

Abstract
Nonalcoholic fatty liver disease (NAFLD) is an increasingly recognized cause of liver-related morbidity and mortality. It can develop secondary to numerous causes but a great majority of NAFLD cases occur in patients who are obese or present with other components of metabolic syndrome (hypertension, dyslipidemia, diabetes). This is called primary NAFLD and insulin resistance plays a key role in its pathogenesis. Obesity is characterized by expanded adipose tissue, which is under a state of chronic inflammation. This disturbs the normal storage and endocrine functions of adipose tissue. In obesity, the secretome (adipokines, cytokines, free fatty acids and other lipid moieties) of fatty tissue is amplified, which through its autocrine, paracrine actions in fat tissue and systemic effects especially in the liver leads to an altered metabolic state with insulin resistance (IR). IR leads to hyperglycemia and reactive hyperinsulinemia, which stimulates lipid-accumulating processes and impairs hepatic lipid metabolism. IR enhances free fatty acid delivery to liver from the adipose tissue storage due to uninhibited lipolysis. These changes result in hepatic abnormal fat accumulation, which may initiate the hepatic IR and further aggravate the altered metabolic state of whole body. Hepatic steatosis can also be explained by the fact that there is enhanced dietary fat delivery and physical inactivity. IR and NAFLD are also seen in various lipodystrophic states in contrary to popular belief that these problems only occur due to excessive adiposity in obesity. Hence, altered physiology of adipose tissue is central to development of IR, metabolic syndrome and NAFLD.
Keywords: Nonalcoholic fatty liver disease, Obesity, Adipose tissue, Adipokines, Insulin resistance
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is a disease of this modern era. It has burst out onto the clinical landscape over the past 25 years[1] and is likely to become the most common cause of chronic liver disease[2]. The prevalence of NAFLD in the general US population is approximately 20% (17% to 33%)[3,4]. Similar data are obtained in the Japanese and Italian populations[5,6]. NAFLD comprises a morphological spectrum of liver lesions ranging from simple triglyceride accumulation in hepatocytes (hepatic steatosis; HS) to inflammatory and hepatocellular ballooning injury (non-alcoholic steatohepatitis; NASH), eventually leading to fibrosis and cirrhosis[7]. Excessive alcohol intake (> 20 mg/d in females and > 40 mg/d in males) must be excluded to diagnose NAFLD[8]. NAFLD is the most frequent cause of abnormal liver function tests (80% of cases) in the US[9]. Although elevated liver enzymes often correlate poorly with degree of chronic liver injury[10], steatosis and fibrosis, a persistently elevated alanine aminotransferase (ALT) level is often the tipping point for further diagnostic evaluation. With adoption of the new ALT standard (≤ 30 U/L in men and ≤ 19 U/L in women)[11,12], NAFLD can be detected in early stages and thus may be useful in counseling patients about further workup. Liver biopsy is subsequently required to confirm the diagnosis of NASH. Recently, a NAFLD activity score (NAS) was proposed[13] to assess natural progression and therapy; however, it requires repeated liver biopsies (Table 1). Recently, it has been suggested that the non-invasive blood test called NASH FibroSURE TMwhich includes quantitative surrogate markers for steatosis (SteatoTest)[14], fibrosis (FibroTest)[15] and NASH (NASH Test)[16] is able to assess the liver status of subjects with NAFLD. These have a sensitivity/specificity of 71%/72% for significant steatosis; 88%/50% for NASH and 83%/78% for advanced fibrosis. Further novel non-invasive tests are needed that will improve the diagnostic accuracy for the presence, grade and stage of NAFLD.
Table 1.
Histological scoring and staging system for NASH
| Steatosis Macrovesicular |
Steatohepatitis |
Stages of NASH Fibrosis | ||
| Lobular Inflammation | Ballooning | |||
| Definition and score | Large fat droplet with signet cell appearance of hepatocytes | Assessment of inflammatory foci on 200 × field | Swollen hepatocytes | On trichome staining |
| 0 | Less than 5% | Less than 2 foci | None | None |
| 1 | 5% to 33% | 2 to 4 foci | Few | Perisinusoidal (zone 3) or periportal 1a-mild, zone 3 1b-moderate, zone 3 1c-portal or periportal |
| 2 | 33% to 66% | More than 4 foci | Many cells or prominent ballooning | Perisinusoidal (zone 3) and portal/periportal |
| 3 | More than 66% | - | - | Bridging |
| 4 | - | - | - | Cirrhosis |
The pathophysiology of NAFLD is complex and available data suggest that environmental factors such as diet, exercise, and/or toxins[17] are likely to be important in its causation. The fact that the prevalence of NAFLD varies among different racial groups[18,19] and that there are variable rates of disease progression within individuals with similar risk factors strongly indicates that genes also play a role[20]. A recent study suggests that abnormalities in at least 23 genes may be involved[21]. However, like other common complex metabolic diseases, there is likely to be an interaction between the environment and genetics that determines phenotypic expression of NAFLD in any individual patient. The great majority of NAFLD occurs in the setting of so-called metabolic syndrome (MS), in which insulin resistance (IR) plays a key role[22,23]. The prevalence of MS (22%) and of NAFLD (20%) in the general US population are amazingly similar[24]. These observations support that primary NAFLD is a hepatic complication of MS. Other much less common causes of NAFLD (secondary NAFLD) are drugs; amiodarone[25] glucocorticoids[26], tamoxifen[27] and isoniazid[28], surgical procedures; gastroplasty and bypass surgeries for morbid obesity[29-31], total parenteral nutrition[32], metabolic genetic disorders; abetalipoproteinemia[33], tyrosinemia and lipodystrophy; congenital disorders[34] and anti-HIV drugs[35].
The Third Report of National Cholesterol Education Program (Adult Treatment Panel III; ATPIII) provides a working definition of MS[36] based on a combination of 5 categorical risk factors: central obesity, hypertension, hypertriglyceridemia, low levels of HDL-cholesterol, and hyperglycemia. The prevalence of MS is on the rise due to obesity and sedentary life style. Obesity is defined by World Health Organization as excess weight gain for a given height. The prevalence of obesity (BMI ≥ 30 kg/m2) in the US is 31%, and the prevalence of morbid obesity (BMI ≥ 40 kg/m2) is growing even at a greater rate than that of obesity[37]. The most obvious body change in obesity is increase in the fat mass, which becomes a key pathological contributor to MS and is related to abnormal production of cytokines, chronic sub-clinical inflammatory state and abnormal coagulation. The prevalence of NAFLD increases to 74% in obese persons and 90% in morbidly obese persons[38,39]. The probability of NAFLD progression increases with the degree of obesity, and about 15%-30% of morbidly obese patients (BMI ≥ 35 kg/m2) have NASH[40].
The presence and severity of NAFLD/NASH are closely related to coexistence of MS components[41], obesity and other risk factors for IR[42]. This paper reviews the chemical and molecular basis of IR, highlighting the role of adipose tissue and adipocytokines in its development especially in obese people. Brief consideration is also given to normal lipid metabolic pathways of liver to explain the involvement of IR in pathogenic mechanisms of NAFLD.
NORMAL LIPID METABOLISM
After meals, dietary triglycerides (TAG) are transported to the liver from intestines (via chylomicrons). In addition, hepatic TAG synthesis from fatty acids (FA) and glycerol occurs under the influence of insulin in the postprandial state[43]. TAG are secreted into blood as very low density lipoproteins (VLDL) that are either stored in adipose tissue as re-esterified TAG or metabolized into FA and used as energy source. Excessive TAG within the liver may be stored as lipid droplets in hepatocytes (Figure 1). The sources of FA for hepatic TAG formation are either from the plasma (nonesterified fatty acid; NEFA) pool or FA newly synthesized within the liver through de novo lipogenesis (DNL). Metabolic steps of DNL are important, precisely regulated and involve mitochondria[44]. In the postprandial state of energy (Adenosine triphosphate; ATP) excess, surplus glucose is used as FA substrate. Glucose via its conversion to pyruvate enters the Kreb's cycle of mitochondria. Citrate formed in the Kreb's cycle is shuttled to cytosol where it is converted to acetyl-CoA by ATP citrate lyase. Acetyl-CoA carboxylase 1 (ACC1) enzyme then converts acetyl-CoA to malonyl-CoA, which is used by fatty acid synthase to form different long chain FA in cytosol. The hepatic uptake of FA from NEFA pool is not regulated and as a result the influx directly relates to plasma FA concentration. The affluent diets of modern age hence promote accumulation of surplus fat in the hepatocytes by providing more dietary TAG[45], FA (expanding NEFA pool) as well as glucose (substrate of DNL). Increased insulin levels after meals promote all of the steps of hepatic fat accumulation. Oxidation of FA occurs in mitochondria, peroxisomes, and microsomes. Short-chain and medium-chain FA are oxidized in mitochondria only[46]. Long-chain and very long-chain FA are shortened by the extramitochondrial (peroxisomes and microsomes) oxidation first and then mitochondrion enzymes complete the process. Entry of FA into mitochondria is the rate-limiting step of oxidation. FA must be activated by fatty acyl-CoA synthase to fatty acyl-CoA in the cytoplasm. The transport of fatty acyl-CoA into the mitochondria is accomplished by the intermediation of carnitine acyltransferaseI(CPT-1), an enzyme that resides in the outer mitochondrial membrane[47].
Figure 1.
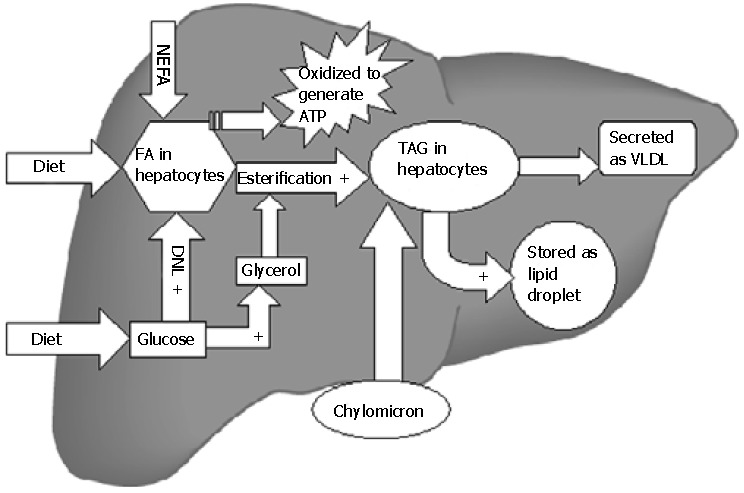
Lipid metabolism in liver. All steps indicated by + are stimulated by insulin. Insulin suppresses the secretion of VLDL and the β-oxidation of fatty acids. Thus hyperinsulinemia in the setting of insulin resistance favors TAG accumulation in liver. TAG: triglycerides; VLDL: very low density lipoprotein; FA: fatty acid; NEFA: nonesterified fatty acids; DNL: de novo lipogenesis; ATP: adenosine triphosphate.
Three molecular mediators of lipid metabolism namely: Sterol regulatory element-binding protein (SREBP1-c), carbohydrate response element-binding protein (ChREBP) and peroxisome proliferative activated receptor (PPAR-γ) are worth mentioning at this point. Insulin acts on SREBP1-c on hepatocyte cell membranes[48], which transcriptionally activates most of the genes involved in DNL. SREBP1-c also activates ACC-2, an isoform of ACC that produces malonyl-CoA at the mitochondrial membrane[49]. Increase in malonyl-CoA decreases β-oxidation because it inhibits CPT-1 at higher concentrations[50] and hence leads to FA build up. Hyperglycemia can also stimulate lipogenesis directly by activating ChREBP[51], which induces gene expression of liver-type pyruvate kinase (L-PK), a key regulatory enzyme in glycolysis[52]. L-PK catalyzes the conversion of phosphoenolpyruvate (PEP) to pyruvate, which enters the Kreb's cycle to generate citrate. Citrate is a principal source of acetyl-CoA used for FA synthesis. ChREBP stimulates gene expression of most enzymes involved in DNL[53]. Hence hyperglycemia stimulates both glycolysis and lipogenesis, thereby facilitating the conversion of glucose to FA under conditions of energy excess. PPAR-γ transcription factor participates in the development of HS in rodents. Normally, PPAR-γ is expressed at very low levels in the liver; however, in animal models with IR and fatty livers its expression is markedly increased[54]. Studies demonstrate that SREBP-1c can also transcriptionally activate PPAR-γ[55]. The genetic deletion of hepatic PPAR-γ in livers of ob/ob mice markedly alleviates the development of HS[56], independent of the presence of hyperinsulinemia or hyperglycemia. The precise molecular events mediated by PPAR-γ that promote hepatic TAG deposition have not been fully defined. It is also not known whether PPAR-γ expression is increased in human fatty livers.
INSULIN RESISTANCE
Insulin resistance (IR) may be defined as altered metabolic condition in which higher than normal insulin levels are needed to achieve normal metabolic responses or normal insulin concentrations fail to achieve a normal metabolic response. IR manifests with a reduced efficiency of insulin to inhibit hepatic glucose production and stimulate glucose utilization in skeletal muscle and adipose tissue. Subsequent high glucose levels result in compensatory hyperinsulinemia, which accounts for the elevation of Homeostasis Model Assessment (HOMA)-IR values[57,58]. Incidence of IR increases with age[59] and is almost always higher in males[60]. Other important conditions associated with IR include hyperuricaemia[61], gallstones[62], thrombophilia[63], endothelial dysfunction and polycystic ovary syndrome[64].
The biological action of insulin depends upon interaction of insulin with its specific receptor (Table 2). The insulin receptor is a glycosylated tetramer consisting of two extra cellular insulin binding (alpha) subunits and two trans-membranous (beta) subunits with tyrosine kinase activity. Insulin activates the receptor and causes subsequent tyrosine phosphorylation of insulin receptor substrate proteins (namely IRS-1 and IRS-2). This initiates a cascade of events leading to translocation of a specific glucose transporter-4 (GLUT-4) from intracellular pool to the cell membrane. GLUT-4 facilitates glucose transport along the concentration gradient from extracellular space into the cytoplasm of myocytes and adipocytes. The mechanisms responsible for IR may involve insulin binding, IRS proteins or even GLUT-4[65]. IR can be central (hepatic) or peripheral (muscle, fat tissue) depending upon the primary site of involvement. Peripheral IR impairs uptake of glucose from blood into skeletal muscles and adipose tissues. It manifests with increased free FA liberation from adipose tissue secondary to the unopposed antilipolytic action of insulin on hormone-sensitive lipase (HSL)[66] and is best measured through the euglycemic clamp technique[67]. Hepatic IR manifests with unrestrained hepatic glucose production resulting from impaired glycogen synthesis and failure of insulin to suppress gluconeogenesis[68]. Hepatic IR may also be caused by fat accumulation in hepatocytes itself[69]. Regarding the primary site of IR in NAFLD (peripheral vs hepatic), recent data indicate the periphery to be the initial site followed by or resulting in HS exacerbating hepatic IR and thus the degree of overall IR[70].
Table 2.
Pathophysiology of insulin resistance
| Actions of insulin | Mechanism of action of insulin | Alterations in insulin resistant states | Net metabolic effect |
| (a) Stimulatory Increases glucose transport: In adipocytes In myocytes | -Insulin binds to its membrane receptor to cause up regulation of GLUT-4 via mediation of IRS-1/2(activated by phosphorylation at tyrosine sites) | -Impaired post receptor signaling involving IRS proteins -Abnormal phosphorylation of IRS-1 makes it inhibitor of the receptor kinase -Activation of IKK-β by free FA and cytokines leads to activation of NF-κB which further inhibits the genes involved in GLUT synthesis | -Hyperglycemia -Decreased glucose utilization as energy source -Reactive hyperinsulinemia |
| Increases glycogenesis In hepatocytes In myocytes | -By providing the building blocks -Increases expression and activity of glycogen synthase and inhibiting the glycogenolytic enzymes | -Decreased glycogen synthesis | -Hyperglycemia -Decreased postprandial glycogen stores in liver |
| Increases lipogenesis In adipose tissue In liver (DNL) | -Increases the supply of substrates -Postprandial stimulation of FAS, ACC and SCD1 -Increases supply of free FA in AT | -Further increase in lipogenesis,esp. DNL -Increased delivery of free FA to liver -Decreased oxidation in hepatocytes | -Excessive fat storage in AT and in other tissues (lipotoxicity) -Hepatic steatosis -Increased adiposity |
| Increases protein synthesis in muscles | -Activates the translational machinery -Activates protein kinase B which activates the protein synthesizing enzymes -In long term exposure increases ribosome in cells | -Decreased protein synthesis | -Sarcopenia |
| Increases glucose oxidative pathways | -Enhances glycolysis and Kreb's cycle by activating all the key regulator enzymes | -Inhibited -Lipid oxidation preferentially used for energy purposes | -Hyperglycemia -Oxidative stress in hepatocytes |
| (b) Inhibitory Decreases gluconeogenesis in liver | -Inhibits pyruvate carboxylase, glucose 6 phosphatase and PEP kinase -Shuttles substrates to lipogenesis | -Enhanced gluconeogenesis -Decreased inhibition of keyregulatory enzymes -Activation of AMPK | -Increased hepatic glucose output -Excessive availability of substrates for lipogenesis -Fasting hyperglycemia |
| Decreases hepatic glucose output | -Decreases gluconeogenesis -Increases glycogen synthesis -Increases oxidation of glucose | -Increased gluconeogenesis -Decreased glycogenesis and oxidative disposal of glucose | -Hyperglycemia |
| Suppresses lipolysis in adipose tissue | -Suppression of HSL | -Increased rate of free FA release in fasting state in lean and obese -When corrected for body weight in obese, postprandial lipolysis may seem to be normal or even decreased | -Increased plasma free FA both in fasting and post-prandial states(May be due to a mass effect of overall expansion of body fat depots in case of obese) -Increased free FA efflux -Increased VLDL |
| Decreases apolipoprotein secretion | -Insulin decreases the synthesis and secretion of Apo-B and Apo-C | -Hyperinsulinemia causes further suppression of expression of apolipoprotein genes and also inhibits post translational modifications and secretion -Enhanced synthesis of Apo-B 48 in intestines | -Trapping of TAG inside the liver -Hepatic steatosis -Increased VLDL |
| Suppresses β oxidation of fatty acids | -Insulin acts via binding to SREBP-1 transcription factor to cause increased expression of ACC-1 leading to generation of FAS substrates for lipogenesis | -Reactive hyperinsulinemia with unrestricted effect on SREBP causes further inhibition of β-oxidation of free FA in hepatocytes mitochondria | -Hepatic steatosis -CYP system over expression and generation of ROS |
Hyperinsulinemia and hyperglycemia are two main net metabolic effects of IR. IR also contributes to expansion of NEFA pool and is now alone considered to be responsible for both the hits of 'two hit hypotheses' of NAFLD. IKK-β: Inhibitor of kappaB kinase beta; NF-κB: Nuclear factor-kappa B; FAS: Fatty acyl synthase; ACC: Acyl-CoA carboxylase; SCD: stearoyl-CoA desaturase; PEP: Phosphoenol pyruvate; CYP: Cytochrome P. FA: fatty acids; AT: adipose tissue; HSL: hormone sensitive lipase; SREBP: sterol regulatory element binding protein;IRS:receptor substrate proteins; GLUT: glucose transporter; AMPK: adenosine monophosphate kinase; ROS: reactive oxygen species.
Organ-specific deposition of fat is a strong predictor of hyperinsulinemia and IR. Analogous to fat in the liver, increased intramyocellular TAG content closely correlates with muscle IR. The aberrantly high availability of NEFA reduces muscle use of glucose (main consumer of glucose in the body)[71]. The resultant hyperglycemia potentiates glucose-stimulated insulin secretion, which chronically burns out β-cells. This explains the link between obesity, IR and development of type 2 diabetes.
The metabolic and cellular mechanism(s) linking IR to NAFLD are complex and are not fully understood. There are several distinct clinical syndromes associated with severe IR, which are also associated with NAFLD, e.g. obesity, diabetes mellitus and lipoatrophy[72,73]. The severity of NAFLD improves after pharmacological and non-pharmacological interventions aiming at restoring insulin sensitivity[74]. This demonstrates the role of IR in pathogenesis of NAFLD and a 'two hit' process has been proposed to explain it[75].
Two hit hypothesis
Non-alcoholic fatty liver disease represents a continuum of hepatic injuries, which progress from simple fatty liver (FL or HS) to hepatocellular ballooning degeneration, formation of Mallory bodies and fibrosis (NASH). The first hit involves accumulation of TAG in hepatocytes as described above. It has also been recognized that HS in itself leads to hepatic IR by activating protein kinase-theta (PKC-theta;) and Jun N-terminal kinase (JNK)[65,76], which interfere with tyrosine phosphorylation of IRS-1 and IRS-2 and impairs insulin action in hepatocytes[77]. Steatosis and IR can cause and potentiate each other creating a vicious cycle of metabolic dysfunction.
Once the presence of hepatic steatosis is established, progression to steatohepatitis involves a 'second hit' and oxidative stress is thought to play a key role. Fatty liver is more susceptible to oxidative injury[78]. Oxidative stress results from an imbalance of prooxidants (enhanced reactive oxygen species; ROS/reactive nitrogen species; RNS) and antioxidants (nutritional deficiencies) and leads to lipid peroxidation[79]. The chemical modification of biologic molecules may be directly toxic to the cells or may stimulate host-immune response that leads to inflammation, collagen production and further progression of disease[80-82]. The factors promoting prooxidant generation include mitochondrial dysfunction (NADPH oxidase, electron transport chain leakage)[83] and induction of hepatic cytochrome (microsomal, peroxisomal CYP2E1)[84,85] resulting from overburden of FA oxidation system (described above)[86]. It is now believed that IR itself may predispose to oxidative stress by stimulating microsomal lipid peroxidases and by decreasing mitochondrial β-oxidation[81]. CYP2E1 is normally suppressed by insulin but is invariably increased in the livers of patients with NASH[87] and in rodent dietary models of steatohepatitis CYP2E1 is the catalyst of microsomal lipid peroxidation[88]. Attention should now shift from considering hepatic steatosis as a benign process to an important unhealthy condition, which leads to IR, oxidative stress and further progression of NAFLD. This is evident from the fact that severe forms of NASH correlate well with presence of hepatic steatosis, visceral obesity and MS[89].
ADIPOSE TISSUE AND OBESITY
Earlier it was generally believed that the purpose of adipose tissue (AT) was simply a repository of fat. The first report suggesting the fat was an active metabolic tissue was published by Von Gierke[90]. Presently it is considered an endocrine organ and a key regulator of both metabolism and inflammation[91]. In adult mammals, the major bulk of AT is a loose association of lipid-filled adipocytes which are held in a framework of collagen (stroma) containing stromal-vascular cells, fibroblastic connective tissue cells, leukocytes, macrophages and pre-adipocytes (not yet filled with lipid). Approximately 60% to 85% of the weight of white adipose tissue is lipid, with 90%-99% being TAG. Small amounts of free FA, diglycerides, cholesterol and phospholipids are also present. AT is the only organ with unlimited growth potential at any stage of our life. The size of adipose tissue mass is a function of both adipocyte number and size. It can expand by hyperplastic (increase in number) and hypertrophic (increase in the size of adipocytes) growth. Hypertrophy occurs primarily by lipid accumulation within the cell and is reversible, however once adipocyte hyperplasia occurs, they remain throughout the life. Adipocytes are specially adapted for the uptake and release of energy in the form of FA. Fatty acids are converted to TAG inside adipocytes which accumulate as surplus fuel during caloric abundance and released as NEFA back to the circulation as needed in the periods of food shortage and calorie debt (e.g. fasting, starvation, exercise). Adipose tissue is extensively supplied with blood circulation. Insulin plays a major role in the control of AT development and function. It not only regulates lipogenesis but also the rate of lipolysis and NEFA efflux. Adipose tissue in adults can be divided into two types depending on its location: subcutaneous and visceral (intraperitoneal: omental and mesenteric fat). Both fat depots differ in pathophysiology. Insulin actions are blunted in omental compared with subcutaneous AT[92] and can be explained by the increased endogenous protein tyrosine phosphatase 1B (which down regulates the insulin receptors) levels in omental adipocytes[93]. Visceral adiposity is more strongly correlated with NALFD/MS and is measured by waist-to-hip ratio[94]. CT and MRI are the gold standards for measuring visceral fat[95].
ROLE OF ADIPOSE TISSUE IN NON-ALCOHOLIC FATTY LIVER DISEASE
Excess adipose tissue predisposes towards the development of IR by virtue of its secreted factors[96]. Well over 100 secreted factors have been identified. Adipokines are biologically active proteins secreted both by cellular and stromal fractions of AT. In addition adipocytes also secrete FA, cytokines, cholesterol, steroid hormones and prostaglandins[97]. Secretion of all factors except adiponectin increases as AT enlarges in obesity[98]. Various stimuli have been proposed to explain the overproduction of adipokines in obesity. These include increase in mass, chronic inflammation with infiltration of AT by macrophages, hypoxia (growth of adipose tissue ahead of angiogenesis), endoplasmic reticulum stress and oxidative stress[99]. This increase in synthesis is however reversible and decreases with weight loss[100].
The links between adiposity and development of IR and NAFLD thereafter can be explained by the following hypotheses.
Portal/visceral hypothesis
From an anatomical perspective, visceral adipocytes can be a crucial source of FA and other factors entering portal circulation[101]. FA are elevated in sera of obese and diabetic patients. Like protein hormones free FA are also thought to be potent signaling molecules[102]. Enhanced delivery of FA from enlarged visceral adipose tissue to the liver leads to reduced hepatic insulin clearance with further increase of circulating insulin levels[103]. FA stimulate hepatic gluconeogenesis, TAG synthesis and impair insulin suppression of hepatic glucose output[103]. Intrahepatic lipids increase by 22% for any 1% increase in total, by 21% for any 1% increase in subcutaneous and by 104% for 1% increase in intra-abdominal adipose tissue[104]. NEFA levels are raised in the peripheral blood of NASH patients[105], however measurements of portal NEFA levels are still needed to justify the portal theory (Figure 2).
Figure 2.
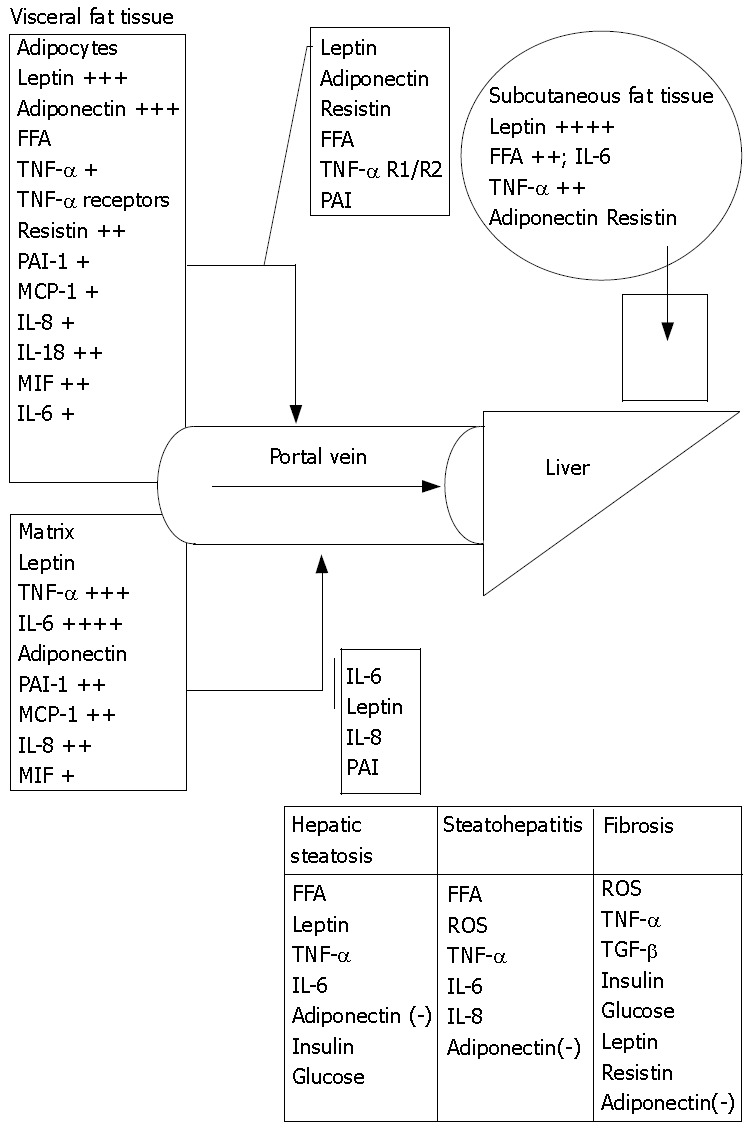
Portal/Visceral hypothesis. Human adipose tissue (AT) is a potent source of inflammatory cytokines and that the majority of this release is due to the nonfat cells in the AT except for leptin and adiponectin that are primarily secreted by adipocytes. Adipocytes secrete at least as much PAI-1 (plasminogen activator inhibitor-1), MCP-1 (macrophage chemotaxis protein), IL-8 (Interleukin), and IL-6 in vitro as they do leptin but the nonfat cells of AT secrete even more of these proteins. The secretion of leptin by the nonfat cells is negligible. Obesity markedly elevates the total release of TNF-α, IL-6, and IL-8 by AT. Visceral fat releases more resistin, IL-6, PAI-1, TGF β1, IL-8 and IL-10 per gram of tissue than subcutaneous fat. (+) indicates fold increase in secretion in obesity; (-) indicates protective effect.
Endocrine paradigm
Adipocytokines have wide-ranging effects on food intake, energy expenditure and metabolism. These are secreted by one cell for the purpose of altering either its own function (autocrine effect) or those of adjacent cells (paracrine effect). In many instances, individual cytokines have multiple biologic activities. Different cytokines have similar activity, which provides biologic redundancy within the inflammatory and immune systems. As a result, it is infrequent that loss or neutralization of one cytokine will markedly interfere with either of these systems. This fact has great significance in the studies of genetic knock out animals. FA released from adipose tissue have a variety of systemic effects including induction of peripheral IR. In myocytes free FA compete with glucose for its entry and utilization[106] and as a consequence, lead to ATP depletion, diminished GLUT-4 translocation and reduced muscular glycogen synthesis[107].
Leptin: Leptin is the first adipokine that was described[108]. Encoded by "ob" gene, it is primarily synthesized and released into the circulation by mature adipocyte in response to changes in body fat mass and nutritional status[109]. In hypothalamus, leptin stimulates anorexigenic pathways and decreases food intake[110]. Circulating levels of leptin are high in obese, proportional to BMI[111,112] and acutely decrease in response to fasting or restriction of energy intake to a much larger extent than would be expected for smaller reductions of adiposity[113] and low levels act as a signal of negative energy balance. Adipocyte size and anatomical location (subcutaneous) appear to be the major determinants of leptin mRNA expression and secretion. In vivo, overfeeding and obesity, glucocorticoid treatments, glucose, and insulin administration increase circulating leptin levels[114,115] , whereas fasting, sustained exercise, cold exposure, and weight loss reduce leptin levels[116,117]. Leptin acts as an insulin-sensitizing hormone and reduces lipid content of myocytes, hepatocytes and pancreatic β-cells[118]. In muscle, insulin sensitization is achieved through inhibition of malonyl-CoA synthesis[119], which increases transport of FA into mitochondria. Leptin directly stimulates adenosine monophosphate kinase (AMPK), which activates ATP-producing catabolic pathways, such as β-oxidation, glycolysis and inhibits ATP-consuming anabolic pathways[120]. Animals lacking leptin effects; ob/ob mice (leptin gene mutation), db/db mice and fa/fa rats (leptin receptor gene mutations) are obese, insulin resistant and have HS[121,122] , while leptin injections attenuate their fatty livers and metabolic abnormalities[123]. In humans with lipoatrophic diabetes, there is little or no fat mass, diminished leptin levels, markedly elevated TAG and also fatty liver[124]. Leptin therapy in them reduces liver enzymes, BMI, hepatic fat content and histological features of steatohepatitis[125]. In obese NAFLD patients, leptin levels are elevated and are directly correlated with the severity of HS[126]. This brings up the concept of leptin resistance[127]. The anti-steatotic and insulin-sensitizing actions of leptin are blunted in obesity. The reason for this phenomenon is currently poorly understood and it may result from defects in leptin signaling or transport across blood brain barrier[128]. In animal models leptin is a critical fibrogenic factor[129]. This action may be mediated by transforming growth factor (TGF)-β or may involve activation of hepatic stellate cells (HSC) directly. HSC, upon activation produce leptin[130], which further stimulates fibrogenesis, as both quiescent and activated HSC express leptin receptors[131].
Adiponectin: Adiponectin, an antilipogenic and insulin-sensitizing protein[132] is almost exclusively expressed in white AT adipocytes and circulates at high levels[133]. In contrast to other adipokines, its expression and serum levels are reduced in obesity and a variety of insulin resistant states[134]. In obese rats the cellular mRNA and circulating levels of adiponectin are low[135,136] and prompt increase is seen with body weight reduction through food restriction[134]. Similar inverse correlation between BMI and adiponectin is also observed in Pima Indians[137]. IL-6 and TNF-α are potent inhibitors of adiponectin expression[138] and high levels of these cytokines in obesity and NAFLD explain this relationship. The decline in adiponectin levels coincide with the onset of IR[135,139] and various links have been suggested between low circulating adiponectin, increased liver fat content and the degree of hepatic IR[140,141]. Administration of adiponectin ameliorates IR in mouse models of obesity and diabetes[142]. The insulin sensitizing properties of adiponectin are due to its ability to activate AMPK in hepatocytes, myocytes and locally in adipocytes. In turn, it enhances hepatic FA oxidation and insulin suppression of glucose production[143], myocellular FA oxidation (by inactivation of ACC-1)[144] and lipolysis in AT. Adiponectin also has anti-inflammatory effects and inhibits the local production of TNF-α and IFN. Low adiponectin levels might predispose liver to cell necrosis and[145,146] correlate with the severity of NASH and liver enzyme abnormalities[147]. Adiponectin therapy improves IR in lipodystrophy, however, complete reversal of IR in the animal models requires co-administration of leptin, further supporting the importance of coordinated roles of different adipokines in causation of this complex disease.
Tumor necrosis factor-α: TNF-α is synthesized and secreted by visceral adipocytes, stromovascular cells and endotoxin-activated macrophages[148]. Unstimulated adipose tissue releases relatively small amounts of TNF-α[149]. Its effects depend upon interaction with TNF receptors (TNF-R1 and TNF-R2) and also with more than 20 different cytokine receptors. TNF receptors are also secreted by adipocytes. TNF-R1 mediates apoptosis and lipolysis while TNF-R2 is involved in the induction of IR[150]. TNF-α mainly acts in an autocrine/paracrine fashion in AT and plays a central role in the generation of IR in rodents. It does so directly by reducing mRNA expression of GLUT-4[148], reducing lipoprotein lipase (LPL) activity and increasing expression of hormone sensitive lipase in AT. Also TNF-α impairs insulin signaling through JNK mediated serine phosphorylation of IRS proteins in the surrounding adipocytes[151]. TNF-α causes activation of PPAR-γ also and reduces expression and secretion of leptin by adipocytes. In vitro studies on human adipose tissue do not show any clear role of TNF-α in causing these direct metabolic derangements. Circulating TNF-α is increased in obese[152] and diabetic subjects while weight loss decreases its levels. Although the correlation between IR and plasma TNF-α is weak, adipocyte TNF-α mRNA levels correlate well with BMI, body fat and hyperinsulinemia. To date no study has demonstrated whether TNF-α, via the portal vein reaches the liver and causes hepatic injury. However, the local production of TNF-α by Kupffer cells has been proposed to play a key role in the pathogenesis of NASH/NAFLD. FL in ob/ob mice is significantly improved by inhibition of hepatic TNF-α production or by infusion of anti-TNF-α antibodies[153]. TNF-α knockout could prevent the development of IR induced by a high fat diet in mice, suggesting a role of TNF-α in mediating free FA-induced IR.
Interleukin-6: IL-6 is predominantly an endocrine cytokine[154] with multiple systemic effects ranging from inflammation to host defense (regulation of B and T cell functions) and tissue injury. Approximately 33% of circulating IL-6 come from the AT, of which the matrix components with immune cells (monocytes), fibroblasts and endothelial cells contribute the major part (90%). Omental fat secretes 3 times more IL-6 than subcutaneous AT. However, IL-6 expression in subcutaneous fat is prone to rapid changes following meals, exercise (mediated via stimulation) and weight changes. Plasma IL-6 levels are increased in obesity[152] and predict the development of type 2 diabetes, metabolic syndrome and cardiovascular diseases[155,156]. It has a weak inhibitory effect on adipogenesis. In paracrine fashion, it decreases adiponectin secretion from the surrounding adipocytes, inhibits lipoprotein lipase (LPL) on endothelial cells and activates lipolysis (post exercise). IL-6 can enter the liver via portal circulation from visceral adipose tissue and mediates its effects on hepatocytes by interacting with its receptor, activating suppressor of cytokines signaling-3 (SOCS-3). This causes inhibition of IRS phosphorylation, which leads to hepatic IR[157]. IL-6 also has pro-inflammatory properties and causes activation of Kupffer cells resulting in fibrogenesis. It also enhances hepatic C-reactive protein synthesis. Paradoxically, administration of IL-6 in the cerebrospinal fluid of rats decreases body weight and IR, probably by enhancing the energy expenditure due to its action on hypothalamus.
Resistin: Resistin is claimed to represent an important link between obesity and IR[158]. In humans the major source of resistin is probably from the peripheral blood macrophages and adipocytes[159] and its levels correlate with IL-6[160] and BMI[161,162]. In mice, recombinant resistin promotes systemic IR by AMPK activation and decreasing upregulation of GLUT-4 in adipocytes whereas anti-resistin antibody administration improves IR[163]. Mice lacking resistin (rstn-/-) exhibit low blood glucose levels after fasting owing to reduced hepatic glucose production[164]. Its serum levels are higher in mouse models of obesity and decrease by PPARγ agonist treatment[165].
Acylation-stimulating protein: Adipose tissue releases substantial amounts of acylation-stimulating protein (ASP), which is derived from the interaction of complement C3, factor B and adipsin[166]. ASP stimulates glucose transport and enhances TAG storage in adipocytes[167]. These stimulatory effects are independent of and complementary to those of insulin[168]. Insulin itself increases the production of ASP precursor C3 in adipocytes[169]. Production of ASP may also be increased by IL-6 [168] and its levels in NAFLD are high correlating with HOMA-IR score[170]. Although ASP levels are increased in obese subjects, resistance to ASP could redirect FA flux away from AT and towards the liver causing HS.
Angiotensinogen: Angiotensinogen is also found in adipocytes[171] and its levels are increased in obesity[172]. Nascent data suggest it may be important in NAFLD, because angiotensin II antagonists improve liver function tests in patients with NAFLD and attenuate fibrosis in animal models[173].
Lipodystrophy-the ectopic fat storage syndrome
Hepatic steatosis is not only confined to obese subjects with excessive fat tissue, but it occurs frequently in lipodystrophy characterized by severe loss of AT from different regions of the body[174] and correlates directly in severity with the extent of fat loss[175]. Patients with congenital or acquired generalized lipodystrophy often are insulin resistant, diabetic and their HS may progress to cirrhosis[176]. The mechanism for HS in lipodystrophy is related to the reduced storage capacity of AT depots and diversion of TAG to other tissues[69]. Also the failure of AT mass to expand and accommodate a high-energy influx causes impaired adipokine production. Experiments in mice and humans demonstrate development of severe IR with lipodystrophy[177,178]. IR in lipoatrophic mice is fully reversed by a combination of physiological doses of adiponectin and leptin, but only partially by either adiponectin or leptin alone. This suggests that reduced adiponectin and leptin levels might be causing NAFLD in subjects with lipodystrophy[179].
ROLE IN NON-ALCOHOLIC STEATOHEPATITIS AND CIRRHOSIS
Subjects with NASH are at significantly increased risk to develop cirrhosis. Once cirrhosis has developed, the characteristic hepatocellular changes, as well as steatosis, are often no longer conspicuous, possibly leading to such cases being mistakenly diagnosed as cryptogenic. Studies suggest that NAFLD is the most important cause of cryptogenic cirrhosis[180,181]. A fraction of NAFLD patients develop hepatocellular carcinoma[182]. The risk of this malignant complication is increased more than 4 times in cirrhotic patients compared to the general population[183]. About 3% of NASH cases may progress to terminal liver failure, requiring liver transplantation[184]. At present, up to 4% to 10% of liver transplants can probably be accounted for by end-stage NASH[4]; this estimate is very likely to rise. The pathogenesis of progression is not well understood and various risk factors are proposed. Low intake of antioxidant vitamins, high intake of saturated fat[185], presence of small bowel bacterial overgrowth[186] and obstructive sleep apnea[187] may contribute to it in different settings. Eventually, various factors (cytokines, hormones, neurotransmitters or FA) through their interactions modulate the necro-inflammatory component of NASH (Table 3).
Table 3.
Serum factors in obesity and potential mechanisms of NASH
| Factors | Increase in obesity | Source: tissue/cells | Basis of increased levels | Role in development of NASH |
| FFA[199] | 41% | Diet Adipose tissue: (adipocytes) visceral/subcutaneous | -Over nutrition -Unopposed peripheral lipolytic activity secondary to IR | -Lipotoxicity -Induce JNK dependent hepatocytes apoptosis -Cause Bax translocation to hepatocyte lysosomes leading to lysosomal degradation and release of cathepsin B -Enhance expression of apoptosis effectors(TNF-α and Fas) -Generation of ROS at ETC of mitochondria -Increase in hepatic lipid peroxidation |
| TNF-α[152] | 28% | Liver: Kupffer cells/macrophages/HSC/hepatocytes Adipose tissue: macrophages in matrix/adipocytes | -Chronic inflammation in adipose tissue with macrophage infiltration -LPS/endotoxins from small bowel overgrowth -Viral infection -Ethanol -Reactive oxygen species | -Receptor mediated mitochondrial injury with release of ROS and caspases -Induction of lipid peroxidation and cell necrosis (intermediation of ceramide) -TNF-α R1 activation leads to Fas induced apoptosis -Causes release of other cytotoxic cytokines(IL-6, TGF-β) from activated Kupffer cells |
| IL-6[152] | 46% | Blood: monocytes/endothelial cells Adipose tissue: subcutaneous/omental Liver: Kupffer cells/HSC/macrophages | -TNF-α mediated activation of Kupffer cells -Pro-inflammatory cytokines release by cells (macrophages) in adipose tissue | -Mediates synthesis of acute phase proteins by hepatocytes -Activates HSC to cause fibrosis and up regulate various genes involved |
| Leptin[111,112] | 4 .2 to 5.8 times | Adipose tissue: mature adipocyte/few matrix cells Liver: activated HSC | -Increased mass of adipose tissue -Chronic inflammatory mediators in adipose tissue -Leptin resistance | -Regulates hepatic fibrosis by activation of HSC(induction of α2 collagen gene) and modulation of Kupffer cell function -Protects HSC from apoptosis and enhances their regeneration -Up regulates profibrogenic TGF-β synthesis |
| Resistin[161,162] | Non significant | Adipose tissue: visceral/subcutaneous (adipocytes) Blood: monocytes Liver: Kupffer cells | -IL-6 and TNF-α release inadipose tissue secondary to inflammation -Chronic liver injury | -NF-κB mediated activation of HSC and release of pro-inflammatory(MCP, IL-8, TNF-α) and fibrogenic(TGF-β, leptin) cytokines |
| IL-8[200] | 33% | Inflammatory cells in adipose tissue,liver and blood | -Pro-inflammatory cytokines | -Mediates inflammatory response in NASH |
| PAI-1[201] | 3.5 times | Liver: (activated HSC) Adipose tissue: visceral/omental (matrix and adipocytes) | -Locally produced TGF-β and TNF-α | -Inhibits the activation of fibrinolytic plasmin during fibrogenesis |
| Angiotensinogen[172] | 14% | Liver: hepatocytes Adipose tissue: visceral/subcutaneous (adipocytes) | -Hyperinsulinemia of IR -FFA | -Activates HSC to secrete TGF-β to cause fibrosis |
FFA: Free fatty acids; JNK: Janus N kinase; ROS: Reactive oxygen species; ETC: Electron transport chain; TNF-α: Tumor necrosis factor-α; HSC: Hepatic stellate cells; IL: Interleukin; TGF-β: Transforming growth factor-β; MCP: Monocyte chemoattractant protein; NF-κB: Nuclear factor-kappa B; PAI-1: Plasminogen activator inhibitor-1; IR: Insulin resistance.
The development of fibrosis indicates further progression of liver injury. Extracellular accumulation of collagen matrix is predominantly localized in pericentral and perisinusoidal areas of the hepatic lobule[188]. However, isolated portal fibrosis (IPF) is also found to be associated with hepatic steatosis in NAFLD[39,189]. HSC, owing to their localization in hepatic lobule play a pivotal role in the deposition of collagen. HSC undergo a phenotypic transition (activation) to myofibroblast-like cells that synthesize different extracellular matrix components[190]. HSC acquire ability to effectively proliferate and migrate to the area of liver injury and express soluble mediators of inflammation, angiogenesis and hepatocyte growth[191]. Obesity and IR are independently associated with the progression and degree of fibrosis[40,192]. This view is supported by the increase in cell proliferation and collagen production by insulin in cultured HSC[193]. Hyperglycemia also acts as a stimulus to secretion of the profibrogenic cytokine (connective tissue growth factor)[194]. Oxidative stress is implicated in activation of HSC and products of oxidative stress have been shown to have profibrogenic actions in in vitro studies[195]. Among adipokines, leptin stimulates collagen expression, proliferation and prevention of apoptosis of HSC[196]. However, association between plasma leptin levels and the degree of fibrosis is not clearly identified. The levels of anti-fibrogenic adiponectin are markedly diminished in obesity. Resistin is recently shown to modulate human HSC activity[197]. Other cytokines like IL-10 and TGF-β also dictate the extent of fibrosis in NASH. It is believed that release of TGF-β1 by necrotic hepatocytes may be one of the first signals to activate adjacent quiescent HSC[198].
CONCLUSION
NAFLD is emerging as one of the most common liver disorders claiming urgent attention of the public, clinicians and researchers. Obesity is an epidemic health disorder and NAFLD can be a major health problem. Lifestyle modifications including calorie restriction, physical activity and weight loss are main therapeutic modalities. Future research is warranted to elucidate the pathogenesis of NAFLD and NASH, thereby developing accurate non-invasive diagnostic tests and novel therapeutic protocols.
Footnotes
S- Editor Zhu LH L- Editor Zhu LH E- Editor Wang HF
References
- 1.Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55:434–438. [PubMed] [Google Scholar]
- 2.Clark JM, Brancati FL, Diehl AM. Nonalcoholic fatty liver disease. Gastroenterology. 2002;122:1649–1657. doi: 10.1053/gast.2002.33573. [DOI] [PubMed] [Google Scholar]
- 3.Ruhl CE, Everhart JE. Epidemiology of nonalcoholic fatty liver. Clin Liver Dis. 2004;8:501–519, vii. doi: 10.1016/j.cld.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 4.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43:S99–S112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 5.Bellentani S, Tiribelli C, Saccoccio G, Sodde M, Fratti N, De Martin C, Cristianini G. Prevalence of chronic liver disease in the general population of northern Italy: the Dionysos Study. Hepatology. 1994;20:1442–1449. doi: 10.1002/hep.1840200611. [DOI] [PubMed] [Google Scholar]
- 6.Jimba S, Nakagami T, Takahashi M, Wakamatsu T, Hirota Y, Iwamoto Y, Wasada T. Prevalence of non-alcoholic fatty liver disease and its association with impaired glucose metabolism in Japanese adults. Diabet Med. 2005;22:1141–1145. doi: 10.1111/j.1464-5491.2005.01582.x. [DOI] [PubMed] [Google Scholar]
- 7.Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467–2474. doi: 10.1111/j.1572-0241.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 8.Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatology. 2003;37:1202–1219. doi: 10.1053/jhep.2003.50193. [DOI] [PubMed] [Google Scholar]
- 9.Skelly MM, James PD, Ryder SD. Findings on liver biopsy to investigate abnormal liver function tests in the absence of diagnostic serology. J Hepatol. 2001;35:195–199. doi: 10.1016/s0168-8278(01)00094-0. [DOI] [PubMed] [Google Scholar]
- 10.de Lédinghen V, Combes M, Trouette H, Winnock M, Amouretti M, de Mascarel A, Couzigou P. Should a liver biopsy be done in patients with subclinical chronically elevated transaminases? Eur J Gastroenterol Hepatol. 2004;16:879–883. doi: 10.1097/00042737-200409000-00011. [DOI] [PubMed] [Google Scholar]
- 11.Prati D, Taioli E, Zanella A, Della Torre E, Butelli S, Del Vecchio E, Vianello L, Zanuso F, Mozzi F, Milani S, et al. Updated definitions of healthy ranges for serum alanine aminotransferase levels. Ann Intern Med. 2002;137:1–10. doi: 10.7326/0003-4819-137-1-200207020-00006. [DOI] [PubMed] [Google Scholar]
- 12.Kunde SS, Lazenby AJ, Clements RH, Abrams GA. Spectrum of NAFLD and diagnostic implications of the proposed new normal range for serum ALT in obese women. Hepatology. 2005;42:650–656. doi: 10.1002/hep.20818. [DOI] [PubMed] [Google Scholar]
- 13.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 14.Poynard T, Ratziu V, Naveau S, Thabut D, Charlotte F, Messous D, Capron D, Abella A, Massard J, Ngo Y, et al. The diagnostic value of biomarkers (SteatoTest) for the prediction of liver steatosis. Comp Hepatol. 2005;4:10. doi: 10.1186/1476-5926-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ratziu V, Massard J, Charlotte F, Messous D, Imbert-Bismut F, Bonyhay L, Tahiri M, Munteanu M, Thabut D, Cadranel JF, et al. Diagnostic value of biochemical markers (FibroTest-FibroSURE) for the prediction of liver fibrosis in patients with non-alcoholic fatty liver disease. BMC Gastroenterol. 2006;6:6. doi: 10.1186/1471-230X-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poynard T, Ratziu V, Charlotte F, Messous D, Munteanu M, Imbert-Bismut F, Massard J, Bonyhay L, Tahiri M, Thabut D, et al. Diagnostic value of biochemical markers (NashTest) for the prediction of non alcoholo steato hepatitis in patients with non-alcoholic fatty liver disease. BMC Gastroenterol. 2006;6:34. doi: 10.1186/1471-230X-6-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cotrim HP, Andrade ZA, Parana R, Portugal M, Lyra LG, Freitas LA. Nonalcoholic steatohepatitis: a toxic liver disease in industrial workers. Liver. 1999;19:299–304. doi: 10.1111/j.1478-3231.1999.tb00053.x. [DOI] [PubMed] [Google Scholar]
- 18.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 19.Solga SF, Clark JM, Alkhuraishi AR, Torbenson M, Tabesh A, Schweitzer M, Diehl AM, Magnuson TH. Race and comorbid factors predict nonalcoholic fatty liver disease histopathology in severely obese patients. Surg Obes Relat Dis. 2005;1:6–11. doi: 10.1016/j.soard.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 20.Willner IR, Waters B, Patil SR, Reuben A, Morelli J, Riely CA. Ninety patients with nonalcoholic steatohepatitis: insulin resistance, familial tendency, and severity of disease. Am J Gastroenterol. 2001;96:2957–2961. doi: 10.1111/j.1572-0241.2001.04667.x. [DOI] [PubMed] [Google Scholar]
- 21.Sreekumar R, Rosado B, Rasmussen D, Charlton M. Hepatic gene expression in histologically progressive nonalcoholic steatohepatitis. Hepatology. 2003;38:244–251. doi: 10.1053/jhep.2003.50290. [DOI] [PubMed] [Google Scholar]
- 22.Diehl AM, Li ZP, Lin HZ, Yang SQ. Cytokines and the pathogenesis of non-alcoholic steatohepatitis. Gut. 2005;54:303–306. doi: 10.1136/gut.2003.024935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bugianesi E, McCullough AJ, Marchesini G. Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology. 2005;42:987–1000. doi: 10.1002/hep.20920. [DOI] [PubMed] [Google Scholar]
- 24.Alberti KG, Zimmet P, Shaw J. The metabolic syndrome--a new worldwide definition. Lancet. 2005;366:1059–1062. doi: 10.1016/S0140-6736(05)67402-8. [DOI] [PubMed] [Google Scholar]
- 25.Poucell S, Ireton J, Valencia-Mayoral P, Downar E, Larratt L, Patterson J, Blendis L, Phillips MJ. Amiodarone-associated phospholipidosis and fibrosis of the liver. Light, immunohistochemical, and electron microscopic studies. Gastroenterology. 1984;86:926–936. [PubMed] [Google Scholar]
- 26.Grieco A, Forgione A, Miele L, Vero V, Greco AV, Gasbarrini A, Gasbarrini G. Fatty liver and drugs. Eur Rev Med Pharmacol Sci. 2005;9:261–263. [PubMed] [Google Scholar]
- 27.Pratt DS, Knox TA, Erban J. Tamoxifen-induced steatohepatitis. Ann Intern Med. 1995;123:236. doi: 10.7326/0003-4819-123-3-199508010-00018. [DOI] [PubMed] [Google Scholar]
- 28.Novaro GM. Isoniazid and nonalcoholic steatohepatitis. J Clin Gastroenterol. 1999;28:180. doi: 10.1097/00004836-199903000-00023. [DOI] [PubMed] [Google Scholar]
- 29.Blackburn GL, Mun EC. Effects of weight loss surgeries on liver disease. Semin Liver Dis. 2004;24:371–379. doi: 10.1055/s-2004-860866. [DOI] [PubMed] [Google Scholar]
- 30.Baddeley RM. An epilogue to jejunoileal bypass. World J Surg. 1985;9:842–849. doi: 10.1007/BF01655388. [DOI] [PubMed] [Google Scholar]
- 31.McFarland RJ, Gazet JC, Pilkington TR. A 13-year review of jejunoileal bypass. Br J Surg. 1985;72:81–87. doi: 10.1002/bjs.1800720202. [DOI] [PubMed] [Google Scholar]
- 32.Reid AE. Nonalcoholic steatohepatitis. Gastroenterology. 2001;121:710–723. doi: 10.1053/gast.2001.27126. [DOI] [PubMed] [Google Scholar]
- 33.Partin JS, Partin JC, Schubert WK, McAdams AJ. Liver ultrastructure in abetalipoproteinemia: Evolution of micronodular cirrhosis. Gastroenterology. 1974;67:107–118. [PubMed] [Google Scholar]
- 34.Powell EE, Searle J, Mortimer R. Steatohepatitis associated with limb lipodystrophy. Gastroenterology. 1989;97:1022–1024. doi: 10.1016/0016-5085(89)91513-8. [DOI] [PubMed] [Google Scholar]
- 35.Fortgang IS, Belitsos PC, Chaisson RE, Moore RD. Hepatomegaly and steatosis in HIV-infected patients receiving nucleoside analog antiretroviral therapy. Am J Gastroenterol. 1995;90:1433–1436. [PubMed] [Google Scholar]
- 36.Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA. 2001;285:2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 37.Mokdad AH, Ford ES, Bowman BA, Dietz WH, Vinicor F, Bales VS, Marks JS. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA. 2003;289:76–79. doi: 10.1001/jama.289.1.76. [DOI] [PubMed] [Google Scholar]
- 38.Angulo P. Treatment of nonalcoholic fatty liver disease. Ann Hepatol. 2002;1:12–19. [PubMed] [Google Scholar]
- 39.Abrams GA, Kunde SS, Lazenby AJ, Clements RH. Portal fibrosis and hepatic steatosis in morbidly obese subjects: A spectrum of nonalcoholic fatty liver disease. Hepatology. 2004;40:475–483. doi: 10.1002/hep.20323. [DOI] [PubMed] [Google Scholar]
- 40.Ratziu V, Giral P, Charlotte F, Bruckert E, Thibault V, Theodorou I, Khalil L, Turpin G, Opolon P, Poynard T. Liver fibrosis in overweight patients. Gastroenterology. 2000;118:1117–1123. doi: 10.1016/s0016-5085(00)70364-7. [DOI] [PubMed] [Google Scholar]
- 41.Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, McCullough AJ, Natale S, Forlani G, Melchionda N. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50:1844–1850. doi: 10.2337/diabetes.50.8.1844. [DOI] [PubMed] [Google Scholar]
- 42.Braillon A, Capron JP, Hervé MA, Degott C, Quenum C. Liver in obesity. Gut. 1985;26:133–139. doi: 10.1136/gut.26.2.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6:1–28. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 45.Brautigan DL, Brown M, Grindrod S, Chinigo G, Kruszewski A, Lukasik SM, Bushweller JH, Horal M, Keller S, Tamura S, et al. Allosteric activation of protein phosphatase 2C by D-chiro-inositol-galactosamine, a putative mediator mimetic of insulin action. Biochemistry. 2005;44:11067–11073. doi: 10.1021/bi0508845. [DOI] [PubMed] [Google Scholar]
- 46.Rao MS, Reddy JK. Peroxisomal beta-oxidation and steatohepatitis. Semin Liver Dis. 2001;21:43–55. doi: 10.1055/s-2001-12928. [DOI] [PubMed] [Google Scholar]
- 47.Fromenty B, Robin MA, Igoudjil A, Mansouri A, Pessayre D. The ins and outs of mitochondrial dysfunction in NASH. Diabetes Metab. 2004;30:121–138. doi: 10.1016/s1262-3636(07)70098-8. [DOI] [PubMed] [Google Scholar]
- 48.Horton JD, Shimomura I. Sterol regulatory element-binding proteins: activators of cholesterol and fatty acid biosynthesis. Curr Opin Lipidol. 1999;10:143–150. doi: 10.1097/00041433-199904000-00008. [DOI] [PubMed] [Google Scholar]
- 49.Horton JD. Sterol regulatory element-binding proteins: transcriptional activators of lipid synthesis. Biochem Soc Trans. 2002;30:1091–1095. doi: 10.1042/bst0301091. [DOI] [PubMed] [Google Scholar]
- 50.McGarry JD, Foster DW. Effects of exogenous fatty acid concentration on glucagon-induced changes in hepatic fatty acid metabolism. Diabetes. 1980;29:236–240. doi: 10.2337/diab.29.3.236. [DOI] [PubMed] [Google Scholar]
- 51.Yamashita H, Takenoshita M, Sakurai M, Bruick RK, Henzel WJ, Shillinglaw W, Arnot D, Uyeda K. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc Natl Acad Sci USA. 2001;98:9116–9121. doi: 10.1073/pnas.161284298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawaguchi T, Osatomi K, Yamashita H, Kabashima T, Uyeda K. Mechanism for fatty acid "sparing" effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J Biol Chem. 2002;277:3829–3835. doi: 10.1074/jbc.M107895200. [DOI] [PubMed] [Google Scholar]
- 53.Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci USA. 2004;101:7281–7286. doi: 10.1073/pnas.0401516101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chao L, Marcus-Samuels B, Mason MM, Moitra J, Vinson C, Arioglu E, Gavrilova O, Reitman ML. Adipose tissue is required for the antidiabetic, but not for the hypolipidemic, effect of thiazolidinediones. J Clin Invest. 2000;106:1221–1228. doi: 10.1172/JCI11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fajas L, Schoonjans K, Gelman L, Kim JB, Najib J, Martin G, Fruchart JC, Briggs M, Spiegelman BM, Auwerx J. Regulation of peroxisome proliferator-activated receptor gamma expression by adipocyte differentiation and determination factor 1/sterol regulatory element binding protein 1: implications for adipocyte differentiation and metabolism. Mol Cell Biol. 1999;19:5495–5503. doi: 10.1128/mcb.19.8.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, Brewer B, Reitman ML, Gonzalez FJ. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Invest. 2003;111:737–747. doi: 10.1172/JCI17223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kahn CR. Insulin resistance, insulin insensitivity, and insulin unresponsiveness: a necessary distinction. Metabolism. 1978;27:1893–1902. doi: 10.1016/s0026-0495(78)80007-9. [DOI] [PubMed] [Google Scholar]
- 58.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 59.Barbieri M, Ragno E, Benvenuti E, Zito GA, Corsi A, Ferrucci L, Paolisso G. New aspects of the insulin resistance syndrome: impact on haematological parameters. Diabetologia. 2001;44:1232–1237. doi: 10.1007/s001250100634. [DOI] [PubMed] [Google Scholar]
- 60.Falkner B, Sherif K, Kushner H. Gender differences in the relationship between insulin-mediated glucose utilization and sex hormones in young African-Americans. J Gend Specif Med. 2000;3:60–65. [PubMed] [Google Scholar]
- 61.Pascual E. Hyperuricemia and gout. Curr Opin Rheumatol. 1994;6:454–458. doi: 10.1097/00002281-199407000-00018. [DOI] [PubMed] [Google Scholar]
- 62.Loria P, Lonardo A, Lombardini S, Carulli L, Verrone A, Ganazzi D, Rudilosso A, D'Amico R, Bertolotti M, Carulli N. Gallstone disease in non-alcoholic fatty liver: prevalence and associated factors. J Gastroenterol Hepatol. 2005;20:1176–1184. doi: 10.1111/j.1440-1746.2005.03924.x. [DOI] [PubMed] [Google Scholar]
- 63.Sakkinen PA, Wahl P, Cushman M, Lewis MR, Tracy RP. Clustering of procoagulation, inflammation, and fibrinolysis variables with metabolic factors in insulin resistance syndrome. Am J Epidemiol. 2000;152:897–907. doi: 10.1093/aje/152.10.897. [DOI] [PubMed] [Google Scholar]
- 64.Yildiz BO, Yarali H, Oguz H, Bayraktar M. Glucose intolerance, insulin resistance, and hyperandrogenemia in first degree relatives of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88:2031–2036. doi: 10.1210/jc.2002-021499. [DOI] [PubMed] [Google Scholar]
- 65.Virkamäki A, Ueki K, Kahn CR. Protein-protein interaction in insulin signaling and the molecular mechanisms of insulin resistance. J Clin Invest. 1999;103:931–943. doi: 10.1172/JCI6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Previs SF, Withers DJ, Ren JM, White MF, Shulman GI. Contrasting effects of IRS-1 versus IRS-2 gene disruption on carbohydrate and lipid metabolism in vivo. J Biol Chem. 2000;275:38990–38994. doi: 10.1074/jbc.M006490200. [DOI] [PubMed] [Google Scholar]
- 67.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237:E214–E223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- 68.Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. 2006;55 Suppl 2:S9–S15. doi: 10.2337/db06-S002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Unger RH. Lipid overload and overflow: metabolic trauma and the metabolic syndrome. Trends Endocrinol Metab. 2003;14:398–403. doi: 10.1016/j.tem.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 70.Marchesini G, Brizi M, Morselli-Labate AM, Bianchi G, Bugianesi E, McCullough AJ, Forlani G, Melchionda N. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med. 1999;107:450–455. doi: 10.1016/s0002-9343(99)00271-5. [DOI] [PubMed] [Google Scholar]
- 71.Forouhi NG, Jenkinson G, Thomas EL, Mullick S, Mierisova S, Bhonsle U, McKeigue PM, Bell JD. Relation of triglyceride stores in skeletal muscle cells to central obesity and insulin sensitivity in European and South Asian men. Diabetologia. 1999;42:932–935. doi: 10.1007/s001250051250. [DOI] [PubMed] [Google Scholar]
- 72.Krssak M, Falk Petersen K, Dresner A, DiPietro L, Vogel SM, Rothman DL, Roden M, Shulman GI. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia. 1999;42:113–116. doi: 10.1007/s001250051123. [DOI] [PubMed] [Google Scholar]
- 73.Reaven G. The metabolic syndrome or the insulin resistance syndrome? Different names, different concepts, and different goals. Endocrinol Metab Clin North Am. 2004;33:283–303. doi: 10.1016/j.ecl.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 74.Hegele RA. Phenomics, lipodystrophy, and the metabolic syndrome. Trends Cardiovasc Med. 2004;14:133–137. doi: 10.1016/j.tcm.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 75.Ueno T, Sugawara H, Sujaku K, Hashimoto O, Tsuji R, Tamaki S, Torimura T, Inuzuka S, Sata M, Tanikawa K. Therapeutic effects of restricted diet and exercise in obese patients with fatty liver. J Hepatol. 1997;27:103–107. doi: 10.1016/s0168-8278(97)80287-5. [DOI] [PubMed] [Google Scholar]
- 76.Day CP, James OF. Steatohepatitis: a tale of two "hits"? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 77.Perseghin G, Petersen K, Shulman GI. Cellular mechanism of insulin resistance: potential links with inflammation. Int J Obes Relat Metab Disord. 2003;27 Suppl 3:S6–S11. doi: 10.1038/sj.ijo.0802491. [DOI] [PubMed] [Google Scholar]
- 78.Combettes-Souverain M, Issad T. Molecular basis of insulin action. Diabetes Metab. 1998;24:477–489. [PubMed] [Google Scholar]
- 79.McCullough AJ. Pathophysiology of nonalcoholic steatohepatitis. J Clin Gastroenterol. 2006;40 Suppl 1:S17–S29. doi: 10.1097/01.mcg.0000168645.86658.22. [DOI] [PubMed] [Google Scholar]
- 80.Sies H, Stahl W, Sevanian A. Nutritional, dietary and postprandial oxidative stress. J Nutr. 2005;135:969–972. doi: 10.1093/jn/135.5.969. [DOI] [PubMed] [Google Scholar]
- 81.Aronis A, Madar Z, Tirosh O. Mechanism underlying oxidative stress-mediated lipotoxicity: exposure of J774.2 macrophages to triacylglycerols facilitates mitochondrial reactive oxygen species production and cellular necrosis. Free Radic Biol Med. 2005;38:1221–1230. doi: 10.1016/j.freeradbiomed.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 82.Videla LA, Rodrigo R, Orellana M, Fernandez V, Tapia G, Quiñones L, Varela N, Contreras J, Lazarte R, Csendes A, et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin Sci (Lond) 2004;106:261–268. doi: 10.1042/CS20030285. [DOI] [PubMed] [Google Scholar]
- 83.Poniachik J, Csendes A, Díaz JC, Rojas J, Burdiles P, Maluenda F, Smok G, Rodrigo R, Videla LA. Increased production of IL-1alpha and TNF-alpha in lipopolysaccharide-stimulated blood from obese patients with non-alcoholic fatty liver disease. Cytokine. 2006;33:252–257. doi: 10.1016/j.cyto.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 84.Schattenberg JM, Wang Y, Singh R, Rigoli RM, Czaja MJ. Hepatocyte CYP2E1 overexpression and steatohepatitis lead to impaired hepatic insulin signaling. J Biol Chem. 2005;280:9887–9894. doi: 10.1074/jbc.M410310200. [DOI] [PubMed] [Google Scholar]
- 85.De Craemer D, Pauwels M, Van den Branden C. Alterations of peroxisomes in steatosis of the human liver: a quantitative study. Hepatology. 1995;22:744–752. doi: 10.1002/hep.1840220309. [DOI] [PubMed] [Google Scholar]
- 86.Lombardo YB, Chicco AG. Effects of dietary polyunsaturated n-3 fatty acids on dyslipidemia and insulin resistance in rodents and humans. A review. J Nutr Biochem. 2006;17:1–13. doi: 10.1016/j.jnutbio.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 87.Woodcroft KJ, Hafner MS, Novak RF. Insulin signaling in the transcriptional and posttranscriptional regulation of CYP2E1 expression. Hepatology. 2002;35:263–273. doi: 10.1053/jhep.2002.30691. [DOI] [PubMed] [Google Scholar]
- 88.Hotamisligil GS. Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes. 2005;54 Suppl 2:S73–S78. doi: 10.2337/diabetes.54.suppl_2.s73. [DOI] [PubMed] [Google Scholar]
- 89.Mathurin P, Gonzalez F, Kerdraon O, Leteurtre E, Arnalsteen L, Hollebecque A, Louvet A, Dharancy S, Cocq P, Jany T, et al. The evolution of severe steatosis after bariatric surgery is related to insulin resistance. Gastroenterology. 2006;130:1617–1624. doi: 10.1053/j.gastro.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 90.Von Gierke E. Ueber fett metabolism. Ver Deutsch Ges Path. 1906;10:182–185. [Google Scholar]
- 91.Ahima RS. Adipose tissue as an endocrine organ. Obesity (Silver Spring) 2006;14 Suppl 5:242S–249S. doi: 10.1038/oby.2006.317. [DOI] [PubMed] [Google Scholar]
- 92.Zierath JR, Livingston JN, Thörne A, Bolinder J, Reynisdottir S, Lönnqvist F, Arner P. Regional difference in insulin inhibition of non-esterified fatty acid release from human adipocytes: relation to insulin receptor phosphorylation and intracellular signalling through the insulin receptor substrate-1 pathway. Diabetologia. 1998;41:1343–1354. doi: 10.1007/s001250051075. [DOI] [PubMed] [Google Scholar]
- 93.Wu X, Hoffstedt J, Deeb W, Singh R, Sedkova N, Zilbering A, Zhu L, Park PK, Arner P, Goldstein BJ. Depot-specific variation in protein-tyrosine phosphatase activities in human omental and subcutaneous adipose tissue: a potential contribution to differential insulin sensitivity. J Clin Endocrinol Metab. 2001;86:5973–5980. doi: 10.1210/jcem.86.12.8109. [DOI] [PubMed] [Google Scholar]
- 94.van der Kooy K, Leenen R, Seidell JC, Deurenberg P, Visser M. Abdominal diameters as indicators of visceral fat: comparison between magnetic resonance imaging and anthropometry. Br J Nutr. 1993;70:47–58. doi: 10.1079/bjn19930104. [DOI] [PubMed] [Google Scholar]
- 95.Iacobellis G. Imaging of visceral adipose tissue: an emerging diagnostic tool and therapeutic target. Curr Drug Targets Cardiovasc Haematol Disord. 2005;5:345–353. doi: 10.2174/1568006054553408. [DOI] [PubMed] [Google Scholar]
- 96.Grimble RF. Inflammatory status and insulin resistance. Curr Opin Clin Nutr Metab Care. 2002;5:551–559. doi: 10.1097/00075197-200209000-00015. [DOI] [PubMed] [Google Scholar]
- 97.Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89:2548–2556. doi: 10.1210/jc.2004-0395. [DOI] [PubMed] [Google Scholar]
- 98.Fain JN, Madan AK, Hiler ML, Cheema P, Bahouth SW. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology. 2004;145:2273–2282. doi: 10.1210/en.2003-1336. [DOI] [PubMed] [Google Scholar]
- 99.Trayhurn P, Wood IS. Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr. 2004;92:347–355. doi: 10.1079/bjn20041213. [DOI] [PubMed] [Google Scholar]
- 100.Krug AW, Ehrhart-Bornstein M. Newly discovered endocrine functions of white adipose tissue: possible relevance in obesity-related diseases. Cell Mol Life Sci. 2005;62:1359–1362. doi: 10.1007/s00018-005-4555-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nielsen S, Guo Z, Johnson CM, Hensrud DD, Jensen MD. Splanchnic lipolysis in human obesity. J Clin Invest. 2004;113:1582–1588. doi: 10.1172/JCI21047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 103.Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest. 2000;106:473–481. doi: 10.1172/JCI10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Thomas EL, Hamilton G, Patel N, O'Dwyer R, Doré CJ, Goldin RD, Bell JD, Taylor-Robinson SD. Hepatic triglyceride content and its relation to body adiposity: a magnetic resonance imaging and proton magnetic resonance spectroscopy study. Gut. 2005;54:122–127. doi: 10.1136/gut.2003.036566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.de Almeida IT, Cortez-Pinto H, Fidalgo G, Rodrigues D, Camilo ME. Plasma total and free fatty acids composition in human non-alcoholic steatohepatitis. Clin Nutr. 2002;21:219–223. doi: 10.1054/clnu.2001.0529. [DOI] [PubMed] [Google Scholar]
- 106.Carey DG, Jenkins AB, Campbell LV, Freund J, Chisholm DJ. Abdominal fat and insulin resistance in normal and overweight women: Direct measurements reveal a strong relationship in subjects at both low and high risk of NIDDM. Diabetes. 1996;45:633–638. doi: 10.2337/diab.45.5.633. [DOI] [PubMed] [Google Scholar]
- 107.Roden M. Non-invasive studies of glycogen metabolism in human skeletal muscle using nuclear magnetic resonance spectroscopy. Curr Opin Clin Nutr Metab Care. 2001;4:261–266. doi: 10.1097/00075197-200107000-00003. [DOI] [PubMed] [Google Scholar]
- 108.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 109.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 110.Ahima RS, Saper CB, Flier JS, Elmquist JK. Leptin regulation of neuroendocrine systems. Front Neuroendocrinol. 2000;21:263–307. doi: 10.1006/frne.2000.0197. [DOI] [PubMed] [Google Scholar]
- 111.Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 112.Corica F, Allegra A, Corsonello A, Buemi M, Calapai G, Ruello A, Nicita Mauro V, Ceruso D. Relationship between plasma leptin levels and the tumor necrosis factor-alpha system in obese subjects. Int J Obes Relat Metab Disord. 1999;23:355–360. doi: 10.1038/sj.ijo.0800826. [DOI] [PubMed] [Google Scholar]
- 113.Ahrén B, Baldwin RM, Havel PJ. Pharmacokinetics of human leptin in mice and rhesus monkeys. Int J Obes Relat Metab Disord. 2000;24:1579–1585. doi: 10.1038/sj.ijo.0801447. [DOI] [PubMed] [Google Scholar]
- 114.Wellhoener P, Fruehwald-Schultes B, Kern W, Dantz D, Kerner W, Born J, Fehm HL, Peters A. Glucose metabolism rather than insulin is a main determinant of leptin secretion in humans. J Clin Endocrinol Metab. 2000;85:1267–1271. doi: 10.1210/jcem.85.3.6483. [DOI] [PubMed] [Google Scholar]
- 115.Mayer-Davis EJ, D'Agostino R, Karter AJ, Haffner SM, Rewers MJ, Saad M, Bergman RN. Intensity and amount of physical activity in relation to insulin sensitivity: the Insulin Resistance Atherosclerosis Study. JAMA. 1998;279:669–674. doi: 10.1001/jama.279.9.669. [DOI] [PubMed] [Google Scholar]
- 116.Dubuc GR, Phinney SD, Stern JS, Havel PJ. Changes of serum leptin and endocrine and metabolic parameters after 7 days of energy restriction in men and women. Metabolism. 1998;47:429–434. doi: 10.1016/s0026-0495(98)90055-5. [DOI] [PubMed] [Google Scholar]
- 117.Boden G, Chen X, Mozzoli M, Ryan I. Effect of fasting on serum leptin in normal human subjects. J Clin Endocrinol Metab. 1996;81:3419–3423. doi: 10.1210/jcem.81.9.8784108. [DOI] [PubMed] [Google Scholar]
- 118.Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, Sanna V, Jebb SA, Perna F, Fontana S, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110:1093–1103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Muoio DM, Dohm GL, Fiedorek FT, Tapscott EB, Coleman RA. Leptin directly alters lipid partitioning in skeletal muscle. Diabetes. 1997;46:1360–1363. doi: 10.2337/diab.46.8.1360. [DOI] [PubMed] [Google Scholar]
- 120.Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Müller C, Carling D, Kahn BB. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415:339–343. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- 121.Kaplan ML, Leveille GA. Obesity: prediction of preobesity among progeny from crosses of ob+ mice. Proc Soc Exp Biol Med. 1973;143:925–928. doi: 10.3181/00379727-143-37442. [DOI] [PubMed] [Google Scholar]
- 122.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 123.Ebihara K, Kusakabe T, Hirata M, Masuzaki H, Miyanaga F, Kobayashi N, Tanaka T, Chusho H, Miyazawa T, Hayashi T, et al. Efficacy and safety of leptin-replacement therapy and possible mechanisms of leptin actions in patients with generalized lipodystrophy. J Clin Endocrinol Metab. 2007;92:532–541. doi: 10.1210/jc.2006-1546. [DOI] [PubMed] [Google Scholar]
- 124.Danforth E. Failure of adipocyte differentiation causes type II diabetes mellitus? Nat Genet. 2000;26:13. doi: 10.1038/79111. [DOI] [PubMed] [Google Scholar]
- 125.Lee JH, Chan JL, Sourlas E, Raptopoulos V, Mantzoros CS. Recombinant methionyl human leptin therapy in replacement doses improves insulin resistance and metabolic profile in patients with lipoatrophy and metabolic syndrome induced by the highly active antiretroviral therapy. J Clin Endocrinol Metab. 2006;91:2605–2611. doi: 10.1210/jc.2005-1545. [DOI] [PubMed] [Google Scholar]
- 126.Margetic S, Gazzola C, Pegg GG, Hill RA. Leptin: a review of its peripheral actions and interactions. Int J Obes Relat Metab Disord. 2002;26:1407–1433. doi: 10.1038/sj.ijo.0802142. [DOI] [PubMed] [Google Scholar]
- 127.Caro JF, Kolaczynski JW, Nyce MR, Ohannesian JP, Opentanova I, Goldman WH, Lynn RB, Zhang PL, Sinha MK, Considine RV. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet. 1996;348:159–161. doi: 10.1016/s0140-6736(96)03173-x. [DOI] [PubMed] [Google Scholar]
- 128.Wang Z, Zhou YT, Kakuma T, Lee Y, Kalra SP, Kalra PS, Pan W, Unger RH. Leptin resistance of adipocytes in obesity: role of suppressors of cytokine signaling. Biochem Biophys Res Commun. 2000;277:20–26. doi: 10.1006/bbrc.2000.3615. [DOI] [PubMed] [Google Scholar]
- 129.Honda H, Ikejima K, Hirose M, Yoshikawa M, Lang T, Enomoto N, Kitamura T, Takei Y, Sato N. Leptin is required for fibrogenic responses induced by thioacetamide in the murine liver. Hepatology. 2002;36:12–21. doi: 10.1053/jhep.2002.33684. [DOI] [PubMed] [Google Scholar]
- 130.Saxena NK, Saliba G, Floyd JJ, Anania FA. Leptin induces increased alpha2(I) collagen gene expression in cultured rat hepatic stellate cells. J Cell Biochem. 2003;89:311–320. doi: 10.1002/jcb.10494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ikejima K, Takei Y, Honda H, Hirose M, Yoshikawa M, Zhang YJ, Lang T, Fukuda T, Yamashina S, Kitamura T, et al. Leptin receptor-mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology. 2002;122:1399–1410. doi: 10.1053/gast.2002.32995. [DOI] [PubMed] [Google Scholar]
- 132.Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem. 1995;270:26746–26749. doi: 10.1074/jbc.270.45.26746. [DOI] [PubMed] [Google Scholar]
- 133.Hu E, Liang P, Spiegelman BM. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J Biol Chem. 1996;271:10697–10703. doi: 10.1074/jbc.271.18.10697. [DOI] [PubMed] [Google Scholar]
- 134.Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257:79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 135.Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, Tataranni PA. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab. 2001;86:1930–1935. doi: 10.1210/jcem.86.5.7463. [DOI] [PubMed] [Google Scholar]
- 136.Statnick MA, Beavers LS, Conner LJ, Corominola H, Johnson D, Hammond CD, Rafaeloff-Phail R, Seng T, Suter TM, Sluka JP, et al. Decreased expression of apM1 in omental and subcutaneous adipose tissue of humans with type 2 diabetes. Int J Exp Diabetes Res. 2000;1:81–88. doi: 10.1155/EDR.2000.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Krakoff J, Funahashi T, Stehouwer CD, Schalkwijk CG, Tanaka S, Matsuzawa Y, Kobes S, Tataranni PA, Hanson RL, Knowler WC, et al. Inflammatory markers, adiponectin, and risk of type 2 diabetes in the Pima Indian. Diabetes Care. 2003;26:1745–1751. doi: 10.2337/diacare.26.6.1745. [DOI] [PubMed] [Google Scholar]
- 138.Louthan MV, Barve S, McClain CJ, Joshi-Barve S. Decreased serum adiponectin: an early event in pediatric nonalcoholic fatty liver disease. J Pediatr. 2005;147:835–838. doi: 10.1016/j.jpeds.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 139.Ruan H, Hacohen N, Golub TR, Van Parijs L, Lodish HF. Tumor necrosis factor-alpha suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3T3-L1 adipocytes: nuclear factor-kappaB activation by TNF-alpha is obligatory. Diabetes. 2002;51:1319–1336. doi: 10.2337/diabetes.51.5.1319. [DOI] [PubMed] [Google Scholar]
- 140.Matsuzawa Y, Funahashi T, Nakamura T. Molecular mechanism of metabolic syndrome X: contribution of adipocytokines adipocyte-derived bioactive substances. Ann N Y Acad Sci. 1999;892:146–154. doi: 10.1111/j.1749-6632.1999.tb07793.x. [DOI] [PubMed] [Google Scholar]
- 141.Kadowaki T, Hara K, Yamauchi T, Terauchi Y, Tobe K, Nagai R. Molecular mechanism of insulin resistance and obesity. Exp Biol Med (Maywood) 2003;228:1111–1117. doi: 10.1177/153537020322801003. [DOI] [PubMed] [Google Scholar]
- 142.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- 144.Fruebis J, Tsao TS, Javorschi S, Ebbets-Reed D, Erickson MR, Yen FT, Bihain BE, Lodish HF. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci USA. 2001;98:2005–2010. doi: 10.1073/pnas.041591798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.López-Bermejo A, Botas P, Funahashi T, Delgado E, Kihara S, Ricart W, Fernández-Real JM. Adiponectin, hepatocellular dysfunction and insulin sensitivity. Clin Endocrinol (Oxf) 2004;60:256–263. doi: 10.1046/j.1365-2265.2004.01977.x. [DOI] [PubMed] [Google Scholar]
- 146.Kamada Y, Tamura S, Kiso S, Matsumoto H, Saji Y, Yoshida Y, Fukui K, Maeda N, Nishizawa H, Nagaretani H, et al. Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology. 2003;125:1796–1807. doi: 10.1053/j.gastro.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 147.Xu A, Yin S, Wong L, Chan KW, Lam KS. Adiponectin ameliorates dyslipidemia induced by the human immunodeficiency virus protease inhibitor ritonavir in mice. Endocrinology. 2004;145:487–494. doi: 10.1210/en.2003-1140. [DOI] [PubMed] [Google Scholar]
- 148.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 149.Kern PA, Saghizadeh M, Ong JM, Bosch RJ, Deem R, Simsolo RB. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J Clin Invest. 1995;95:2111–2119. doi: 10.1172/JCI117899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Van der Poll T, Romijn JA, Endert E, Borm JJ, Büller HR, Sauerwein HP. Tumor necrosis factor mimics the metabolic response to acute infection in healthy humans. Am J Physiol. 1991;261:E457–E465. doi: 10.1152/ajpendo.1991.261.4.E457. [DOI] [PubMed] [Google Scholar]
- 151.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271:665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 152.Panagiotakos DB, Pitsavos C, Yannakoulia M, Chrysohoou C, Stefanadis C. The implication of obesity and central fat on markers of chronic inflammation: The ATTICA study. Atherosclerosis. 2005;183:308–315. doi: 10.1016/j.atherosclerosis.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 153.Ofei F, Hurel S, Newkirk J, Sopwith M, Taylor R. Effects of an engineered human anti-TNF-alpha antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM. Diabetes. 1996;45:881–885. doi: 10.2337/diab.45.7.881. [DOI] [PubMed] [Google Scholar]
- 154.Papanicolaou DA, Vgontzas AN. Interleukin-6: the endocrine cytokine. J Clin Endocrinol Metab. 2000;85:1331–1333. doi: 10.1210/jcem.85.3.6582. [DOI] [PubMed] [Google Scholar]
- 155.Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000;101:1767–1772. doi: 10.1161/01.cir.101.15.1767. [DOI] [PubMed] [Google Scholar]
- 156.Spranger J, Kroke A, Möhlig M, Hoffmann K, Bergmann MM, Ristow M, Boeing H, Pfeiffer AF. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes. 2003;52:812–817. doi: 10.2337/diabetes.52.3.812. [DOI] [PubMed] [Google Scholar]
- 157.Senn JJ, Klover PJ, Nowak IA, Mooney RA. Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes. 2002;51:3391–3399. doi: 10.2337/diabetes.51.12.3391. [DOI] [PubMed] [Google Scholar]
- 158.Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 159.Patel L, Buckels AC, Kinghorn IJ, Murdock PR, Holbrook JD, Plumpton C, Macphee CH, Smith SA. Resistin is expressed in human macrophages and directly regulated by PPAR gamma activators. Biochem Biophys Res Commun. 2003;300:472–476. doi: 10.1016/s0006-291x(02)02841-3. [DOI] [PubMed] [Google Scholar]
- 160.Stejskal D, Adamovská S, Bartek J, Juráková R, Prosková J. Resistin - concentrations in persons with type 2 diabetes mellitus and in individuals with acute inflammatory disease. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2003;147:63–69. [PubMed] [Google Scholar]
- 161.Janowska J, Zahorska-Markiewicz B, Olszanecka-Glinianowicz M. Relationship between serum resistin concentration and proinflammatory cytokines in obese women with impaired and normal glucose tolerance. Metabolism. 2006;55:1495–1499. doi: 10.1016/j.metabol.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 162.Silha JV, Krsek M, Skrha JV, Sucharda P, Nyomba BL, Murphy LJ. Plasma resistin, adiponectin and leptin levels in lean and obese subjects: correlations with insulin resistance. Eur J Endocrinol. 2003;149:331–335. doi: 10.1530/eje.0.1490331. [DOI] [PubMed] [Google Scholar]
- 163.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 164.Banerjee RR, Rangwala SM, Shapiro JS, Rich AS, Rhoades B, Qi Y, Wang J, Rajala MW, Pocai A, Scherer PE, et al. Regulation of fasted blood glucose by resistin. Science. 2004;303:1195–1198. doi: 10.1126/science.1092341. [DOI] [PubMed] [Google Scholar]
- 165.Way JM, Görgün CZ, Tong Q, Uysal KT, Brown KK, Harrington WW, Oliver WR, Willson TM, Kliewer SA, Hotamisligil GS. Adipose tissue resistin expression is severely suppressed in obesity and stimulated by peroxisome proliferator-activated receptor gamma agonists. J Biol Chem. 2001;276:25651–25653. doi: 10.1074/jbc.C100189200. [DOI] [PubMed] [Google Scholar]
- 166.Xia Z, Cianflone K. Acylation-stimulating protein precursor proteins in adipose tissue in human obesity. Metabolism. 2003;52:1360–1366. doi: 10.1016/s0026-0495(03)00254-3. [DOI] [PubMed] [Google Scholar]
- 167.Maslowska M, Sniderman AD, Germinario R, Cianflone K. ASP stimulates glucose transport in cultured human adipocytes. Int J Obes Relat Metab Disord. 1997;21:261–266. doi: 10.1038/sj.ijo.0800396. [DOI] [PubMed] [Google Scholar]
- 168.Cianflone K, Xia Z, Chen LY. Critical review of acylation-stimulating protein physiology in humans and rodents. Biochim Biophys Acta. 2003;1609:127–143. doi: 10.1016/s0005-2736(02)00686-7. [DOI] [PubMed] [Google Scholar]
- 169.Maslowska M, Scantlebury T, Germinario R, Cianflone K. Acute in vitro production of acylation stimulating protein in differentiated human adipocytes. J Lipid Res. 1997;38:1–11. [PubMed] [Google Scholar]
- 170.Saleh J, Christou N, Cianflone K. Regional specificity of ASP binding in human adipose tissue. Am J Physiol. 1999;276:E815–E821. doi: 10.1152/ajpendo.1999.276.5.E815. [DOI] [PubMed] [Google Scholar]
- 171.Massiéra F, Bloch-Faure M, Ceiler D, Murakami K, Fukamizu A, Gasc JM, Quignard-Boulange A, Negrel R, Ailhaud G, Seydoux J, et al. Adipose angiotensinogen is involved in adipose tissue growth and blood pressure regulation. FASEB J. 2001;15:2727–2729. doi: 10.1096/fj.01-0457fje. [DOI] [PubMed] [Google Scholar]
- 172.Umemura S, Nyui N, Tamura K, Hibi K, Yamaguchi S, Nakamaru M, Ishigami T, Yabana M, Kihara M, Inoue S, et al. Plasma angiotensinogen concentrations in obese patients. Am J Hypertens. 1997;10:629–633. doi: 10.1016/s0895-7061(97)00053-8. [DOI] [PubMed] [Google Scholar]
- 173.Dixon JB, Bhathal PS, Jonsson JR, Dixon AF, Powell EE, O'Brien PE. Pro-fibrotic polymorphisms predictive of advanced liver fibrosis in the severely obese. J Hepatol. 2003;39:967–971. doi: 10.1016/s0168-8278(03)00459-8. [DOI] [PubMed] [Google Scholar]
- 174.Reitman ML, Arioglu E, Gavrilova O, Taylor SI. Lipoatrophy revisited. Trends Endocrinol Metab. 2000;11:410–416. doi: 10.1016/s1043-2760(00)00309-x. [DOI] [PubMed] [Google Scholar]
- 175.Gavrilova O, Marcus-Samuels B, Graham D, Kim JK, Shulman GI, Castle AL, Vinson C, Eckhaus M, Reitman ML. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J Clin Invest. 2000;105:271–278. doi: 10.1172/JCI7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Addy CL, Gavrila A, Tsiodras S, Brodovicz K, Karchmer AW, Mantzoros CS. Hypoadiponectinemia is associated with insulin resistance, hypertriglyceridemia, and fat redistribution in human immunodeficiency virus-infected patients treated with highly active antiretroviral therapy. J Clin Endocrinol Metab. 2003;88:627–636. doi: 10.1210/jc.2002-020795. [DOI] [PubMed] [Google Scholar]
- 177.Reue K, Phan J. Metabolic consequences of lipodystrophy in mouse models. Curr Opin Clin Nutr Metab Care. 2006;9:436–441. doi: 10.1097/01.mco.0000232904.82038.db. [DOI] [PubMed] [Google Scholar]
- 178.Estrada V, Martínez-Larrad MT, González-Sánchez JL, de Villar NG, Zabena C, Fernández C, Serrano-Ríos M. Lipodystrophy and metabolic syndrome in HIV-infected patients treated with antiretroviral therapy. Metabolism. 2006;55:940–945. doi: 10.1016/j.metabol.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 179.Wolfsdorf J, Sadeghi-Nejad A, Senior B. Leptin-replacement therapy in lipodystrophy. N Engl J Med. 2002;346:2008–2009; author reply 2009-2010. [PubMed] [Google Scholar]
- 180.Clark JM, Diehl AM. Nonalcoholic fatty liver disease: an underrecognized cause of cryptogenic cirrhosis. JAMA. 2003;289:3000–3004. doi: 10.1001/jama.289.22.3000. [DOI] [PubMed] [Google Scholar]
- 181.Caldwell SH, Oelsner DH, Iezzoni JC, Hespenheide EE, Battle EH, Driscoll CJ. Cryptogenic cirrhosis: clinical characterization and risk factors for underlying disease. Hepatology. 1999;29:664–669. doi: 10.1002/hep.510290347. [DOI] [PubMed] [Google Scholar]
- 182.Bugianesi E, Leone N, Vanni E, Marchesini G, Brunello F, Carucci P, Musso A, De Paolis P, Capussotti L, Salizzoni M, et al. Expanding the natural history of nonalcoholic steatohepatitis: from cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology. 2002;123:134–140. doi: 10.1053/gast.2002.34168. [DOI] [PubMed] [Google Scholar]
- 183.Ekstedt M, Franzén LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, Kechagias S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44:865–873. doi: 10.1002/hep.21327. [DOI] [PubMed] [Google Scholar]
- 184.Fassio E, Alvarez E, Domínguez N, Landeira G, Longo C. Natural history of nonalcoholic steatohepatitis: a longitudinal study of repeat liver biopsies. Hepatology. 2004;40:820–826. doi: 10.1002/hep.20410. [DOI] [PubMed] [Google Scholar]
- 185.Musso G, Gambino R, De Michieli F, Cassader M, Rizzetto M, Durazzo M, Fagà E, Silli B, Pagano G. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology. 2003;37:909–916. doi: 10.1053/jhep.2003.50132. [DOI] [PubMed] [Google Scholar]
- 186.Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut. 2001;48:206–211. doi: 10.1136/gut.48.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Singh H, Pollock R, Uhanova J, Kryger M, Hawkins K, Minuk GY. Symptoms of obstructive sleep apnea in patients with nonalcoholic fatty liver disease. Dig Dis Sci. 2005;50:2338–2343. doi: 10.1007/s10620-005-3058-y. [DOI] [PubMed] [Google Scholar]
- 188.Angulo P, Keach JC, Batts KP, Lindor KD. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology. 1999;30:1356–1362. doi: 10.1002/hep.510300604. [DOI] [PubMed] [Google Scholar]
- 189.Dixon JB, Bhathal PS, O'Brien PE. Nonalcoholic fatty liver disease: predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology. 2001;121:91–100. doi: 10.1053/gast.2001.25540. [DOI] [PubMed] [Google Scholar]
- 190.Cassiman D, Libbrecht L, Desmet V, Denef C, Roskams T. Hepatic stellate cell/myofibroblast subpopulations in fibrotic human and rat livers. J Hepatol. 2002;36:200–209. doi: 10.1016/s0168-8278(01)00260-4. [DOI] [PubMed] [Google Scholar]
- 191.Reeves HL, Friedman SL. Activation of hepatic stellate cells--a key issue in liver fibrosis. Front Biosci. 2002;7:d808–d826. doi: 10.2741/reeves. [DOI] [PubMed] [Google Scholar]
- 192.Bugianesi E, Manzini P, D'Antico S, Vanni E, Longo F, Leone N, Massarenti P, Piga A, Marchesini G, Rizzetto M. Relative contribution of iron burden, HFE mutations, and insulin resistance to fibrosis in nonalcoholic fatty liver. Hepatology. 2004;39:179–187. doi: 10.1002/hep.20023. [DOI] [PubMed] [Google Scholar]
- 193.Svegliati-Baroni G, Ridolfi F, Di Sario A, Casini A, Marucci L, Gaggiotti G, Orlandoni P, Macarri G, Perego L, Benedetti A, et al. Insulin and insulin-like growth factor-1 stimulate proliferation and type I collagen accumulation by human hepatic stellate cells: differential effects on signal transduction pathways. Hepatology. 1999;29:1743–1751. doi: 10.1002/hep.510290632. [DOI] [PubMed] [Google Scholar]
- 194.Paradis V, Perlemuter G, Bonvoust F, Dargere D, Parfait B, Vidaud M, Conti M, Huet S, Ba N, Buffet C, et al. High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: a potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology. 2001;34:738–744. doi: 10.1053/jhep.2001.28055. [DOI] [PubMed] [Google Scholar]
- 195.Parola M, Robino G. Oxidative stress-related molecules and liver fibrosis. J Hepatol. 2001;35:297–306. doi: 10.1016/s0168-8278(01)00142-8. [DOI] [PubMed] [Google Scholar]
- 196.Aleffi S, Petrai I, Bertolani C, Parola M, Colombatto S, Novo E, Vizzutti F, Anania FA, Milani S, Rombouts K, et al. Upregulation of proinflammatory and proangiogenic cytokines by leptin in human hepatic stellate cells. Hepatology. 2005;42:1339–1348. doi: 10.1002/hep.20965. [DOI] [PubMed] [Google Scholar]
- 197.Meier U, Gressner AM. Endocrine regulation of energy metabolism: review of pathobiochemical and clinical chemical aspects of leptin, ghrelin, adiponectin, and resistin. Clin Chem. 2004;50:1511–1525. doi: 10.1373/clinchem.2004.032482. [DOI] [PubMed] [Google Scholar]
- 198.Leu JI, Crissey MA, Taub R. Massive hepatic apoptosis associated with TGF-beta1 activation after Fas ligand treatment of IGF binding protein-1-deficient mice. J Clin Invest. 2003;111:129–139. doi: 10.1172/JCI16712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Jansson PA, Larsson A, Lönnroth PN. Relationship between blood pressure, metabolic variables and blood flow in obese subjects with or without non-insulin-dependent diabetes mellitus. Eur J Clin Invest. 1998;28:813–818. doi: 10.1046/j.1365-2362.1998.00360.x. [DOI] [PubMed] [Google Scholar]
- 200.Straczkowski M, Dzienis-Straczkowska S, Stêpieñ A, Kowalska I, Szelachowska M, Kinalska I. Plasma interleukin-8 concentrations are increased in obese subjects and related to fat mass and tumor necrosis factor-alpha system. J Clin Endocrinol Metab. 2002;87:4602–4606. doi: 10.1210/jc.2002-020135. [DOI] [PubMed] [Google Scholar]
- 201.De Pergola G, De Mitrio V, Giorgino F, Sciaraffia M, Minenna A, Di Bari L, Pannacciulli N, Giorgino R. Increase in both pro-thrombotic and anti-thrombotic factors in obese premenopausal women: relationship with body fat distribution. Int J Obes Relat Metab Disord. 1997;21:527–535. doi: 10.1038/sj.ijo.0800435. [DOI] [PubMed] [Google Scholar]