

Abstract
Gastrointestinal stromal tumor (GIST) represents the most common mesenchymal malignancy of the gastrointestinal (GI) tract. In neurofibromatosis (NF), the increased incidence of tumor needs to be considered even in non-symptomatic individuals. Patients with neurofibromatosis NF type 1 have an increased risk of developing GI tumors including rare types such as GIST. We report a case of GIST in a 53-year-old male patient with neurofibromatosis. The patient was diagnosed with NF four years ago and his medical history revealed that he was hospitalized 5 times with a provisional diagnosis of massive lower gastrointestinal bleeding. GIST was diagnosed at explorative laparotomy and the tumor was 21 cm × 13 cm × 7 cm in size. Immunohistochemical examination showed that vimentin, actin and CD117 were positive. Computerized tomography showed peritoneal implants three months later. Imatinib mesylate (600 mg/d) was initiated. However, control computerized tomography revealed liver and omental metastasis. The dosage was elevated to 800 mg/d. Despite high dosage, the progression of the metastatic lesions continued in the liver and omentum. The patient started oral sunitinib malate (Sutent® Pfizer Inc, New York, NY, USA) 50 mg per day for 4 consecutive weeks, followed by 2 wk off per treatment cycle. The metastatic lesions in the liver and omentum were decreased in size after four courses, suggesting that sunitinib is also an effective treatment modality for metastatic GIST in NF patients.
Keywords: Neurofibromatosis, Gastrointestinal stromal tumors, Imatinib, Sunitinib
INTRODUCTION
Gastrointestinal stromal tumor (GIST) represents the most common mesenchymal malignancy of the gastrointestinal (GI) tract. Before 2000 this neoplasm accounted for approximately 1%-3% of all malignant GI tumors. A population-based study showed that GIST occurs approximately in 15 of one million individuals[1]. Neurofibromatosis (NF) is divided into two groups: NF Type I(Von Recklinghausen's disease) and NF type 2. Neurofibromatosis type 1 (NF1) is among the most common neurogenetic disorders, affecting approximately one in 3000 individuals worldwide. The most common features of NF1 are abnormal pigmentation manifested as café-au-lait macules, freckle on skinfold, and iris hamartomas (Lisch nodules). Patients with NF2 have a predisposition for the development of meningiomas, bilateral acoustic neuromas, and spinal nerve root tumors. Neurofibromas may also undergo secondary malignant degeneration and become sarcomatous. GIST and neurofibromatosis have been reported together in a few cases[2-4]. Here we report a case of GIST in a patient with neurofibromatosis who responded well to sunitinib after imatinib failure.
CASE REPORT
A 53-year-old male patient was admitted to our hospital with complaints of weakness and vertigo. His physical examination findings were normal except for pigmented lesions on his face and body (café-au-lait spots), and neurofibromas (Figure 1A and B). He was diagnosed with NF four years ago. His medical history revealed that the patient was hospitalized 5 times with a provisional diagnosis of massive lower gastrointestinal bleeding. Abdominal computerized tomography (CT) showed a well-defined heterogeneous mass consisting of an external solid area (50 mm × 40 mm) and internal cystic component adjacent to the anterior abdominal wall (Figure 2A and B). Solid portions of the tumor were intensely stained on contrast imaging. Magnetic resonance imaging (MRI) revealed low and markedly high intensity areas in the tumor on T1-weighted and T2-weighted sequences, respectively (Figure 3).
Figure 1.
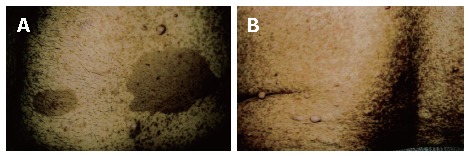
Photos of patients's tumour. Pigmented lesions (café–au-lait spots) and neurofibromas on the anterior abdominal wall (A) and on the back region (B).
Figure 2.
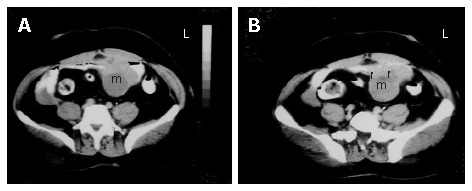
A: Axial sequential contrast-enhanced CT of the abdomen showing a well-defined heterogeneous mass consisting of an external solid area and internal cystic componenet adjacent to the anterior abdominal wall; B: the solid portions of the tumor were intensely stained on contrast imaging (arrows).
Figure 3.
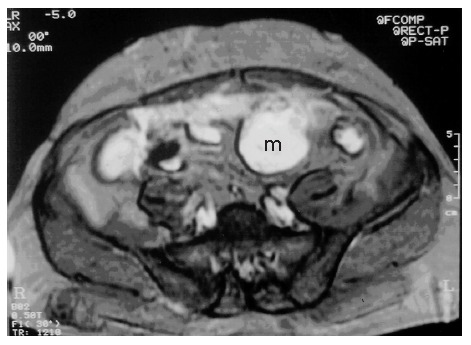
T2-weighted MRI shows heterogeneous hyperintense cystic and solid mass adjacent to the anterior abdominal wall.
Laboratory examination findings were as follows: hemoglobin 8.2 g/dL, hematocrit 28.2%, platelets 763000/mm3, WBC 13900/mm3, erythrocyte sedimentation rate 70 mm/h, creatinine 1.02 mg/dL, sodium 142 mEq/L, potassium 4.06 mmol/L, calcium 8.7 mg/dL, AST 18 U/L, ALT 33 U/L, total bilirubin 0.3 mg/dL, LDH 95 U/L, ALP 235 U/L, GGT 54 U/L, CRP 39.1, CA-19-9 < 0.6 U/mL (normal range: 0-23), CEA 1.49 ng/mL (normal range: 0-4.6), AFP 4.9 IU/mL (normal range: 0-8.5). Fecal occult blood test was negative.
Oesephagogastroduodenoscopy showed alkaline reflux gastritis and colonoscopy findings were normal. Abdominal ultrasonogram showed a mass with wall thickness of 9 cm, mimicking small intestine at the lower left quadrant. An explorative laparotomy was performed. The intra-abdominal mass was resected with the small intestine and sigmoid colon segments. Pathological gross examination showed that the tumor size was 21 cm × 13 cm × 7 cm with negative margins. Its external surface was nodular and cross-section was bloody, necrotic and dirty. Immunohistochemical examination showed that vimentin and actin were positive and S100, desmin, EMA, CD34, chromogranin were negative. At a later examination CD 117 was determined to be positive. The patient was diagnosed as gastrointestinal stromal tumor (Figures 4 and 5). Control computerized tomography showed peritoneal implants three months later. Imatinib mesylate (600 mg/d) was administered for 1.5 years. Later on, the dose was increased to 800 mg/d as metastasis of the liver and omentum was discovered. However, the progression of the metastatic lesions continued despite high dosage. The longest diameter of the lesions was 6 cm in the liver and 4 cm in the omentum. The patient started oral sunitinib, 50 mg per day for 4 consecutive weeks followed by 2 wk off per treatment cycle. After two courses of treatment, the metastasis in the liver was decreased from 6 cm to 5 cm and in the omentum from 4 cm to 3 cm. He was continued on sunitinib treatment. After four courses, the lesions were 4 cm and 2 cm, respectively. However, the number of the lesions both in the liver and omentum was not decreased while the sizes of the lesions were decreased. The patient's performance was well and the disease was accepted to be stabilized with partial response. He was still using sunitinib at the time when he was reported.
Figure 4.
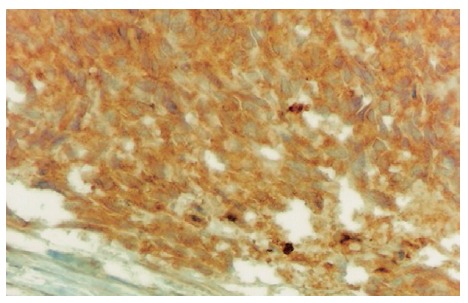
Immunohisto-chemical CD117 reactivity in tumor cells (DAB, × 400).
Figure 5.
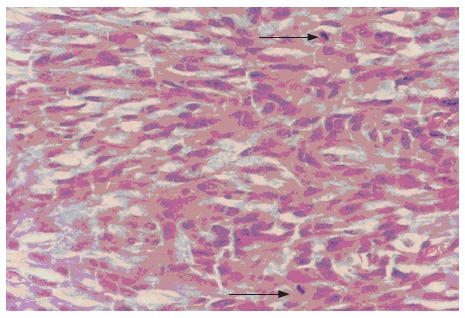
Spindle tumor cells with two mitoses marked with arrow (HE, × 400).
DISCUSSION
GIST represents the most common mesenchymal malignancy of the gastrointestinal tract. GIST occurs predominantly in adults at a median age of 58 years but can occur at any time throughout life. The majority of GIST cases (60% to 70%) arise from stomach. These tumors bear certain histopathological similarities to a specific cell type inherent in the GI tract, known as interstitial cells of Cajal (ICC)[5]. ICCs are the unique 'pacemaker cells' that are normally present in the myenteric plexus. CD117 antigen can be detected by immunohistochemical staining as a marker. Expert pathologists can define a rare subset (fewer than 5%) of GIST cases that fail to express CD117, and these cases are most likely to be driven by alternative kinases such as PDGFRA[6].
Neurofibroma is a benign peripheral nerve tumor composed of proliferating Schwann cells and fibroblasts. It presents as multiple, palpable, rubbery, cutaneous tumors. Patients with NF1 are at increased risk of developing nervous system neoplasms. Mutation of NF1 gene on chromosome 17 causes Von Recklinghausen's disease. The NF1 gene is a tumor-supressor gene encoding a protein, neurofibromin, which modulates signal transduction through the ras GTPase pathway. However, individuals with NF1 are also prone to development of a wide variety of nervous system abnormalities[7]. NF2 is caused by mutation of the NF2 gene on chromosome 22q and much less frequent than NF1. NF2 encodes a protein called merlin. Patients with NF2 have a predisposition to developing meningiomas, bilateral acoustic neuromas and spinal nerve root tumors[8].
The most common tumor in individuals with NF1 is the benign peripheral nerve sheath tumor (neurofibroma), which is typically first noted around puberty. Although these tumors are benign, a subtype of benign neurofibroma (plexiform neurofibroma) can undergo transformation into a malignant peripheral nerve sheath tumor (MPNST). The latter is a highly aggressive cancer, which is associated with poor survival and a dismal response to conventional cancer therapies[7].
The entities of NF1- associated gastrointestinal lesions include: hyperplastic lesion of intestinal neural tissue and its supporting structures, GIST, endocrine cell tumor of the duodenum and periampullary region, and a miscellaneous group of other tumors[9]. GIST has been reported in relation with NF1, and most of the patients with NF1-associated GIST have multiple GISTs[10,11]. Miettinen et al[12] analyzed the clinicopathologic and molecular genetic features of 45 cases of NF and GIST, showing that the great majority of tumors occur in the jejunum or ileum with multiple tumors, the most common clinical presentations are gastrointestinal bleeding and anemia, the majority of the tumors are small and mitotically inactive.
GIST is a rare neoplasm accounting for about 1%-3% of all malignant neoplasms of the gastrointestinal tract. GIST has been classified as a myogenic, neurogenic, or non-differentiated tumor[13]. The malignancy is commonly determined by the tumor size and the mitotic ratio. Tumor larger than 5 cm in diameter or having a mitotic count greater than 5 per high-power field is classified as malignant[14]. In our case the tumor size was 21 cm × 13 cm and diagnosed as malignant GIST.
NF1-associated GIST has been observed in older adults (the mean age of one series being 56.2 years, and nearly all of them were multiple in the small intestine), while GIST in non-NF1 patients is most commonly found in the stomach (60%-70%), followed by the small intestine (20%-30%), colorectum and oesophagus (cumulative, 10%)[15]. GIST that occurs in patients with NF commonly originates from the proximal small intestine and is often multiple[16]. In our case the GIST was located in the small intestine and colon.
Imatinib mesylate, an inhibitor of kinase activity of KIT and PDGFR, has improved the clinical outcomes of metastatic GIST patients. Demetri et al[17] showed that 5% of patients with advanced GIST demonstrate primary resistance to imatinib mesylate and 14% develop early resistance. Sunitinib malate (Sutent®, Pfizer Inc, New York, NY, USA) is a tyrosine kinase inhibitor of oral multi-targeted receptors and has anti-angiogenic and anti-tumor activities. Sunitinib and imatinib bind to the ATP-binding domain of both KIT and PDGFR, but they are members of different chemical classes and have different binding characteristics and affinities. A phase I/II study showed that sunitinib has clinical activity in patients with imatinib-resistant GIST[18]. Another study randomized 312 GIST patients with imatinib mesylate resistance to receive sunitinib (n = 207) and placebo (n = 105) and showed that the time to progression (median 27.3 wk), progression-free survival (median 24.1 wk), disease control and tumour response are superior with sunitinib compared with placebo, and the most common treatment-related adverse events with sunitinib are fatigue, diarrhoea, skin discolouration, and nausea[19].
Patients with neurofibromatosis type 1 have an increased risk of developing gastrointestinal tumors, including rare types of GIST. GIST can occur at any localization in the GI system. When located in the small intestine, conventional endoscopy may be rather difficult and further diagnostic means such as ultrasound, computerized tomography or possibly capsule endoscopy should be considered. In neurofibromatosis, the increased incidence of tumor needs to be considered even in non-symptomatic individuals.
Footnotes
S- Editor Liu Y L- Editor Wang XL E- Editor Chen GJ
References
- 1.Kindblom LG, Meis-Kindblo J, Bümming P, et al. Incidence, prevalence, phenotype and biologic spectrum of gastrointestinal stromal cell tumors (GIST)- a population based study of 600 cases (abstract 577O) Ann Oncol. 2002;13 Suppl 5:157. [Google Scholar]
- 2.Sinha R, Verma R, Kong A. Mesenteric gastrointestinal stromal tumor in a patient with neurofibromatosis. AJR Am J Roentgenol. 2004;183:1844–1846. doi: 10.2214/ajr.183.6.01831844. [DOI] [PubMed] [Google Scholar]
- 3.Köppen S, Wejda B, Dormann A, Hoffmeister D, Stolte M, Huchzermeyer H. Gastrointestinal stromal tumours (GIST) of the jejunum in a patient with neurofibromatosis type 1 (von Recklingshausen's disease) Z Gastroenterol. 2004;42:1183–1187. doi: 10.1055/s-2004-813587. [DOI] [PubMed] [Google Scholar]
- 4.Nakamura H, Satoh Y, Ikeda T, Endoh T, Imai K. A case of upper jejunal gastrointestinal stromal tumor (GIST) accompanied with von Recklinghausen's disease. Nihon Shokakibyo Gakkai Zasshi. 2000;97:1385–1390. [PubMed] [Google Scholar]
- 5.Perez-Atayde AR, Shamberger RC, Kozakewich HW. Neuroectodermal differentiation of the gastrointestinal tumors in the Carney triad. An ultrastructural and immunohistochemical study. Am J Surg Pathol. 1993;17:706–714. doi: 10.1097/00000478-199307000-00008. [DOI] [PubMed] [Google Scholar]
- 6.Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A, Town A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–710. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- 7.Arun D, Gutmann DH. Recent advances in neurofibromatosis type 1. Curr Opin Neurol. 2004;17:101–105. doi: 10.1097/00019052-200404000-00004. [DOI] [PubMed] [Google Scholar]
- 8.Lee JH, David M. Cancers of Childhood. Cancer Principles & Practice of Oncology. 7th ed. 1889-1897. [Google Scholar]
- 9.Fuller CE, Williams GT. Gastrointestinal manifestations of type 1 neurofibromatosis (von Recklinghausen's disease) Histopathology. 1991;19:1–11. doi: 10.1111/j.1365-2559.1991.tb00888.x. [DOI] [PubMed] [Google Scholar]
- 10.Miettinen M, Kopczynski J, Makhlouf HR, Sarlomo-Rikala M, Gyorffy H, Burke A, Sobin LH, Lasota J. Gastrointestinal stromal tumors, intramural leiomyomas, and leiomyosarcomas in the duodenum: a clinicopathologic, immunohistochemical, and molecular genetic study of 167 cases. Am J Surg Pathol. 2003;27:625–641. doi: 10.1097/00000478-200305000-00006. [DOI] [PubMed] [Google Scholar]
- 11.Min KW, Balaton AJ. Small intestinal stromal tumors with skeinoid fibers in neurofibromatosis: report of four cases with ultrastructural study of skeinoid fibers from paraffin blocks. Ultrastruct Pathol. 1993;17:307–314. doi: 10.3109/01913129309027777. [DOI] [PubMed] [Google Scholar]
- 12.Miettinen M, Fetsch JF, Sobin LH, Lasota J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol. 2006;30:90–96. doi: 10.1097/01.pas.0000176433.81079.bd. [DOI] [PubMed] [Google Scholar]
- 13.Bagnolo F, Bonassi U, Scelsi R, Testoni PA. Gastric stromal tumour: a rare neoplasm presenting with gastrointestinal bleeding. Eur J Gastroenterol Hepatol. 1998;10:791–794. [PubMed] [Google Scholar]
- 14.Pidhorecky I, Cheney RT, Kraybill WG, Gibbs JF. Gastrointestinal stromal tumors: current diagnosis, biologic behavior, and management. Ann Surg Oncol. 2000;7:705–712. doi: 10.1007/s10434-000-0705-6. [DOI] [PubMed] [Google Scholar]
- 15.Miettinen M, Sarlomo-Rikala M, Lasota J. Gastrointestinal stromal tumors: recent advances in understanding of their biology. Hum Pathol. 1999;30:1213–1220. doi: 10.1016/s0046-8177(99)90040-0. [DOI] [PubMed] [Google Scholar]
- 16.Levy AD, Patel N, Abbott RM, Dow N, Miettinen M, Sobin LH. Gastrointestinal stromal tumors in patients with neurofibromatosis: imaging features with clinicopathologic correlation. AJR Am J Roentgenol. 2004;183:1629–1636. doi: 10.2214/ajr.183.6.01831629. [DOI] [PubMed] [Google Scholar]
- 17.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 18.Heinrich MC, Maki RG, Corless CL, Antonescu CR, Fletcher JA, Fletcher CD, Huang X, Baum CM, Demetri GD. Sunitinib (SU) response in imatinib-resistant (IM-R) GIST correlates with KIT and PDGFRA mutation status (abstract 9502) J Clin Oncol. 2006;24 suppl:520. [Google Scholar]
- 19.Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–1338. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]