected patient termed as the quasispecies population. Quasispecies are populations of different but closely related genomes and data have suggested that Interferon alpha exerts a selective pressure on HCV quasispecies[21,22]. Thus, these molecular polymorphisms are clinically relevant and are one of the major factors in determining the outcome of the Interferon therapy.
Since 1986, before the discovery of HCV, the efficacy of Interferon alpha has been demonstrated in the treat-ment of chronic non A non B hepatitis[23,24]. Interferon alpha is a member of the cytokine family produced by cells in response to viral infections or other stimuli by binding to their specific receptors on the surface of target cells, Interferons stimulate a cascade of intracellular signalling pathways that result in the suppression of numerous Interferon-Stimulated Genes (ISGs) whose products (proteins) can mediate the effects of Interferon. Among these, are the INF induced, double stranded (ds) RNA dependant protein kinase (PKR), the 2' 5' oligoadenylate synthetase and the Mx proteins, of which the antiviral activities have been well demonstrated[25]. Interferon also acts as a stimulus of immunity and modulates cell growth, differentiation and apoptosis[26].
In an effort to understand the role of different factors on the outcome of Interferon therapy, numerous studies have been performed to define predictors of response and both viral and host factors have been studied.
Among the viral factors examined, viral genotype seems to be the most important predicting factor; it appears to be a predictive parameter of the SVR as strong as well-established predictive parameters such as the pre-treatment viral load, the stage of fibrosis on liver, and the age and the sex of the patient. Trials clearly reported a high SVR rate of 76% to 84% in patients infected with HCV genotypes 2 or 3 whereas a weaker SVR of 42% to 52% was obtained in HCV genotype 1-infected patients[27-29]. The SVR is likely to be higher for genotype 2 than for genotype 3 where a shortened treatment period was experienced, but ongoing studies will determine the role of the initial viral load and a rapid virologic response[30,31]. Therapeutic outcomes are well known for these three genotypes because of their geographic distribution; genotype 1 represents more than 70% of the HCV infections in the Western world, genotypes 2 and 3 infect 10% to 20% of the other patients. Studies focused on the outcome of the antiviral therapy conducted in patients infected by HCV genotype 4 are mainly provided by Egyptian cohorts and results showed an intermediate SVR rate comprised between 55% to 69%[32,33]. Genotypes 5 and 6 are less studied because of their minor distribution, patients infected by these genotypes may achieve a SVR at a level between the SVRs of genotype 1 and genotypes 2-3[34].
Differences in the SVR rate observed among different HCV genotypes have highlighted a presumptive role of the viral genetic determinants and have suggested that some virus-encoded functions are involved in the response to Interferon. Although a very efficient in vitro or in vivo or animal models of HCV infection is not available, several studies have been undertaken to identify which viral gene may interfere with antiviral molecules. Both in vivo and in vitro studies demonstrated that the structural proteins E1 and E2, the non-structural proteins NS3, NS5A and NS5B may contribute to the resistance of the combined Interferon alpha and ribavirin therapy. Moreover, all these proteins share a genetic polymorphism involved in the resistance mechanisms as shown in Figure 1.
Figure 1.
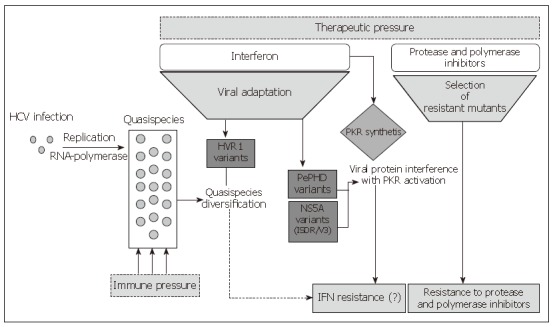
Mechanisms involved in HCV resistance to antiviral therapies.
GENOMIC DOMAINS CONTRIBUTING TO RESISTANCE
As previously described by Pawlotsky in this issue of the journal, HCV presents a high genetic diversity. The intra-genotype analysis of this diversity along the viral genome shows different degrees of variation; regions such as the 5'UTR and the core are highly conserved, the non-structural regions 2, 3, 5b and the 3'UTR are relatively variable whereas the envelope regions E1 and E2 and the NS4 and the NS5A genes exhibit the highest sequence diversity.
Polymorphism and significance of amino acid substitu-tions within E2 regions in Interferon alpha resistance
E2 glycoprotein is a type I transmembrane protein of 70 kDa, with an N-terminal ectodomain and a C-terminal hydrophobic anchor. It assembles with E1 to form a heterodimer. It has been shown to interact with two potential HCV receptors, the human tetraspanin CD81 and the human scavenger receptor SR-BI, in experiments based on soluble E2 (sE2) binding (Figure 2). The role of E2 in the cell entry of HCV is extensively reviewed in this issue by Dubuisson et al.
Figure 2.
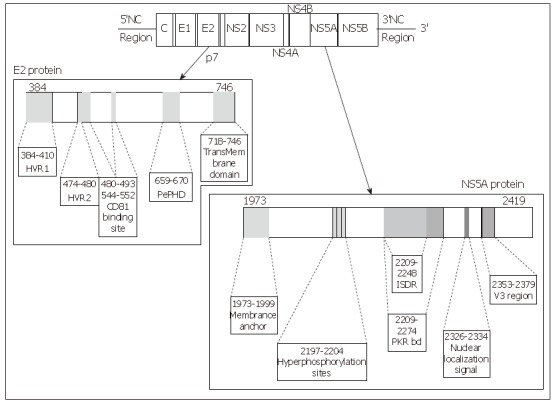
Linear representation of the E2 and NS5A proteins of the HCV, location of the main interesting domains of each protein. Each site of interest is represented by a grey zone on the protein and is linked to a box containing its name and amino acid positions.
One of the major ways by which cellular Interferon alpha inhibits viral replication involves the Interferon-alpha-inducible double-stranded RNA-activated Protein Kinase R (PKR). Indeed the Interferon alpha binds to the PKR and leads to its autophosphorylation which in turn initiates the phosphorylation of eukaryotic initiation factor 2 alpha (eIF2α) by the PKR. This phosphorylation inhibits the RNA transcription. Indeed, eIF2α is necessary to initiate the translation by forming a complex with GTP and met-tRNA and then allowing binding to the 40S ribosomal subunit. The E2 glycoprotein, or more precisely, a 12-amino acid domain of this protein located between residues 659 and 670 and known as PePHD (PKR-eIF2α phosphorylation homology domain), is involved in PKR inhibition. Taylor et al[35] showed that PePHD was the main domain of E2 able to bind PKR in vitro. The PePHD motif sequence is very similar to that of the auto-phosphorylation sites of PKR and its target, eIF2α, and this similarity is more marked for genotype 1 viruses than for genotype 2 and 3 viruses (Figure 3). E2 glycoproteins of genotype 1 viruses (HCV-1 E2) behave in vitro as pseudosubstrates, inhibiting the kinase activity of PKR. In mammalian cells, the stimulation of translation by HCV-1 E2 is consistent with the hypothesis of PKR inhibition. This inhibitory activity has also been observed in a yeast model (Saccharomyces cerevisiae expressing HCV1 E2). The replacement of HCV-1 E2 by proteins identical to those of genotype 2 and 3 viruses abolishes this inhibitory effect. An interaction between genotype 1 strains and PKR, via PePHD, has therefore been proposed to account for the intrinsic resistance of the strains of this genotype to Interferon alpha[35].
Figure 3.

Sequence homology between HCV E2-PePHD domain and PKR. PePHD domains from various HCV genotypes (1a, 1b, 2a, 2b, 3a) are aligned and compared with the PKR sequence. Identical and similar amino acids to PKR are shown by (.) and (+), respectively. PKR auto-phosphorylation sites are underlined.
The clinical relationship between aminoacid sequence of PePHD and the outcome of Interferon therapy has been a matter of controversy. A few studies have add-ressed polymorphism of the PePHD region in patients carrying strains of genotypes 1, 2 and 3. Abid et al studied a small number of patients infected with genotype 1b HCV. They initially showed that in some patients responding to treatment, the virus had a PePHD sequence identical to that of the HCV-J strain (RSELSPLLLSTT), calling Taylor's hypothesis into question[36]. For HCV 2a/b isolates, conflicting results about the association of PePHD mutations and treatment response have been published[37,38]. A number of studies focusing exclusively on the diversity of PePHD sequences in genotype 1b HCV before treatment have since shown strong conservation of this motif, regardless of the response subsequently obtained[39,40].
The genetic heterogeneity of the E2-HVR1 region can also be used to describe the composition of qua-sispecies precisely, provided a large number of clones are analysed. The preliminary studies in this area were based on analysis of single-strand polymorphisms or of a restricted number of clones (less than 10). A correlation between high levels of quasispecies complexity before Interferon alpha monotherapy and a lack of response to treatment was reported[41,42]. This led to the suggestion that genetic variability ensured a reservoir of potentially resistant strains. However, more recent studies with larger numbers of sequenced clones or based on SSCP enhanced sensitivity protocols have not confirmed the link between the genetic complexity of HCV and virological response[43]. These conflicting results illustrate the problems of methodological standardisation associated with studies of quasispecies.
Polymorphism of the HCV non-structural regions and impact on the treatment response
The NS2, NS3 and NS4A/B proteins may not be im-plicated in the antiviral treatment resistance with the current combination pegylated Interferon alpha plus ribavirin. However, new therapies are in development targeting the protease encoded by NS3 and the polymerase encoded the NS5B. As already observed in the anti-HIV HAART treatment, the genetic polymorphism of NS3 and NS5B will be analysed in relation to therapy. HCV presents a high mutation rate and the antiviral pressure may favour the emergence of resistant strains. Mutation points have been already observed in vitro (replicon system).
Polymorphism of NS5A and Interferon resistance
The NS5A protein is the non structural HCV protein which is the protein most reported to be implicated in the Interferon resistance Many interactions between NS5A and different molecules from several intracellular pathways have been also demonstrated in cellular in vitro systems. An overview of the key roles of the genetic heterogeneity of this protein will be detailed in this paragraph.
Structure of the NS5A protein: NS5A is a phospho-protein found in a basally phosphorylated form of 56kDa and a hyperphosphorylated form of 58 kDa. It varies in length, from 445 amino acids in genotype 4a, 447 amino acids in genotype 1b, 448 in genotypes 1a and 1c, 450 in genotype 5a, 451 in genotype 6a, 452 in genotype 3 to 452 amino acids in genotype 2a and b. The amino acid sequence varies depending on the genotype. The NS5A protein is a pleïotropic protein involved both in the viral replication and in many interactions with cellular signalling pathways. Although its function remains unclear, many domains of interest have been described in this protein (Figure 2).
Many interactions between NS5A and cellular signall-ing pathways have been reported and the interacting sequence has been identified for some of them. Although studies have been conducted in vitro, results strongly suggest a potential involvement of the NS5A protein in the establishment of a chronic hepatitis and in the carcinogenesis and outcome of a liver tumour (for review, see[44]). In this review, we will focus only on the protein interaction of NS5A with the Interferon pathway.
Interaction with the cellular Interferon pathway: Pho-sphorylation of PKR triggered a global translastional repression comprising the viral replication. Gale et al[45] identified in the C-terminal NS5A a Protein Kinase R binding domain. They demonstrated in vitro that the NS5A protein is able to bind and inhibit the Interferon-alpha-inducible double-stranded RNA-activated PKR, the interaction occuring via the NS5A PKR binding domain. In that way, the NS5A protein has been suggested to balance the Interferon cellular antiviral pathway and to be involved in the resistance to the Interferon based-therapy.
The NS5A protein is also able to interact with the Interferon pathway in a PKR-independent manner. It has been demonstrated in vitro that NS5A induces the expression of the pro-inflammatory chemokine interleukin 8 (IL-8) at both the mRNA and protein levels. This chemokine is known to inhibit directly the Interferon alpha activity. The clinical relevance of these in vitro studies has been reported and it appears that the IL-8 serum levels were higher in infected patients than in healthy controls. A second study showed an increase of the pre-treatment IL-8 level in the non-responder than in the SVR[46,47].
Mutations in the NS5A protein and relationship with the Interferon resistance: The 40 first amino acids of the PKR binding domain present a high level of variability. It was termed the Interferon sensitivity determining region (ISDR) reported to play a key role in the Interferon therapy response[48,49]. Molecular analysis of this 40 amino acid region showed the potential role of mutations in the resistance to Interferon therapy. They demonstrated a correlation between the success of Interferon therapy and the variability of the ISDR domain in Japanese patients with HCV genotype 1b or 2 infection. Patients infected with viral strains whose ISDR harbours more than four mutations different from the HCV-J sequence (the Japanese prototype strain defined as the wild-type) were responders to Interferon therapy whereas patients infected with a wild-type or strain harbouring less than four mutations in ISDR were non-responders. Numerous studies investigating the correlation between the mutations in ISDR in genotype 1b HCV and the outcome of the Interferon-based therapy have been undertaken. Japanese results were concordant with the initial study whereas European and north-American groups did not describe such a correlation and debate remains controversial. A recent meta-analysis has been conducted on 1230 ISDR sequences from HCV 1b-infected patients by Pascu et al[50]. Sequences were provided from Japanese and European studies. Analyses were realised by logistic regression and clearly demonstrated an association between number of ISDR mutations and response to the treatment both in Japanese and European patients, irrespective from a geographical distribution. First, controversial results may be explained in part by the HCV European strains, most of the European HCV 1b strains present less than 3 mutations and it was difficult to identify a statistical difference between the Interferon therapy response among European infected patients. It is also important to note that treatment schedules were not the same in Europe and in Japan. A correlation between the variability in the ISDR region and a SVR has been demonstrated in the genotype 2a-infected patients. Conversely, investigations in the patients infected by genotypes 2b or 3a did not find any relation between the successful therapy and genetic variability of the NS5A gene[51,52]. Additionally, in vitro studies did not report any interaction between NS5A genotype 3a and PKR in viral resistant strains to the Interferon therapy[53].
The major implication of the variability in the re-sistance to antiviral agents has been recently pointed out, but few studies focused on the entire NS5A protein at the quasispecies level and its kinetics under therapy. Inshauspe et al[54] identified another domain localised in the C-terminal region of NS5A and termed it the V3 domain. It is 27 amino acids in length and it harbours a high variability level. Duverlie et al[55] demonstrated a relationship between the mutation level in V3 and the response to Interferon therapy. Resistant strains were highly conserved whereas sensitive strains were variable. Our group and others confirmed this correlation[56]. We followed HCV quasispecies diversity at baseline and under Interferon alpha-ribavirin combined treatment in the entire NS5A gene and in each region of interest (PKR-bd, ISDR and V3). As reported by Puig-Basoiti et al[57], the V3 domain showed a higher quasispecies diversity in responder patients than in non-responders in pre-treatment samples and these data confirmed the potential role of the genetic diversity in the success of the Interferon-based therapy.
NS3 protease as a target for specific antiviral therapy
Previous studies on the full-length genome sequence have led to the HCV NS3 domain as being classified as one of the less variable regions of the genome[58,59]. Nevertheless, it appears important to consider the diversity of HCV protease, because it is an attractive target for specific anti-HCV therapy.
Catalytic functions and the three-dimensional struc-ture of HCV NS3 protease have been reviewed in this issue by Dubuisson et al. Some of the structural and functional constraints affecting the NS3 protease have been demonstrated by the definition of major domains, including the catalytic triad, the substrate binding pocket, the NS4A binding domain and the residues binding to the zinc ion[60].
Natural polymorphism of NS3 protease: The diversity of the HCV NS3 protease gene in clinical samples has been studied[61,62]. Although the protease NS3 is considered to be one of the less variable genes in the HCV genome, variability of both nucleotide and amino acid sequences exists. The proportion of synonymous substitutions affecting the region is significantly higher than the proportion of non synonymous ones suggesting that the NS3 protease mutations are the products of random genetic drift rather than of positive selection. NS3 protease structural and chemical integrity is required for it to process the HCV polyprotein. The perpetuation of substitutions depends in large part on the extent to which the viral protease can tolerate them while remaining functional. The NS3 protease appears to be tolerant, particularly in its loop regions between helices and sheets (Figure 4). Sporadic mutations affecting the catalytic residues have already been observed from different patients. These catalytic-triad amino acid mutations probably lead to a loss of function, and therefore may be lethal. In that sense, they are not crucial for resistance to NS3 protease inhibitors[61].
Figure 4.
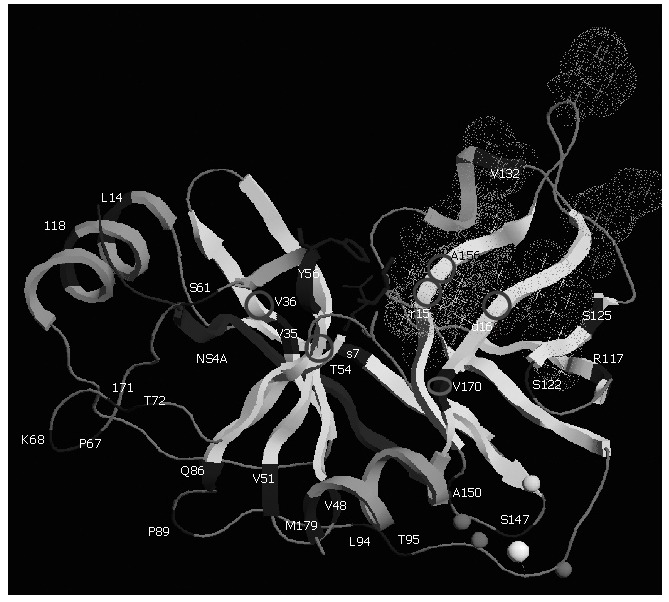
Three-dimensional structure of NS3 protease (PDB accession code 1NS3). Polymorphism and main residues implicated in resistance are shown. The protease is shown based on its secondary structure in light grey. Main polymorphic residues are shown in dark grey. The side chains of the residues forming the catalytic triad (H57, D81, and S139) are displayed in dark grey ball-and-stick representation. The NS4A cofactor is shown in dark grey and Zinc ion as a white ball. Zn2+ ligands (C97, C99 and C149) and H145 residue are modeled in light grey spheres. Stars correspond to the side chains of the residues forming the S1 to S6 substrate binding pockets (grey dots cloud) of the enzyme. Residues implicated in resistance to protease NS3 inhibitors are shown with circles. This figure was prepared using the RasTop programme version 2.0.3-VF.
The NS3 region is known to contain a large number of T-cell epitopes. T-cells directed against the NS3 domain seem to be of particular importance both during the acute and chronic phase of the disease as the CD4+ and CD8+ T-cell response to the NS3 protease correlates with clearance and control of the infection[63]. As reviewed by Dubuisson et al[64,65] in this issue, the NS3 serine protease influences the Interferon-dependent innate cellular host defence by inhibition of RIG-1 and TLR3 signalling. As use of Interferon alpha is the standard of care for HCV, the potential of protease inhibition to enhance Interferon therapy has attracted much interest.
NS3 Polymorphism and impact on response to antiviral specific treatment: The NS3 protease three-dimensional structure description has provided the necessary detailed insight to permit rational inhibitor design. Therapies based on such inhibitors are still at the development and testing stage[66,67]. The generation of such therapies based on the inhibition of site-specific proteolysis has been clearly illustrated in the development of effective inhibitors of human immunodeficiency virus type 1 (HIV-1)[68].
In vitro and in vivo phase I and II studies have shown that evaluated NS3 protease inhibitors (BILN 2061, VX-950 and SCH 503034) developed drug resistant mutations[69,70]. These new drugs meet the same problems of viral resistance that exist in the specific anti-viral treatment of HIV and HBV infected individuals. In untreated patients, less than five percent of protease variants present resistant mutations to inhibitors SCH6, SCH 503034 and VX-950[61,71,72]. The reduced fitness of the most resistant variant may explain that such variants are rarely found in naïve patients[69,70,73]. The replicative fitness of resistant viruses is a critical parameter of viral resistance and is an important factor to consider in achieving sustained virological response. It is underestimated due to low replicative resistant strains not being detected.
Both HCV protease inhibitors evaluated in humans, VX-950 and SCH-503034, have demonstrated a very strong correlation between their antiviral effect, their serum trough levels, and the development of resistance[67,74]. Changing in viral quasispecies with a selection of clones resistant to VX-950 has been observed in patients who experienced viral load breakthrough to VX-950 mono-therapy[66]. Resistant variants may have poorer replicative fitness than wild-type viruses. However, VX-950-resistant clones isolated from patient plasma from the initial phase 1b study were thought to be associated with the lower doses of monotherapy, and selection for these clones was based on inadequate suppression of virus[70]. Four residues in the NS3 protease, when substituted, are known to be associated with resistance/reduced sensitivity to VX-950. R155K/T/S/M and A156T/V, located close to the VX-950 binding groove, a domain of NS3 protease, confer moderate to high level resistance to VX-950; V36A/M and T54A located further from the binding groove confer low-level of resistance to VX-950 (Figure 4).
Three mutations, T54A, V170A and A156S muta-tions conferred low to moderate levels of resistance to SCH-503034 in a replicon model. Mutants with A156T substitution are highly resistant to this compound[73]. Combinations of VX-950 and SCH-503034 may be limited by the selection of cross-resistance mutations.
The polymorphic outside residues central to NS3 protease activity and substrate binding sites, may prove to be accessory substitutions of resistance mutations con-tributing to clinical resistance to HCV protease inhibitor therapy. This has been supposed for 3 separate second site mutations P89L, Q86R and G16R in vitro with the protease SCH6 inhibitor[75]. Moreover, resistance to future NS3 protease inhibitors could occur through mutations arising either in the protease gene itself or, as has been shown for HIV-1, in cleavage sites of the protein[68].
Resistance to specific HCV inhibitors will probably emerge in case of insufficient antiviral pressure. Viral load monitoring and checking for emergence of NS3 protease resistance mutations could contribute to surveillance of sustained viral response.
NS5B polymorphism and impact of the anti-polymerase therapy
Structure of the protein: The last gene of the HCV is the NS5B which encodes the HCV polymerase, which is an RNA-dependent RNA polymerase (RdRP). This 68 kDa protein plays a central role in HCV replication and in viral diversity because of the lack of a proof-reading activity. The NS5B protein belongs to the tail-anchored proteins as previously detailed by Dubuisson et al. The crystal structure of this enzyme has been fully determined. The three dimensional structure is similar to other polymerases; it shows a typical right-hand arrangement with fingers, palm and thumb domains. This organisation forms a cavity called a NTP tunnel in which the traffic of the nucleotides to the active site occurs[76,77] (Figure 5).
Figure 5.

Three-dimensional structure of NS5B RNA-dependent RNA polymerase (PDB accession code 1QUV). The three subdomains of the polymerase are shown (Fingers, Thumb and Palm) as well as the main residues targeted by each class of polymerase inhibitors (NNI 1: Non nucleoside class 1, NNI 2: Non nucleoside class 2, NNI 3: Non nucleoside class 3, NI: Nucleoside Inhibitor). This figure was prepared using the RasTop programme version 2.0.3-VF.
Genetic polymorphism and enzymatic activity: As all enzymatic proteins, the nucleic acid sequence is a fundamental determinant of the three-dimensional structure and it determines good enzymatic activity. It has been shown that the active site does not tolerate any mutations. Its amino acid sequence is highly conserved among all HCV genotypes and any mutation may inhibit viral replication. Several consensus sequence motifs have also been described along the NS5B polymerase; five of them are in the palm sub-domains (motifs A, B, C, D and E) and one is in the finger domain (motif F)[78,79]. Most of the single amino-acid substitutions introduced into the conserved motifs by directed mutagenesis resulted in decreased or abolished viral replication and enzymatic activity[80,81].
NS5B and viral genotype: Although the NS5B protein remains a relatively well conserved protein because of its enzymatic function, the NS5B domain, along with other domains of the HCV, supports a slight variability which provides a basis for distinguishing viral type and subtype. Indeed, the nucleic acid sequence is less conserved than the 5'NC but sufficient to define type and viral subtype. The relation between the structure of the NS5B and the response to the Interferon therapy has rarely been studied and most studies were focused on genotype 1b. Results were contradictory. Vuillermoz et al[82] did not find any mutation differences (point and rate mutations) between responders and non-responders to the HCV NS5B strain; conversely Kumagai et al[83] reported two polymorphisms in NS5B associated with an early viral clearance in the treatment of HCV genotype 1b-infected patients.
New antiviral treatments targeting the HCV poly-merase: The HCV polymerase represents an important target for the new inhibitors of HCV replication and numerous assays are currently in development using the in vitro subgenomic HCV replicon system. Thanks to the knowledge of the three dimensional structure of NS5B, screening of many chemical molecules has been undertaken. Three classes of inhibitors are now reported and include nucleoside analogues, non nucleoside analogues and pyrophosphate mimics. Drugs belonging to the nucleoside inhibitors are in competition with the substrate of the RdRP and inhibit polymerase elongation. The binding site is localised in the fingers sub-domain of the polymerase. Molecules of this class have a 2'methylribose structure and some of them are now well characterised[84,85]. 2'-C-Methyl-Cytidine, 2'-C-Methyl-Adenosine and 2'-C-Methyl-Guanosine have been shown to be effective against genotype 1a and 1b in Con1 subgenomic replicons. A single mutation, S282T, confers a cross resistance to all the 2'-C-Methyl-Nucleoside, and to their prodrug as such the NM283 (prodrug of the 2'-C-Methyl-Cytidine known as the Valopicitabine)[86,87]. The R1479 (4'-Azido-Cytidine), which is another nucleoside inhibitor, is under development[88] and a recent study reported the mutation resistance points as S96T and S96T/N142T. This work showed no cross resistance between R1479 and the 2'C methyl nucleosides[89]. The NS5B genes of non genotype 1 (2a, 2b, 3, 4 and 6) have been studied in a genotype 1 replicon background; most of them reported a weak replication that enabled drug sensitivity assays. It is important to note that prototype sequences of genotype 4a and 6a encoded T282 and L495 which are implicated respectively in the in vitro resistance to the 2'-C-Methyl nucleoside inhibitors and to the benzimidazole non-nucleoside inhibitors[87]. NM283 (Idenix®) has demonstrated an anti-HCV activity in the trials of phases I and II a, it is now in the phase II b clinical development. Results of the phase I trial of R1626 (Roche®), another nucleoside inhibitor, were also encouraging. The first phase II develop-ment study is now starting[90].
Compounds belonging to the non-nucleoside inhibitors (NNI) act as allosteric inhibitors that may affect the initiation step of the polymerisation (inhibition of the conforma-tional transition prior to the formation of an efficient pre-elongation complex, inhibition of RNA binding to NS5B or non-competitive inhibitors of NTP incorporation). Different molecules belong to this class and have distinct targets. Indeed, three binding pockets have been localised and are used to define each class of NNI (1 to 3). Two of them are located in the thumb domain. The benzothiazidines group (NNI 1) binds to one pocket located at the interface between the thumb and palm domains and centralised around Met414. In vitro studies have been carried out mainly with the genotype 1b subgenomic replicon and substitu-tions have been shown to decrease susceptibility (M414T, C451R, G558R, H95R)[91] or to confer resistance (H95Q, N411S, M414L/T, Y448H)[92] to benzothiazine drugs from different pharmaceutical companies. The second pocket, located at the base of the thumb domain, binds to the large group of molecules comprising thiophene 2-carboxylic acids, phenylalanine derivatives and substituted pyranones (NNI 2) and the binding central amino acid is Met423. In vitro assays reported differences in the inhibitory activity between the different genotypes tested. For example, Howe et al[93] showed that the susceptibility level to the HCV-371 (NNI 2) was equal among the genotype 1b isolates but varied with genotypes 1a, 3a and 4 (5 to 59 fold less susceptible than genotype 1b). The last pocket is located at the top of the thumb around Pro495 and is targeted by the benzimidazole 5-carboxamide inhibitors (NNI 3)[94]. As with the other nucleoside inhibitors, sensitivity has been studied in vitro in the subgenomic replicon; Tomei et al[95] have recently reported that HCV strains resistant to benzimidazole molecules harbored a substitution in Pro495. Interestingly, it has been shown that a chimaeric 1b:2b HCV replicon, harbouring a genotype 2b NS5B, was sensitive to the 2'-C-Methyl-Adenosine but completely insensitive to the NNI 1 (benzimidazole) and NNI 3 (thiophene)[96]. These results underlined that a genotype specificity may exist for each anti-polymerase inhibitor.
All of these results showed the importance of genetic diversity in the HCV therapeutic strategy. Preliminary results, both in vitro and in vivo trials, strongly suggest a relationship between genetic polymorphism and sensitivity to drug. Identification of the mutations set corresponding to each antiviral drug will be very helpful. It will represent a chance to anticipate treatment regimes at baseline based on the viral genotype, an approach currently only done for HIV therapy, many years after the beginning of HAART[97].
Finally, the genetic diversity of the HCV largely im-pacts in the global approach of this viral disease as in the treatment management as well as in the development of new HCV antiviral therapies.
ACKNOWLEDGMENTS
The authors thank the Fontevraud Group Study that largely contributes to the study of the HCV infection. Studies focused on the NS5A and the E2-PePHD diversity were realised with grants from the Agence Nationale de Recherche sur le SIDA et les Hépatites (ANRS). We thank Professor David Archer, School of Biology, University of Nottingham, UK, for careful reading of the manuscript.
Footnotes
S- Editor Liu Y E- Editor Ma WH
References
- 1.Steinhauer DA, Domingo E, Holland JJ. Lack of evidence for proofreading mechanisms associated with an RNA virus polymerase. Gene. 1992;122:281–288. doi: 10.1016/0378-1119(92)90216-c. [DOI] [PubMed] [Google Scholar]
- 2.Ogata N, Alter HJ, Miller RH, Purcell RH. Nucleotide sequence and mutation rate of the H strain of hepatitis C virus. Proc Natl Acad Sci USA. 1991;88:3392–3396. doi: 10.1073/pnas.88.8.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Okamoto H, Kojima M, Okada S, Yoshizawa H, Iizuka H, Tanaka T, Muchmore EE, Peterson DA, Ito Y, Mishiro S. Genetic drift of hepatitis C virus during an 8.2-year infection in a chimpanzee: variability and stability. Virology. 1992;190:894–899. doi: 10.1016/0042-6822(92)90933-g. [DOI] [PubMed] [Google Scholar]
- 4.Bukh J, Purcell RH, Miller RH. Sequence analysis of the 5' noncoding region of hepatitis C virus. Proc Natl Acad Sci USA. 1992;89:4942–4946. doi: 10.1073/pnas.89.11.4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simmonds P, Smith DB, McOmish F, Yap PL, Kolberg J, Urdea MS, Holmes EC. Identification of genotypes of hepatitis C virus by sequence comparisons in the core, E1 and NS-5 regions. J Gen Virol. 1994;75(Pt 5):1053–1061. doi: 10.1099/0022-1317-75-5-1053. [DOI] [PubMed] [Google Scholar]
- 6.Smith DB. Evolution of the hypervariable region of hepatitis C virus. J Viral Hepat. 1999;6 Suppl 1:41–46. doi: 10.1046/j.1365-2893.1999.00010.x. [DOI] [PubMed] [Google Scholar]
- 7.Penin F, Combet C, Germanidis G, Frainais PO, Deléage G, Pawlotsky JM. Conservation of the conformation and positive charges of hepatitis C virus E2 envelope glycoprotein hypervariable region 1 points to a role in cell attachment. J Virol. 2001;75:5703–5710. doi: 10.1128/JVI.75.12.5703-5710.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robertson B, Myers G, Howard C, Brettin T, Bukh J, Gaschen B, Gojobori T, Maertens G, Mizokami M, Nainan O, et al. Classification, nomenclature, and database development for hepatitis C virus (HCV) and related viruses: proposals for standardization. International Committee on Virus Taxonomy. Arch Virol. 1998;143:2493–2503. doi: 10.1007/s007050050479. [DOI] [PubMed] [Google Scholar]
- 9.Simmonds P, Bukh J, Combet C, Deléage G, Enomoto N, Feinstone S, Halfon P, Inchauspé G, Kuiken C, Maertens G, et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology. 2005;42:962–973. doi: 10.1002/hep.20819. [DOI] [PubMed] [Google Scholar]
- 10.Kuiken C, Mizokami M, Deleage G, Yusim K, Penin F, Shin-I T, Charavay C, Tao N, Crisan D, Grando D, et al. Hepatitis C databases, principles and utility to researchers. Hepatology. 2006;43:1157–1165. doi: 10.1002/hep.21162. [DOI] [PubMed] [Google Scholar]
- 11.Laperche S, Lunel F, Izopet J, Alain S, Dény P, Duverlie G, Gaudy C, Pawlotsky JM, Plantier JC, Pozzetto B, et al. Comparison of hepatitis C virus NS5b and 5' noncoding gene sequencing methods in a multicenter study. J Clin Microbiol. 2005;43:733–739. doi: 10.1128/JCM.43.2.733-739.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laperche S, Saune K, Dény P, Duverlie G, Alain S, Chaix ML, Gaudy C, Lunel F, Pawlotsky JM, Payan C, et al. Unique NS5b hepatitis C virus gene sequence consensus database is essential for standardization of genotype determinations in multicenter epidemiological studies. J Clin Microbiol. 2006;44:614–616. doi: 10.1128/JCM.44.2.614-616.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalinina O, Norder H, Mukomolov S, Magnius LO. A natural intergenotypic recombinant of hepatitis C virus identified in St. Petersburg. J Virol. 2002;76:4034–4043. doi: 10.1128/JVI.76.8.4034-4043.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noppornpanth S, Lien TX, Poovorawan Y, Smits SL, Osterhaus AD, Haagmans BL. Identification of a naturally occurring recombinant genotype 2/6 hepatitis C virus. J Virol. 2006;80:7569–7577. doi: 10.1128/JVI.00312-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holmes EC, Worobey M, Rambaut A. Phylogenetic evidence for recombination in dengue virus. Mol Biol Evol. 1999;16:405–409. doi: 10.1093/oxfordjournals.molbev.a026121. [DOI] [PubMed] [Google Scholar]
- 16.Worobey M, Holmes EC. Homologous recombination in GB virus C/hepatitis G virus. Mol Biol Evol. 2001;18:254–261. doi: 10.1093/oxfordjournals.molbev.a003799. [DOI] [PubMed] [Google Scholar]
- 17.Simmonds P. The origin and evolution of hepatitis viruses in humans. J Gen Virol. 2001;82:693–712. doi: 10.1099/0022-1317-82-4-693. [DOI] [PubMed] [Google Scholar]
- 18.Pybus OG, Charleston MA, Gupta S, Rambaut A, Holmes EC, Harvey PH. The epidemic behavior of the hepatitis C virus. Science. 2001;292:2323–2325. doi: 10.1126/science.1058321. [DOI] [PubMed] [Google Scholar]
- 19.Jeannel D, Fretz C, Traore Y, Kohdjo N, Bigot A, Pê Gamy E, Jourdan G, Kourouma K, Maertens G, Fumoux F, et al. Evidence for high genetic diversity and long-term endemicity of hepatitis C virus genotypes 1 and 2 in West Africa. J Med Virol. 1998;55:92–97. [PubMed] [Google Scholar]
- 20.Fretz C, Jeannel D, Stuyver L, Hervé V, Lunel F, Boudifa A, Mathiot C, de Thé G, Fournel JJ. HCV infection in a rural population of the Central African Republic (CAR): evidence for three additional subtypes of genotype 4. J Med Virol. 1995;47:435–437. doi: 10.1002/jmv.1890470423. [DOI] [PubMed] [Google Scholar]
- 21.Okada S, Akahane Y, Suzuki H, Okamoto H, Mishiro S. The degree of variability in the amino terminal region of the E2/NS1 protein of hepatitis C virus correlates with responsiveness to interferon therapy in viremic patients. Hepatology. 1992;16:619–624. doi: 10.1002/hep.1840160302. [DOI] [PubMed] [Google Scholar]
- 22.Enomoto N, Kurosaki M, Tanaka Y, Marumo F, Sato C. Fluctuation of hepatitis C virus quasispecies in persistent infection and interferon treatment revealed by single-strand conformation polymorphism analysis. J Gen Virol. 1994;75(Pt 6):1361–1369. doi: 10.1099/0022-1317-75-6-1361. [DOI] [PubMed] [Google Scholar]
- 23.Kakumu S, Arao M, Yoshioka K, Hayashi H, Kusakabe A, Hirofuji H, Kawabe M. Recombinant human alpha-interferon therapy for chronic non-A, non-B hepatitis: second report. Am J Gastroenterol. 1990;85:655–659. [PubMed] [Google Scholar]
- 24.Hoofnagle JH, Mullen KD, Jones DB, Rustgi V, Di Bisceglie A, Peters M, Waggoner JG, Park Y, Jones EA. Treatment of chronic non-A,non-B hepatitis with recombinant human alpha interferon. A preliminary report. N Engl J Med. 1986;315:1575–1578. doi: 10.1056/NEJM198612183152503. [DOI] [PubMed] [Google Scholar]
- 25.Knapp S, Yee LJ, Frodsham AJ, Hennig BJ, Hellier S, Zhang L, Wright M, Chiaramonte M, Graves M, Thomas HC, et al. Polymorphisms in interferon-induced genes and the outcome of hepatitis C virus infection: roles of MxA, OAS-1 and PKR. Genes Immun. 2003;4:411–419. doi: 10.1038/sj.gene.6363984. [DOI] [PubMed] [Google Scholar]
- 26.Conrad B. Potential mechanisms of interferon-alpha induced autoimmunity. Autoimmunity. 2003;36:519–523. doi: 10.1080/08916930310001602137. [DOI] [PubMed] [Google Scholar]
- 27.Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, Goodman ZD, Koury K, Ling M, Albrecht JK. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358:958–965. doi: 10.1016/s0140-6736(01)06102-5. [DOI] [PubMed] [Google Scholar]
- 28.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Gonçales FL, Häussinger D, Diago M, Carosi G, Dhumeaux D, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 29.Hadziyannis SJ, Sette H, Morgan TR, Balan V, Diago M, Marcellin P, Ramadori G, Bodenheimer H, Bernstein D, Rizzetto M, et al. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann Intern Med. 2004;140:346–355. doi: 10.7326/0003-4819-140-5-200403020-00010. [DOI] [PubMed] [Google Scholar]
- 30.Dalgard O, Mangia A. Short-term therapy for patients with hepatitis C virus genotype 2 or 3 infection. Drugs. 2006;66:1807–1815. doi: 10.2165/00003495-200666140-00003. [DOI] [PubMed] [Google Scholar]
- 31.von Wagner M, Huber M, Berg T, Hinrichsen H, Rasenack J, Heintges T, Bergk A, Bernsmeier C, Häussinger D, Herrmann E, et al. Peginterferon-alpha-2a (40KD) and ribavirin for 16 or 24 weeks in patients with genotype 2 or 3 chronic hepatitis C. Gastroenterology. 2005;129:522–527. doi: 10.1016/j.gastro.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 32.El-Zayadi AR, Attia M, Barakat EM, Badran HM, Hamdy H, El-Tawil A, El-Nakeeb A, Selim O, Saied A. Response of hepatitis C genotype-4 naïve patients to 24 weeks of Peg-interferon-alpha2b/ribavirin or induction-dose interferon-alpha2b/ribavirin/amantadine: a non-randomized controlled study. Am J Gastroenterol. 2005;100:2447–2452. doi: 10.1111/j.1572-0241.2005.00253.x. [DOI] [PubMed] [Google Scholar]
- 33.Kamal SM, El Tawil AA, Nakano T, He Q, Rasenack J, Hakam SA, Saleh WA, Ismail A, Aziz AA, Madwar MA. Peginterferon {alpha}-2b and ribavirin therapy in chronic hepatitis C genotype 4: impact of treatment duration and viral kinetics on sustained virological response. Gut. 2005;54:858–866. doi: 10.1136/gut.2004.057182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen MH, Keeffe EB. Prevalence and treatment of hepatitis C virus genotypes 4, 5, and 6. Clin Gastroenterol Hepatol. 2005;3:S97–S101. doi: 10.1016/s1542-3565(05)00711-1. [DOI] [PubMed] [Google Scholar]
- 35.Taylor DR, Shi ST, Romano PR, Barber GN, Lai MM. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science. 1999;285:107–110. doi: 10.1126/science.285.5424.107. [DOI] [PubMed] [Google Scholar]
- 36.Abid K, Quadri R, Negro F. Hepatitis C virus, the E2 envelope protein, and alpha-interferon resistance. Science. 2000;287:1555. doi: 10.1126/science.287.5458.1555a. [DOI] [PubMed] [Google Scholar]
- 37.Saito T, Ito T, Ishiko H, Yonaha M, Morikawa K, Miyokawa A, Mitamura K. Sequence analysis of PePHD within HCV E2 region and correlation with resistance of interferon therapy in Japanese patients infected with HCV genotypes 2a and 2b. Am J Gastroenterol. 2003;98:1377–1383. doi: 10.1111/j.1572-0241.2003.07469.x. [DOI] [PubMed] [Google Scholar]
- 38.Watanabe H, Nagayama K, Enomoto N, Itakura J, Tanabe Y, Sato C, Izumi N, Watanabe M. Amino acid substitutions in PKR-eIF2 phosphorylation homology domain (PePHD) of hepatitis C virus E2 protein in genotype 2a/2b and 1b in Japan and interferon efficacy. Hepatol Res. 2003;26:268–274. doi: 10.1016/s1386-6346(03)00164-5. [DOI] [PubMed] [Google Scholar]
- 39.Gaudy C, Lambelé M, Moreau A, Veillon P, Lunel F, Goudeau A. Mutations within the hepatitis C virus genotype 1b E2-PePHD domain do not correlate with treatment outcome. J Clin Microbiol. 2005;43:750–754. doi: 10.1128/JCM.43.2.750-754.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Polyak SJ, Nousbaum JB, Larson AM, Cotler S, Carithers RL, Gretch DR. The protein kinase-interacting domain in the hepatitis C virus envelope glycoprotein-2 gene is highly conserved in genotype 1-infected patients treated with interferon. J Infect Dis. 2000;182:397–404. doi: 10.1086/315720. [DOI] [PubMed] [Google Scholar]
- 41.Moribe T, Hayashi N, Kanazawa Y, Mita E, Fusamoto H, Negi M, Kaneshige T, Igimi H, Kamada T, Uchida K. Hepatitis C viral complexity detected by single-strand conformation polymorphism and response to interferon therapy. Gastroenterology. 1995;108:789–795. doi: 10.1016/0016-5085(95)90452-2. [DOI] [PubMed] [Google Scholar]
- 42.Toyoda H, Kumada T, Nakano S, Takeda I, Sugiyama K, Osada T, Kiriyama S, Sone Y, Kinoshita M, Hadama T. Quasispecies nature of hepatitis C virus and response to alpha interferon: significance as a predictor of direct response to interferon. J Hepatol. 1997;26:6–13. doi: 10.1016/s0168-8278(97)80002-5. [DOI] [PubMed] [Google Scholar]
- 43.López-Labrador FX, Ampurdanès S, Giménez-Barcons M, Guilera M, Costa J, Jiménez de Anta MT, Sánchez-Tapias JM, Rodés J, Sáiz JC. Relationship of the genomic complexity of hepatitis C virus with liver disease severity and response to interferon in patients with chronic HCV genotype 1b infection [correction of interferon] Hepatology. 1999;29:897–903. doi: 10.1002/hep.510290306. [DOI] [PubMed] [Google Scholar]
- 44.Macdonald A, Harris M. Hepatitis C virus NS5A: tales of a promiscuous protein. J Gen Virol. 2004;85:2485–2502. doi: 10.1099/vir.0.80204-0. [DOI] [PubMed] [Google Scholar]
- 45.Gale MJ, Korth MJ, Tang NM, Tan SL, Hopkins DA, Dever TE, Polyak SJ, Gretch DR, Katze MG. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997;230:217–227. doi: 10.1006/viro.1997.8493. [DOI] [PubMed] [Google Scholar]
- 46.Polyak SJ, Khabar KS, Paschal DM, Ezelle HJ, Duverlie G, Barber GN, Levy DE, Mukaida N, Gretch DR. Hepatitis C virus nonstructural 5A protein induces interleukin-8, leading to partial inhibition of the interferon-induced antiviral response. J Virol. 2001;75:6095–6106. doi: 10.1128/JVI.75.13.6095-6106.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Polyak SJ, Khabar KS, Rezeiq M, Gretch DR. Elevated levels of interleukin-8 in serum are associated with hepatitis C virus infection and resistance to interferon therapy. J Virol. 2001;75:6209–6211. doi: 10.1128/JVI.75.13.6209-6211.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Enomoto N, Sakuma I, Asahina Y, Kurosaki M, Murakami T, Yamamoto C, Izumi N, Marumo F, Sato C. Comparison of full-length sequences of interferon-sensitive and resistant hepatitis C virus 1b. Sensitivity to interferon is conferred by amino acid substitutions in the NS5A region. J Clin Invest. 1995;96:224–230. doi: 10.1172/JCI118025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Enomoto N, Sakuma I, Asahina Y, Kurosaki M, Murakami T, Yamamoto C, Ogura Y, Izumi N, Marumo F, Sato C. Mutations in the nonstructural protein 5A gene and response to interferon in patients with chronic hepatitis C virus 1b infection. N Engl J Med. 1996;334:77–81. doi: 10.1056/NEJM199601113340203. [DOI] [PubMed] [Google Scholar]
- 50.Pascu M, Martus P, Höhne M, Wiedenmann B, Hopf U, Schreier E, Berg T. Sustained virological response in hepatitis C virus type 1b infected patients is predicted by the number of mutations within the NS5A-ISDR: a meta-analysis focused on geographical differences. Gut. 2004;53:1345–1351. doi: 10.1136/gut.2003.031336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murakami T, Enomoto N, Kurosaki M, Izumi N, Marumo F, Sato C. Mutations in nonstructural protein 5A gene and response to interferon in hepatitis C virus genotype 2 infection. Hepatology. 1999;30:1045–1053. doi: 10.1002/hep.510300405. [DOI] [PubMed] [Google Scholar]
- 52.Bagaglio S, Bruno R, Lodrini S, De Mitri MS, Andreone P, Loggi E, Galli L, Lazzarin A, Morsica G. Genetic heterogeneity of hepatitis C virus (HCV) in clinical strains of HIV positive and HIV negative patients chronically infected with HCV genotype 3a. J Biol Regul Homeost Agents. 2003;17:153–161. [PubMed] [Google Scholar]
- 53.Castelain S, Khorsi H, Roussel J, François C, Jaillon O, Capron D, Penin F, Wychowski C, Meurs E, Duverlie G. Variability of the nonstructural 5A protein of hepatitis C virus type 3a isolates and relation to interferon sensitivity. J Infect Dis. 2002;185:573–583. doi: 10.1086/339051. [DOI] [PubMed] [Google Scholar]
- 54.Inchauspe G, Zebedee S, Lee DH, Sugitani M, Nasoff M, Prince AM. Genomic structure of the human prototype strain H of hepatitis C virus: comparison with American and Japanese isolates. Proc Natl Acad Sci USA. 1991;88:10292–10296. doi: 10.1073/pnas.88.22.10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duverlie G, Khorsi H, Castelain S, Jaillon O, Izopet J, Lunel F, Eb F, Penin F, Wychowski C. Sequence analysis of the NS5A protein of European hepatitis C virus 1b isolates and relation to interferon sensitivity. J Gen Virol. 1998;79(Pt 6):1373–1381. doi: 10.1099/0022-1317-79-6-1373. [DOI] [PubMed] [Google Scholar]
- 56.Veillon P, Payan C, Le Guillou-Guillemette H, Gaudy C, Lunel F. Quasispecies evolution in NS5A region of hepatitis C virus genotype 1b during interferon or combined interferon-ribavirin therapy. World J Gastroenterol. 2007;13:1195–1203. doi: 10.3748/wjg.v13.i8.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Puig-Basagoiti F, Forns X, Furcić I, Ampurdanés S, Giménez-Barcons M, Franco S, Sánchez-Tapias JM, Saiz JC. Dynamics of hepatitis C virus NS5A quasispecies during interferon and ribavirin therapy in responder and non-responder patients with genotype 1b chronic hepatitis C. J Gen Virol. 2005;86:1067–1075. doi: 10.1099/vir.0.80526-0. [DOI] [PubMed] [Google Scholar]
- 58.Houghton M. Hepatitis C virus. In: Howley DMKPM, editor. Fields Virology. Philadelphia: Lippincott-Raven; 1996. pp. 1035–1058. [Google Scholar]
- 59.Farci P, Purcell RH. Clinical significance of hepatitis C virus genotypes and quasispecies. Semin Liver Dis. 2000;20:103–126. [PubMed] [Google Scholar]
- 60.Bartenschlager R. The NS3/4A proteinase of the hepatitis C virus: unravelling structure and function of an unusual enzyme and a prime target for antiviral therapy. J Viral Hepat. 1999;6:165–181. doi: 10.1046/j.1365-2893.1999.00152.x. [DOI] [PubMed] [Google Scholar]
- 61.Vallet S, Gouriou S, Nousbaum JB, Legrand-Quillien MC, Goudeau A, Picard B. Genetic heterogeneity of the NS3 protease gene in hepatitis C virus genotype 1 from untreated infected patients. J Med Virol. 2005;75:528–537. doi: 10.1002/jmv.20302. [DOI] [PubMed] [Google Scholar]
- 62.Vallet S, Gouriou S, Nkontchou G, Hotta H, Vilerio M, Legrand-Quillien MC, Beaugrand M, Trinchet JC, Nousbaum JB, Dény P, et al. Is hepatitis C virus NS3 protease quasispecies heterogeneity predictive of progression from cirrhosis to hepatocellular carcinoma? J Viral Hepat. 2007;14:96–106. doi: 10.1111/j.1365-2893.2006.00773.x. [DOI] [PubMed] [Google Scholar]
- 63.Söderholm J, Ahlén G, Kaul A, Frelin L, Alheim M, Barnfield C, Liljeström P, Weiland O, Milich DR, Bartenschlager R, et al. Relation between viral fitness and immune escape within the hepatitis C virus protease. Gut. 2006;55:266–274. doi: 10.1136/gut.2005.072231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gale M. Effector genes of interferon action against hepatitis C virus. Hepatology. 2003;37:975–978. doi: 10.1053/jhep.2003.50201. [DOI] [PubMed] [Google Scholar]
- 65.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 66.Kieffer T, Sarrazin C, Bartels D, Hanzelka B, Müh U, Welker M, Wincheringer D, Lin C, Purdy S, Weegink C, et al. Wild-type HCV NS3 protease re-emerges during follow-up after 14 days of dosing with VX-950 in patients with genotype 1 HCV. In: Colombo M. Journal of Hepatology. EASL; 2006. pp. Proceedings of the 41th Annual Meeting of the European Association for the Study of the Liver; 2006 Apr 26–30; Vienna, Austria. Shannon, Ireland: Elsevier, 2006: 44: S7. [Google Scholar]
- 67.Zeuzem S, Sarrazin C, Rouzier R, Tarral A, Brion N, Forestier N, Gupta S, Deckman D, Fellows K, Hussain M, et al. Anti- viral activity of SCH503034, a HCV protease inhibitor, administrated as monotherapy in hepatitis C genotype-1 (HCV-1) patients refractory to pegylated interferon (peg-IFN-α) In: Blei AT. Hepatology. AASLD; 2005. pp. Proceedings of the 56th Annual Meeting of the American Association for the Study of Liver Diseases.; 11–15 November 2005; San Francisco, USA. Hoboken: John Wiley & Sons, 2005: 42: 233A. [Google Scholar]
- 68.Clavel F, Hance AJ. HIV drug resistance. N Engl J Med. 2004;350:1023–1035. doi: 10.1056/NEJMra025195. [DOI] [PubMed] [Google Scholar]
- 69.Lin C, Lin K, Luong YP, Rao BG, Wei YY, Brennan DL, Fulghum JR, Hsiao HM, Ma S, Maxwell JP, et al. In vitro resistance studies of hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061: structural analysis indicates different resistance mechanisms. J Biol Chem. 2004;279:17508–17514. doi: 10.1074/jbc.M313020200. [DOI] [PubMed] [Google Scholar]
- 70.Sarrazin C, Kieffer T, Bartels D, Hanzelka B, Müh U, Welker M, Wincheringer D, Lin C, Grossman T, Purdy S, et al. Characterization of viral variants in the HCV NS3 protease domain of genotype 1 patients that are selected during 14 days of dosing with VX-950. In: Blei AT. Hepatology. AASLD; 2005. pp. Proceedings of the 56th Annual Meeting of the American Association for the Study of Liver Diseases; 2005 Nov 11–15; San Francisco, USA. Hoboken: John Wiley & Sons, 2005: 42: 751A. [Google Scholar]
- 71.Franco S, Clotet B, Martinez M. High genotypic and phenotypic resolution of HCV NS3/4 protease quasispecies: mutations associated with drug resistance in treatment-naive patients. In: Lange J RD. Antiviral Therapy. HIV Drug Resistance Worshop; 2006. pp. Proceedings of the 15th International HIV Drug resistance Workshop; 2006 Jun 13–17; Sitges, Spain. London: International Medical Press, 2006: 11: S14. [Google Scholar]
- 72.Winters MA, Welles SL, Holodniy M. Hepatitis C virus protease gene diversity in patients coinfected with human immunodeficiency virus. J Virol. 2006;80:4196–4199. doi: 10.1128/JVI.80.8.4196-4199.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tong X, Chase R, Skelton A, Chen T, Wright-Minogue J, Malcolm BA. Identification and analysis of fitness of resistance mutations against the HCV protease inhibitor SCH 503034. Antiviral Res. 2006;70:28–38. doi: 10.1016/j.antiviral.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 74.Reesink H, Zeuzem S, Weegink C, Forestier N, van Vliet A, van de Wetering de Rooij J, McNair L, Purdy S, Chu H, Jansen P. Final results of a phase 1b, multiple-dose study of VX-950, a hepatitis C virus protease inhibitor. In: Blei AT. Hepatology. AASLD; 2005. pp. Proceedings of the 56th Annual Meeting of the American association for the Study of Liver Diseases; 2005 Nov 11–15; San Francisco, USA. Hoboken: John Wiley & Sons, 2005: 42: 234A. [Google Scholar]
- 75.Yi M, Tong X, Skelton A, Chase R, Chen T, Prongay A, Bogen SL, Saksena AK, Njoroge FG, Veselenak RL, et al. Mutations conferring resistance to SCH6, a novel hepatitis C virus NS3/4A protease inhibitor. Reduced RNA replication fitness and partial rescue by second-site mutations. J Biol Chem. 2006;281:8205–8215. doi: 10.1074/jbc.M510246200. [DOI] [PubMed] [Google Scholar]
- 76.Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat Struct Biol. 1999;6:937–943. doi: 10.1038/13305. [DOI] [PubMed] [Google Scholar]
- 77.Bressanelli S, Tomei L, Roussel A, Incitti I, Vitale RL, Mathieu M, De Francesco R, Rey FA. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc Natl Acad Sci USA. 1999;96:13034–13039. doi: 10.1073/pnas.96.23.13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lohmann V, Körner F, Herian U, Bartenschlager R. Biochemical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. J Virol. 1997;71:8416–8428. doi: 10.1128/jvi.71.11.8416-8428.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lohmann V, Roos A, Körner F, Koch JO, Bartenschlager R. Biochemical and kinetic analyses of NS5B RNA-dependent RNA polymerase of the hepatitis C virus. Virology. 1998;249:108–118. doi: 10.1006/viro.1998.9311. [DOI] [PubMed] [Google Scholar]
- 80.Cheney IW, Naim S, Lai VC, Dempsey S, Bellows D, Walker MP, Shim JH, Horscroft N, Hong Z, Zhong W. Mutations in NS5B polymerase of hepatitis C virus: impacts on in vitro enzymatic activity and viral RNA replication in the subgenomic replicon cell culture. Virology. 2002;297:298–306. doi: 10.1006/viro.2002.1461. [DOI] [PubMed] [Google Scholar]
- 81.Labonté P, Axelrod V, Agarwal A, Aulabaugh A, Amin A, Mak P. Modulation of hepatitis C virus RNA-dependent RNA polymerase activity by structure-based site-directed mutagenesis. J Biol Chem. 2002;277:38838–38846. doi: 10.1074/jbc.M204657200. [DOI] [PubMed] [Google Scholar]
- 82.Vuillermoz I, Khattab E, Sablon E, Ottevaere I, Durantel D, Vieux C, Trepo C, Zoulim F. Genetic variability of hepatitis C virus in chronically infected patients with viral breakthrough during interferon-ribavirin therapy. J Med Virol. 2004;74:41–53. doi: 10.1002/jmv.20144. [DOI] [PubMed] [Google Scholar]
- 83.Kumagai N, Takahashi N, Kinoshita M, Tsunematsu S, Tsuchimoto K, Saito H, Ishii H. Polymorphisms of NS5B protein relates to early clearance of hepatitis C virus by interferon plus ribavirin: a pilot study. J Viral Hepat. 2004;11:225–235. doi: 10.1111/j.1365-2893.2004.00501.x. [DOI] [PubMed] [Google Scholar]
- 84.Carroll SS, Tomassini JE, Bosserman M, Getty K, Stahlhut MW, Eldrup AB, Bhat B, Hall D, Simcoe AL, LaFemina R, et al. Inhibition of hepatitis C virus RNA replication by 2'-modified nucleoside analogs. J Biol Chem. 2003;278:11979–11984. doi: 10.1074/jbc.M210914200. [DOI] [PubMed] [Google Scholar]
- 85.Carroll SS, Olsen DB. Nucleoside analog inhibitors of hepatitis C virus replication. Infect Disord Drug Targets. 2006;6:17–29. doi: 10.2174/187152606776056698. [DOI] [PubMed] [Google Scholar]
- 86.Migliaccio G, Tomassini JE, Carroll SS, Tomei L, Altamura S, Bhat B, Bartholomew L, Bosserman MR, Ceccacci A, Colwell LF, et al. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J Biol Chem. 2003;278:49164–49170. doi: 10.1074/jbc.M305041200. [DOI] [PubMed] [Google Scholar]
- 87.Ludmerer SW, Graham DJ, Boots E, Murray EM, Simcoe A, Markel EJ, Grobler JA, Flores OA, Olsen DB, Hazuda DJ, et al. Replication fitness and NS5B drug sensitivity of diverse hepatitis C virus isolates characterized by using a transient replication assay. Antimicrob Agents Chemother. 2005;49:2059–2069. doi: 10.1128/AAC.49.5.2059-2069.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Klumpp K, Lévêque V, Le Pogam S, Ma H, Jiang WR, Kang H, Granycome C, Singer M, Laxton C, Hang JQ, et al. The novel nucleoside analog R1479 (4'-azidocytidine) is a potent inhibitor of NS5B-dependent RNA synthesis and hepatitis C virus replication in cell culture. J Biol Chem. 2006;281:3793–3799. doi: 10.1074/jbc.M510195200. [DOI] [PubMed] [Google Scholar]
- 89.Le Pogam S, Jiang WR, Leveque V, Rajyaguru S, Ma H, Kang H, Jiang S, Singer M, Ali S, Klumpp K, et al. In vitro selected Con1 subgenomic replicons resistant to 2'-C-methyl-cytidine or to R1479 show lack of cross resistance. Virology. 2006;351:349–359. doi: 10.1016/j.virol.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 90.Roberts S, Cooksley G, Shaw D, Berns H, Brandl M, Fettner S, Hill G, Ipe D, Klumpp K, Mannino M, et al. Interim results of a multiple ascending dose study of R1626, a novel nucleoside analog targeting HCV polymerase in chronic HCV patients. In: Colombo M. Journal of Hepatology. EASL; 2006. pp. Proceedings of the 41th European Association for the Study of the Liver; 2006 Apr 26–30; Vienna, Austria. Shannon: Elsevier, 2006: 44: S269. [Google Scholar]
- 91.Chan L, Pereira O, Reddy TJ, Das SK, Poisson C, Courchesne M, Proulx M, Siddiqui A, Yannopoulos CG, Nguyen-Ba N, et al. Discovery of thiophene-2-carboxylic acids as potent inhibitors of HCV NS5B polymerase and HCV subgenomic RNA replication. Part 2: tertiary amides. Bioorg Med Chem Lett. 2004;14:797–800. doi: 10.1016/j.bmcl.2003.10.068. [DOI] [PubMed] [Google Scholar]
- 92.Mo H, Lu L, Pilot-Matias T, Pithawalla R, Mondal R, Masse S, Dekhtyar T, Ng T, Koev G, Stoll V, et al. Mutations conferring resistance to a hepatitis C virus (HCV) RNA-dependent RNA polymerase inhibitor alone or in combination with an HCV serine protease inhibitor in vitro. Antimicrob Agents Chemother. 2005;49:4305–4314. doi: 10.1128/AAC.49.10.4305-4314.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Howe AY, Bloom J, Baldick CJ, Benetatos CA, Cheng H, Christensen JS, Chunduru SK, Coburn GA, Feld B, Gopalsamy A, et al. Novel nonnucleoside inhibitor of hepatitis C virus RNA-dependent RNA polymerase. Antimicrob Agents Chemother. 2004;48:4813–4821. doi: 10.1128/AAC.48.12.4813-4821.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kukolj G, McGibbon GA, McKercher G, Marquis M, Lefèbvre S, Thauvette L, Gauthier J, Goulet S, Poupart MA, Beaulieu PL. Binding site characterization and resistance to a class of non-nucleoside inhibitors of the hepatitis C virus NS5B polymerase. J Biol Chem. 2005;280:39260–39267. doi: 10.1074/jbc.M506407200. [DOI] [PubMed] [Google Scholar]
- 95.Tomei L, Altamura S, Paonessa G, De Francesco R, Migliaccio G. HCV antiviral resistance: the impact of in vitro studies on the development of antiviral agents targeting the viral NS5B polymerase. Antivir Chem Chemother. 2005;16:225–245. doi: 10.1177/095632020501600403. [DOI] [PubMed] [Google Scholar]
- 96.Graham DJ, Stahlhut M, Flores O, Olsen DB, Hazuda DJ, Lafemina RL, Ludmerer SW. A genotype 2b NS5B polymerase with novel substitutions supports replication of a chimeric HCV 1b: 2b replicon containing a genotype 1b NS3-5A background. Antiviral Res. 2006;69:24–30. doi: 10.1016/j.antiviral.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 97.De Francesco R, Migliaccio G. Challenges and successes in developing new therapies for hepatitis C. Nature. 2005;436:953–960. doi: 10.1038/nature04080. [DOI] [PubMed] [Google Scholar]