

Abstract
The thrombophilia in adult life has major implications in the hepatic vessels. The resulting portal vein thrombosis has various outcomes and complications. Esophageal varices, portal gastropathy, ascites, severe hypersplenism and liver failure needing liver transplantation are known well. The newly formed collateral venous circulation showing itself as pseudocholangicarcinoma sign and its possible clinical reflection as cholestasis are also known from a long time. The management strategies for these complications of portal vein thrombosis are not different from their counterpart which is cirrhotic portal hypertension, but the prognosis is unquestionably better in former cases. In this review we present and discuss the portal vein thrombosis, etiology and the resulting clinical pictures. There are controversial issues in nomenclature, management (including anticoagulation problems), follow up strategies and liver transplantation. In the light of the current knowledge, we discuss some controversial issues in literature and present our experience and our proposals about this group of patients.
Keywords: Portal vein thrombosis, Pseudocholangiocarcinoma sign, Thrombophilia
INTRODUCTION
Portal vein thrombosis (PVT) refers to total or near-total obstruction of blood flow. This obstruction may result from invasion of primary or secondary tumors of liver and thrombosis of portal vein. The obstruction may extend towards liver involving intrahepatic portal branches or may extend distally to involve splenic or mesenteric branches. In some occasions extensive involvement of all of these vessels may occur with intestinal ischemia. Therefore, the involved segment of portal venous system and the degree of compensatory mechanisms determines the clinical presentation. Although PVT has been observed most commonly in the setting of cirrhosis or liver tumors, the discussion of PVT from this point highlights chronic, noncirrhotic and nontumoral PVT unless otherwise mentioned.
PVT has important outcomes for the liver. On cessation of blood flow liver loses about two thirds of its blood supply. Interestingly, while acute arterial blockage usually results in severe hepatic failure or death, this condition is tolerated well and patients are almost asymptomatic because of compensatory mechanisms. First compensatory mechanism is the well-known "arterial vasodilation" of the hepatic artery (which is a vascular reflex seen in every dual-vessel supplied organ) also observed during portal vein clamping in liver surgery[1]. This "arterial rescue" stabilizes the liver functions at a normal level in the acute stages of PVT. The second rescue mechanism is the "venous rescue" involving rapid development of collateral vessels to bypass the obstructed part. These vessels begin to form in few days after the obstruction and organize into a cavernomatous transformation in 3-5 wk[2,3]. This condition has acutely developing harmful effects over intestinal circulation compromising the patient's life before development of portal hypertension and its results.
Beside these compensatory mechanisms, liver bears the burden of decreased blood to a some extent. The decreased portal blood flow stimulates apoptosis in hepatocytes of rats when portal vein is obliterated in a gradual fashion[4] and increases mitotic activity of hepatocytes in the unaffected lobe. The latter effect is well-known from selective presurgery portal vein obliteration performed in intent to stimulate the hypertrophy of the opposite lobe of the liver used in cancer surgery. Gradual loss of hepatic mass may be responsible for the case of mild to moderate degree of hepatic synthetic dysfunction noted in advanced stages.
PREVALENCE OF PVT
The prevalence of PVT in the general population was studied recently in an autopsy study by Ogren M et al[5]. They found the population prevalence of PVT as high as 1%. And they reported that in PVT patients 28% had cirrhosis, 67% had liver malignancy (primary or metastatic), 10% had inflammatory intraabdominal disease and only 3% had myeloproliferative disease (MPD). This result points out the overall prevalence of PVT is higher than expected. It can be speculated that PVT is not always symptomatic or result in complications like variceal bleeding and ascites. The pathophysiological mechanisms under these "silent PVT patients" are not clear.
ETIOLOGY
PVT has various causes. The design of the studies and the limits evaluating the cause of PVT has changed parallel to the evolution in medical and genetic technology. In previous studies between 1979-1997, PVT were attributed mostly to trauma (5%-17%), intraabdominal sepsis (5%-36%), umbilical sepsis (5%-12%)[6] (important cause of PVT in children), pancreatitis (4%-5%) and prothrombotic disorders (2%-28%), but in nearly 50% of the patients etiology remained unidentified[7-11]. But with the advent of better medical care, potent antibiotic treatment, advanced medical technology, discovery of various causes of genetic thrombophilia and with better understanding of the coagulation system, this profile has also changed.
Identification of etiology usually starts by exclusion of local causes such as cirrhosis, primary or metastatic cancer of liver, pylephlebitis, liver cysts, vascular abnormalities (like webs or aneurysms) and pancreatitis. Beside routine investigations such as liver biopsies or Doppler-USG studies, magnetic resonance or CT based (3 dimensional angiographic images also available) techniques are helpful at this stage. If no local lesion is found, then the studies are directed for a possible trombophilic condition. After all, if no cause can be found, then the condition should be named as "idiopathic PVT".
Denninger et al[12] reported that underlying prothrombotic condition was identified in 72% of PVT patients. There was one or more prothrombotic condition in 26 out of 36 patients studied and primary MPDs were found to be the leading cause with a prevalence of 30%. Other studies by Valla et al[13-15] and De Stefano et al[16] reported similar figures that as a cause of PVT, MPD (overt or occult) rank first in the thrombophilic conditions. Valla et al also proposed that overt or latent MPD may be responsible for thrombophilic milieu for PVT in 48% of patients.
An interesting improvement in the last decade is the discovery of a mutation in Janus kinase 2 (V617F activating tyrosine kinase mutation, JAK2) which leads to MPDs. This discovery of JAK2 mutation is a milestone in early diagnosis of overt or silent MPD[17-19]. The prevalence of JAK2 mutation in PVT (splenic and mesenteric vein thrombosis were also included) is studied in two studies recently[20,21]. They found that JAK2 mutation was present in 17.2%-35.6% of PVT patients. Although it is a promising discovery, there are some drawbacks in JAK2 mutation analysis particularly in PVT (also effective for Budd-Chiari syndrome). First, the diagnostic criteria for MPD in PVT patient population should be refined and a guideline should be developed. Second, further studies with large number of patients should be performed. Lastly, JAK2 mutation is positive only in 50%-90% of patients with overt or occult MPDs (depending on the subtype of MPD) and should not be appointed as a gold standard screening test for MPD. This topic is hot and open to further studies.
To investigate PC, PS and AT deficiencies in patients with PVT is a challenge for the clinician. It is difficult to interpret when the results are found to be lower than normal. There are studies researching whether the low values are result of liver dysfunction or signal a frank deficiency. One of the most important studies is performed in children. Mack et al[22] studied the coagulation parameters (including factors synthesized only in liver) in 11 children with PVT who underwent a surgical correction (mesenterico-left portal vein bypass: Rex Shunt) of the portal venous system before and after surgery. The investigators found a significant correction of both coagulation factors (factors II, V and VII) suggested also by improvement in prothrombin time and of PC and PS levels after surgery. The same parameters studied in other 2 children receiving distal splenorenal shunt in this study and 7 children in an other study receiving H-type mesocaval shunt[23] did not improve as the Rex shunting supporting the importance of decreased portal vein flow in the development of secondary PC and PS deficiencies. Fisher et al[24] studied the same topic by combining the family investigation of PC and PS deficiencies in patients with PVT. They found out there were only 3 family cases of natural anticoagulant deficiencies out of 18 patients marking a secondary finding. Another support to this finding and the previous results is that they have found significant decreases in AT, PC and PS concentrations after distal splenorenal shunt surgery (3 patients). All the mentioned studies confirm the decreased portal blood flow have a great impact over the synthesis of natural anticoagulant and coagulant proteins. The shunt procedures directing the blood from portal system towards systemic circulation may worsen this as shown from comparison of results of splenorenal shunt vs Rex shunt. We think this phenomenon of secondary loss of coagulant and anticoagulant proteins must be considered carefully in clinal practice when anticoagulation with warfarin is considered.
Secondary deficiencies of genetic thrombophilia causes like AT, PC and PS is still a matter of diagnotic challenge. To overcome this problem, there seems to be a narrow range of solutions such as family study (first-degree relatives) of deficient proteins in an attempt to detect familial aggregation of mutant genes and primary genetic studies of the patient to detect the mutant gene(s). These studies are important but not practical and not applicable in every facility. A practical screening method for detection of natural anticoagulant deficiencies in patients with liver disease was proposed by Pabinger et al[25] and used first by Janssen et al[26] in a clinical study which uses the ratio of natural anticoagulant factor to coagulation factors (synthesized only in liver). The formula is the ratio of PS or PC or AT (whichever tested) to [(factor II + factor X)/2]. If the result of this ratio is lower than 70%, then this may signal a shortage of the tested natural anticoagulant protein significantly disproportionate to decreased synthesis of proteins from liver. This formula is easy to use in clinics and is practical to find out a possible genetic deficiency.
PVT cannot be solely charged to one cause. It is now widely accepted that the PVT occurs from both a primary thrombophilic milieu and a cause that triggers the formation of the pathological thrombus in portal circulation. The aforementioned factors are considered as thrombophilic background rather than the primary etiologic reason according to multifactorial theory of thrombogenesis. The other cofactors required to develop a thrombus can be grouped as local and systemic factors most of which are potentially curable or preventable factors.
DIAGNOSIS
The PVT can be defined as presence of portal hypertension (shown by presence of splenomegaly, esophageal or gastric varices) and identification of tubular and serpentine vascular structures resembling cavernoma at the site of hepatic hilum replacing the portal vein. This anatomical description can be made either by use of gray scale ultrasound (findings also shown by Doppler-US) and the diagnosis was confirmed by noninvasive radiological imaging techniques such as computed tomography angiography (Figure 1) and magnetic resonance angiography or digital splenoportography. All the patients should undergo a thorough medical evaluation including a detailed physical examination and questioning of the history.
Figure 1.
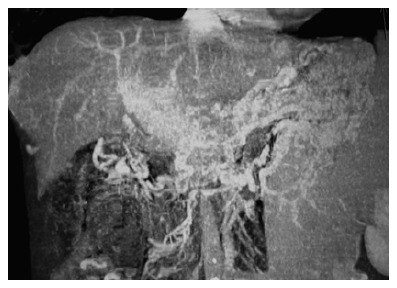
A case of chronic PVT in which typical cavernous transformation is observed. The computed tomography angiography of the portal vein shows intensive collaterals around portal hilus and midline abdominal structures. Portal vein is not visible and replaced by collaterals.
CLINICAL OUTCOMES
The outcomes of the PVT vary from one patient to another and depend on some important factors. The condition may present itself as one of its complications. But because of lack of convincing evidence in the literature, the most common early presentation of PVT (not related to cirrhosis) is still not known. In our institutional experience, most common presentation is variceal bleeding followed by pancytopenia due to hypersplenism. Undefined cholestasis related to pseudocholangiocarcinoma sign (without icterus) is also one of the rare presentations.
With the advent and wide distribution of USG and Doppler-USG, the condition is becoming diagnosed earlier and a patient presenting with ascites (which is a late finding in course of PVT) is almost not seen. Webb and Sherlock[6] reported in 1979 that out of 97 patients with PVT 13 presented with ascites in a high rate (13.5%). There are still some PVT cases presenting as ascites in underdeveloped parts of the world related to absence of early diagnosis with similar rates around 13%[27].
Esophageal and gastric varices related bleeding contribute to the most important cause of morbidity and hospitalization in this group of patients. When the characteristics of variceal bleeding are considered, major differences between cirrhosis vs PVT related bleeding must be pointed out. First, the risk of variceal bleeding in cirrhosis is roughly 80-120 times more than PVT (noncirrhotic) related variceal bleeding[28-30] and esophageal varices (irrespective of size) are noted in about 90% percent of patients. Gastric varices are mostly seen concomitantly with esophageal varices in about 40% of the patients in PVT while portal gastropathy is also a rare feature of this condition[30,31]. Compared with portosystemic varices noted in cirrhotic patients, the varices in PVT patients have some special charateristics like; 1-"varice-on-varice" finding is less commonly observed, 2-the sizes of the varices are smaller, 3-associated portal gastropathy is less commonly present[32]. The retrospective study (examined the determinants of survival in a heterogenous group of patients including 124 noncirrhotic vs 48 cirrhotic PVT cases) of Janssen et al[28] showed that either the presence of varices or bleeding episodes of varices had no impact on survival rates and the most common cause of death was malignancy related causes. Twenty four percent of the study population had malignancies like hepatocellular cancer, pancreatic cancer, or other malignancy elsewhere metastatic to the liver which presented itself in liver as PVT pointing to end stage malignancies.
Although the risk of variceal bleeding is low in non-cirrhotic patients, managing bleeding varices is not different from cirrhotic patients. Unfortunately there are no studies in the literature comparing endoscopic procedures vs surgical procedures in acutely bleeding varices and about the topic of primary prophylaxis. We believe that primary prophylaxis with endoscopic procedures in effort to clear varices should be applied with careful surveying and secondary prophylaxis with combination of endoscopic procedures and medical treatment despite beta-blokers may work although there is not objective and suffient evidence.
In contrast, beta-blockers may result in more sluggish portal flow as is same issue for nitrates and terlipressin increasing the risk of progression of thrombosis and even worsening of portal hypertension. This decision must be adjusted with the patient's condition rather than a standart medical care.
One interesting and misleading clinical condition that may occur during the course of the PVT is "pseudocholangiocarcinoma sign" (PSCS). After the obstruction in the portal system, the "venous rescue" begins to occur immediately which takes time about 5 wk[2] forming new vessels around intrahepatic, extrahepatic biliary tracts and around gallbladder (majorly, vascular plexus of Saint and Petren enlarge and dilate to become large serpentine vessels) named as "portal cavernomatous transformation". Sometimes these vessels can be small in caliber to visualize depending on the extent of portal flow and capacity of new vessel formation, but if a careful ERCP is performed the direct effects of these vessels over biliary ducts or main bile duct as strictures, displacements, thumprinting effects or irregularity can be seen in at least 80% of the patients[30]. This condition mimics a cholangiocellular cancer in ERCP and therefore called PSCS[32,33].
The clinical impact of these changes results in a wide spectrum of findings ranging from normal biochemical and physical findings to overt cholestasis with increased cholestatic enzymes and liver injury (rarely observed). Although a the term called "portal biliopathy" and a classification system is proposed[34] we believe that this term and classification system are impracticable to use because; (1) new collaterals form depending on the site and level of portal vein obstruction, (2) wide range of anatomical variations between human beings in this anatomical site resulting in different types of cavernous transformations, (3) the nature of new vessel formation is unpredictable and not standart in all portal vein thrombosis patients, d) the classification system does not have impact over treatment or follow up strategy. We propose that only defining the presence or absence of PSCS is a satisfactory practice not to complicate the picture further because the clinically obvious cholestasis or jaundice is observed in only about 5%[35]. Therefore this physiological compensatory response is not a "-pathy" but just an adaptive change with a low degree of clinical importance.
The clinically obvious cholestasis either presenting itself as repeating biliary stones or cholangitis or liver injury can be treated with conventional methods including stenting, papillotomy or even surgery.
Hypersplenism is a finding of latent or chronic PVT cases and this complication is important when anti-coagulation treatment is considered. Hypersplenism is almost always present except in the rare patient with a partial thrombus in one branch of the portal vein with the other branch remained intact presenting with a mild degree of portal hypertension. The levels of all blood elements begin to decrease as the condition worsens and severe hypersplenism is now considered one of the most important indications in this group of patients to undergo a shunt surgery combined with or without a splenectomy. In the clinical setting of hypersplenism with low platelet counts combined with esophageal varices raises concerns about the safety of anticoagulation. Unfortunately, the literature lacks information about the safety and long-term results of anticoagulation in well designed prospective controlled studies. A study to clarify this controversial topic was a retrospective analysis. In this study[29] Condat et al included 136 PVT patients in whom 84 was receiving anticoagulant treatment. The study has some limits like heterogenous groups (pretreatment endoscopies of all patients and standardization of a homogeneous varice distribution between two groups is not carried out) and lack of information about comorbid conditions of the engaging patients, but the study has a low statistical degree of evidence to favor anticoagulation treatment. Anticoagulation was not found to be a risk factor for bleeding in this study while nonanticoagulation resulted in more thrombotic recurrences as expected. From our institutional experience, we believe that it may be a good practice to select patients according to their co-morbid conditions, degree of hypersplenism (low platelet counts may be a relative contraindication for anticoagulation) and the condition of varices (eradication combined with or without medical prophylaxis before start of treatment may be considered to decrese bleeding related risks).
LIVER TRANSPLANTATION IN PVT
Previously (during the 1990s) PVT was an established contrain-dication for liver transplantation. But this myth has been vanished with great innovations in both medical-surgical care and radiological interventions. Currently, chronic PVT itself may present as an indication for liver transplantation because of synthetic failure of liver. Although either living donor liver transplanation (LDLT) and deceased donor liver transplantation (DDLT) can be performed, it is the experience and ability of the medical care center that should make the final decision about the type of transplantation. In various studies[36-41] it has been shown that both surgical techniques (thrombectomy, thromboendvenectomy with venous reconstruction, interposition of vein graft, porto-caval hemitransposition and others) and radiological endovascular interventions can be used to overcome venous obstruction in the recipient[42]. A major drawback about these studies is that most of the patient population is formed of PVT patients who had underlying cirrhosis or hepatocellular cancer. Noncirrhotic and nonmalignant chronic PVT patients form a minority.
CONCLUSION
We need further studies to outline and describe the indi-cations for liver transplantation in this patient population. Also it is essential to form universal anatomical classi-fication system of PVT for a standard language in litera-ture. In this regard, the system proposed by Yerdel et al[43] can be suggested.
Footnotes
S- Editor Wang J L- Editor Kremer M E- Editor Wang HF
References
- 1.Henderson JM, Gilmore GT, Mackay GJ, Galloway JR, Dodson TF, Kutner MH. Hemodynamics during liver transplantation: the interactions between cardiac output and portal venous and hepatic arterial flows. Hepatology. 1992;16:715–718. doi: 10.1002/hep.1840160316. [DOI] [PubMed] [Google Scholar]
- 2.Ohnishi K, Okuda K, Ohtsuki T, Nakayama T, Hiyama Y, Iwama S, Goto N, Nakajima Y, Musha N, Nakashima T. Formation of hilar collaterals or cavernous transformation after portal vein obstruction by hepatocellular carcinoma. Observations in ten patients. Gastroenterology. 1984;87:1150–1153. [PubMed] [Google Scholar]
- 3.De Gaetano AM, Lafortune M, Patriquin H, De Franco A, Aubin B, Paradis K. Cavernous transformation of the portal vein: patterns of intrahepatic and splanchnic collateral circulation detected with Doppler sonography. AJR Am J Roentgenol. 1995;165:1151–1155. doi: 10.2214/ajr.165.5.7572494. [DOI] [PubMed] [Google Scholar]
- 4.Ogren M, Bergqvist D, Wåhlander K, Eriksson H, Sternby NH. Trousseau's syndrome - what is the evidence? A population-based autopsy study. Thromb Haemost. 2006;95:541–545. doi: 10.1160/TH05-10-0694. [DOI] [PubMed] [Google Scholar]
- 5.Bilodeau M, Aubry MC, Houle R, Burnes PN, Ethier C. Evaluation of hepatocyte injury following partial ligation of the left portal vein. J Hepatol. 1999;30:29–37. doi: 10.1016/s0168-8278(99)80005-1. [DOI] [PubMed] [Google Scholar]
- 6.Webb LJ, Sherlock S. The aetiology, presentation and natural history of extra-hepatic portal venous obstruction. Q J Med. 1979;48:627–639. [PubMed] [Google Scholar]
- 7.Cardin F, Graffeo M, McCormick PA, McIntyre N, Burroughs A. Adult "idiopathic" extrahepatic venous thrombosis. Importance of putative "latent" myeloproliferative disorders and comparison with cases with known etiology. Dig Dis Sci. 1992;37:335–339. doi: 10.1007/BF01307724. [DOI] [PubMed] [Google Scholar]
- 8.Stringer MD, Heaton ND, Karani J, Olliff S, Howard ER. Patterns of portal vein occlusion and their aetiological significance. Br J Surg. 1994;81:1328–1331. doi: 10.1002/bjs.1800810923. [DOI] [PubMed] [Google Scholar]
- 9.Orozco H, Takahashi T, Mercado MA, Prado E, Chan C. Surgical management of extrahepatic portal hypertension and variceal bleeding. World J Surg. 1994;18:246–250. doi: 10.1007/BF00294409. [DOI] [PubMed] [Google Scholar]
- 10.Orloff MJ, Orloff MS, Rambotti M. Treatment of bleeding esophagogastric varices due to extrahepatic portal hypertension: results of portal-systemic shunts during 35 years. J Pediatr Surg. 1994;29:142–151; discussion 151-154. doi: 10.1016/0022-3468(94)90309-3. [DOI] [PubMed] [Google Scholar]
- 11.Vleggaar FP, van Buuren HR, Schalm SW. Endoscopic sclerotherapy for bleeding oesophagogastric varices secondary to extrahepatic portal vein obstruction in an adult Caucasian population. Eur J Gastroenterol Hepatol. 1998;10:81–85. doi: 10.1097/00042737-199801000-00015. [DOI] [PubMed] [Google Scholar]
- 12.Denninger MH, Chaït Y, Casadevall N, Hillaire S, Guillin MC, Bezeaud A, Erlinger S, Briere J, Valla D. Cause of portal or hepatic venous thrombosis in adults: the role of multiple concurrent factors. Hepatology. 2000;31:587–591. doi: 10.1002/hep.510310307. [DOI] [PubMed] [Google Scholar]
- 13.Valla D, Casadevall N, Huisse MG, Tulliez M, Grange JD, Muller O, Binda T, Varet B, Rueff B, Benhamou JP. Etiology of portal vein thrombosis in adults. A prospective evaluation of primary myeloproliferative disorders. Gastroenterology. 1988;94:1063–1069. doi: 10.1016/0016-5085(88)90567-7. [DOI] [PubMed] [Google Scholar]
- 14.Valla D, Casadevall N, Lacombe C, Varet B, Goldwasser E, Franco D, Maillard JN, Pariente EA, Leporrier M, Rueff B. Primary myeloproliferative disorder and hepatic vein thrombosis. A prospective study of erythroid colony formation in vitro in 20 patients with Budd-Chiari syndrome. Ann Intern Med. 1985;103:329–334. doi: 10.7326/0003-4819-103-3-329. [DOI] [PubMed] [Google Scholar]
- 15.Valla DC, Condat B. Portal vein thrombosis in adults: pathophysiology, pathogenesis and management. J Hepatol. 2000;32:865–871. doi: 10.1016/s0168-8278(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 16.De Stefano V, Teofili L, Leone G, Michiels JJ. Spontaneous erythroid colony formation as the clue to an underlying myeloproliferative disorder in patients with Budd-Chiari syndrome or portal vein thrombosis. Semin Thromb Hemost. 1997;23:411–418. doi: 10.1055/s-2007-996117. [DOI] [PubMed] [Google Scholar]
- 17.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 18.James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, Garçon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 19.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 20.Colaizzo D, Amitrano L, Tiscia GL, Scenna G, Grandone E, Guardascione MA, Brancaccio V, Margaglione M. The JAK2 V617F mutation frequently occurs in patients with portal and mesenteric venous thrombosis. J Thromb Haemost. 2007;5:55–61. doi: 10.1111/j.1538-7836.2006.02277.x. [DOI] [PubMed] [Google Scholar]
- 21.Primignani M, Barosi G, Bergamaschi G, Gianelli U, Fabris F, Reati R, Dell'Era A, Bucciarelli P, Mannucci PM. Role of the JAK2 mutation in the diagnosis of chronic myeloproliferative disorders in splanchnic vein thrombosis. Hepatology. 2006;44:1528–1534. doi: 10.1002/hep.21435. [DOI] [PubMed] [Google Scholar]
- 22.Mack CL, Superina RA, Whitington PF. Surgical restoration of portal flow corrects procoagulant and anticoagulant deficiencies associated with extrahepatic portal vein thrombosis. J Pediatr. 2003;142:197–199. doi: 10.1067/mpd.2003.93. [DOI] [PubMed] [Google Scholar]
- 23.Dubuisson C, Boyer-Neumann C, Wolf M, Meyer D, Bernard O. Protein C, protein S and antithrombin III in children with portal vein obstruction. J Hepatol. 1997;27:132–135. doi: 10.1016/s0168-8278(97)80292-9. [DOI] [PubMed] [Google Scholar]
- 24.Fisher NC, Wilde JT, Roper J, Elias E. Deficiency of natural anticoagulant proteins C, S, and antithrombin in portal vein thrombosis: a secondary phenomenon? Gut. 2000;46:534–539. doi: 10.1136/gut.46.4.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pabinger I, Allaart CF, Hermans J, Briët E, Bertina RM. Hereditary protein C-deficiency: laboratory values in transmitters and guidelines for the diagnostic procedure. Report on a study of the SSC Subcommittee on Protein C and Protein S. Protein C Transmitter Study Group. Thromb Haemost. 1992;68:470–474. [PubMed] [Google Scholar]
- 26.Janssen HL, Meinardi JR, Vleggaar FP, van Uum SH, Haagsma EB, van Der Meer FJ, van Hattum J, Chamuleau RA, Adang RP, Vandenbroucke JP, et al. Factor V Leiden mutation, prothrombin gene mutation, and deficiencies in coagulation inhibitors associated with Budd-Chiari syndrome and portal vein thrombosis: results of a case-control study. Blood. 2000;96:2364–2368. [PubMed] [Google Scholar]
- 27.Sarin SK, Aggarwal SR. Idiopathic portal hypertension. Digestion. 1998;59:420–423. doi: 10.1159/000007502. [DOI] [PubMed] [Google Scholar]
- 28.Janssen HL, Wijnhoud A, Haagsma EB, van Uum SH, van Nieuwkerk CM, Adang RP, Chamuleau RA, van Hattum J, Vleggaar FP, Hansen BE, et al. Extrahepatic portal vein thrombosis: aetiology and determinants of survival. Gut. 2001;49:720–724. doi: 10.1136/gut.49.5.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Condat B, Pessione F, Hillaire S, Denninger MH, Guillin MC, Poliquin M, Hadengue A, Erlinger S, Valla D. Current outcome of portal vein thrombosis in adults: risk and benefit of anticoagulant therapy. Gastroenterology. 2001;120:490–497. doi: 10.1053/gast.2001.21209. [DOI] [PubMed] [Google Scholar]
- 30.Sarin SK, Agarwal SR. Extrahepatic portal vein obstruction. Semin Liver Dis. 2002;22:43–58. doi: 10.1055/s-2002-23206. [DOI] [PubMed] [Google Scholar]
- 31.Bayraktar Y, Balkanci F, Uzunalimoglu B, Gokoz A, Koseoglu T, Batman F, Gurakar A, Van Thiel DH, Kayhan B. Is portal hypertension due to liver cirrhosis a major factor in the development of portal hypertensive gastropathy? Am J Gastroenterol. 1996;91:554–558. [PubMed] [Google Scholar]
- 32.Bayraktar Y, Balkanci F, Kayhan B, Ozenç A, Arslan S, Telatar H. Bile duct varices or "pseudo-cholangiocarcinoma sign" in portal hypertension due to cavernous transformation of the portal vein. Am J Gastroenterol. 1992;87:1801–1806. [PubMed] [Google Scholar]
- 33.Bayraktar Y, Balkanci F, Ozenc A, Arslan S, Koseoglu T, Ozdemir A, Uzunalimoglu B, Telatar H, Gurakar A, Van Thiel DH. The "pseudo-cholangiocarcinoma sign" in patients with cavernous transformation of the portal vein and its effect on the serum alkaline phosphatase and bilirubin levels. Am J Gastroenterol. 1995;90:2015–2019. [PubMed] [Google Scholar]
- 34.Chandra R, Kapoor D, Tharakan A, Chaudhary A, Sarin SK. Portal biliopathy. J Gastroenterol Hepatol. 2001;16:1086–1092. doi: 10.1046/j.1440-1746.2001.02562.x. [DOI] [PubMed] [Google Scholar]
- 35.Dilawari JB, Chawla YK. Pseudosclerosing cholangitis in extrahepatic portal venous obstruction. Gut. 1992;33:272–276. doi: 10.1136/gut.33.2.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manzanet G, Sanjuán F, Orbis P, López R, Moya A, Juan M, Vila J, Asensi J, Sendra P, Ruíz J, et al. Liver transplantation in patients with portal vein thrombosis. Liver Transpl. 2001;7:125–131. doi: 10.1053/jlts.2001.21295. [DOI] [PubMed] [Google Scholar]
- 37.Dumortier J, Czyglik O, Poncet G, Blanchet MC, Boucaud C, Henry L, Boillot O. Eversion thrombectomy for portal vein thrombosis during liver transplantation. Am J Transplant. 2002;2:934–938. doi: 10.1034/j.1600-6143.2002.21009.x. [DOI] [PubMed] [Google Scholar]
- 38.Gerunda GE, Merenda R, Neri D, Angeli P, Barbazza F, Valmasoni M, Feltracco P, Zangrandi F, Gangemi A, Miotto D, et al. Cavoportal hemitransposition: a successful way to overcome the problem of total portosplenomesenteric thrombosis in liver transplantation. Liver Transpl. 2002;8:72–75. doi: 10.1053/jlts.2002.30404. [DOI] [PubMed] [Google Scholar]
- 39.Molmenti EP, Roodhouse TW, Molmenti H, Jaiswal K, Jung G, Marubashi S, Sanchez EQ, Gogel B, Levy MF, Goldstein RM, et al. Thrombendvenectomy for organized portal vein thrombosis at the time of liver transplantation. Ann Surg. 2002;235:292–296. doi: 10.1097/00000658-200202000-00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Egawa H, Tanaka K, Kasahara M, Takada Y, Oike F, Ogawa K, Sakamoto S, Kozaki K, Taira K, Ito T. Single center experience of 39 patients with preoperative portal vein thrombosis among 404 adult living donor liver transplantations. Liver Transpl. 2006;12:1512–1518. doi: 10.1002/lt.20777. [DOI] [PubMed] [Google Scholar]
- 41.Selvaggi G, Weppler D, Nishida S, Moon J, Levi D, Kato T, Tzakis AG. Ten-year experience in porto-caval hemitransposition for liver transplantation in the presence of portal vein thrombosis. Am J Transplant. 2007;7:454–460. doi: 10.1111/j.1600-6143.2006.01649.x. [DOI] [PubMed] [Google Scholar]
- 42.Gómez-Gutierrez M, Quintela J, Marini M, Gala B, Suarez F, Cao I, Sellés CC, Aguirrezabalaga J, Otero A, Mosteiro S. Portal vein thrombosis in patients undergoing orthotopic liver transplantation: intraoperative endovascular radiological procedures. Transplant Proc. 2005;37:3906–3908. doi: 10.1016/j.transproceed.2005.10.063. [DOI] [PubMed] [Google Scholar]
- 43.Yerdel MA, Gunson B, Mirza D, Karayalçin K, Olliff S, Buckels J, Mayer D, McMaster P, Pirenne J. Portal vein thrombosis in adults undergoing liver transplantation: risk factors, screening, management, and outcome. Transplantation. 2000;69:1873–1881. doi: 10.1097/00007890-200005150-00023. [DOI] [PubMed] [Google Scholar]