

Abstract
AIM: To evaluated the therapeutic and prophylactic effect of thalidomide on 2, 4, 6-trinitrobenzene sulfonic acid (TNBS)-induced colitis. Thalidomide has been reported to downregulate the expression of tumor necrosis factor α (TNF-α), IL-12, and vascular endothelial growth factor (VEGF), hallmarks of intestinal inflammation in Crohn’s disease (CD).
METHODS: Male Wistar rats were divided in five groups of ten animals each. Four groups received a rectal infusion of TNBS in ethanol. The first group was sacrificed 7 d after colitis induction. The second and third groups received either thalidomide or placebo by gavage and were sacrificed at 14 d. The fourth group received thalidomide 6 h before TNBS administration, and was sacrificed 7 d after induction. The fifth group acted as the control group and colitis was not induced. Histological inflammatory scores of the colon were performed and lamina propria CD4+ T cells, macrophages, and VEGF+ cells were detected by immunohistochemistry. TNF-α and IL-12 were quantified in the supernatant of organ cultures by ELISA.
RESULTS: Significant reduction in the inflammatory score and in the percentage of VEGF+ cells was observed in the group treated with thalidomide compared with animals not treated with thalidomide. Both TNF-α and IL-12 levels were significantly reduced among TNBS induced colitis animals treated with thalidomide compared with animals that did not receive thalidomide. TNF-α levels were also significantly reduced among the animals receiving thalidomide prophylaxis compared with untreated animals with TNBS-induced colitis. Intestinal levels of TNF-α and IL-12 were significantly correlated with the inflammatory score and the number of VEGF+ cells.
CONCLUSION: Thalidomide significantly attenuates TNBS-induced colitis by inhibiting the intestinal production of TNF-α, IL-12, and VEGF. This effect may support the use of thalidomide as an alternate approach in selected patients with CD.
Keywords: Thalidomide, Experimental colitis, Inflammatory bowel disease, Tumor necrosis factor α, IL-12, Vascular endothelial growth factor
INTRODUCTION
The etiopathogenesis of both forms of inflammatory bowel disease (IBD), Crohn’s disease (CD) and ulcerative colitis (UC) is complex and multifactorial, involving genetic predisposition, environmental triggers, microbial and immune factors. The chronic inflammatory process in IBD is thought to be the result of an imbalance between an excess of proinflammatory cytokines, such as TNF-α, IL-12, IL1-α, IL-8, and IFN-γ, and a relative deficiency of immunoregulatory cytokines, such as IL-1Ra, IL-4, IL-10, IL-13 and transforming growth factor-b[1,2]. In CD, it is well established that the intestinal inflammation is characterized by T helper cell type 1 (Th1) phenotype[3,4] whereas in UC, an atypical Th2 seems to constitute the predominant response[5].
TNF-α appears to play a central role in the inflammatory process of CD. Levels of TNF-α were found to be increased in the circulation[6] as well as in the stool[7] of patients with active CD. Moreover, the serum level of TNF-α has been proposed as an indicator for the risk of relapse after remission of active CD[8]. Additional support for the role of TNF-α in CD emerges from the beneficial effects observed with the use of antibodies against TNF-α in clinical studies[9,10]. It has been demonstrated that IL-12 is expressed and released by lamina propria mononuclear cells from patients with active CD[11]. In an animal model of colitis, treatment with anti-IL-12 was shown to prevent inflammation suggesting a possible role for IL-12 in the pathogenesis of colitis[12].
In addition to the accumulation of inflammatory cells, ulceration, and regeneration, the inflammatory process of IBD is also characterized by neovascularization of the intestinal mucosa. Elevated circulating levels of vascular endothelial growth factor (VEGF) have been shown in patients with IBD[13,14]. Increased spontaneous production of VEGF was also found in the inflamed mucosa of IBD patients[15]. In a recent clinical study, a reduction of serum VEGF levels was observed in CD patients who responded to anti-TNF-α treatment[16].
Novel therapies using antibody against TNF-α are still expensive and some patients do not respond. Thalidomide, another anti-TNF-α agent, has been effectively used in the treatment of diseases believed to be mediated by pro-inflammatory cytokines, such as rheumatoid arthritis[17], chronic graft-versus-host disease[18], and chronic cutaneous lupus erythematosus[19]. Although the mechanisms of the immune-modulating action of thalidomide are not completely understood, results from in vitro studies indicated that thalidomide is capable of inhibiting both bacterial lipopolysacharide–stimulated IL-12 and TNF-α production by monocytes[20,21].
Preliminary data from open label trials suggest that thalidomide may have a role in the treatment of refractory CD[22,23]. However, selective inhibition of TNF-α using oxpentifyline may not be effective in CD[24]. Therefore, it is relevant to study the mechanism by which thalidomide exerts its therapeutic effect on intestinal inflammation. In the present study, we investigated both the therapeutic and the prophylactic effect of thalidomide on histologic inflammation, distribution of immune competent cells and VEGF-producing cells in the intestinal lamina propria, and the production of TNF-α and IL-12 in TNBS-induced colitis.
MATERIALS AND METHODS
Animals
Male Wistar rats (each weighing between 250 and 300 g) obtained from a local supplier, were maintained on a 12-h/12-h light/dark cycle in a temperature-controlled room (24°C). Animals were housed in rack-mounted, wire cages with 3 animals per cage. Standard laboratory pelleted formula and tap water were provided ad libitum. The care and use of animals, as well as all procedures reported in this study were approved by the institutional animal care committee of the Federal University of Rio de Janeiro and are in accordance with the guidelines of the Institutional Care and Use Committee of the National Institutes of Health, and the Guide for the Care and Use of Laboratory Animals[25].
Induction of colitis
On day (d) 0, rats were lightly anesthetized with ether, and colitis was induced by intracolonic instillation of 0.8ml of a solution containing 20mg of 2,4,6-TNBS (Sigma Chemical Co., St. Louis, MO) in 17.5% ethanol (Merck, Darmstadt, Germany) using a rubber cannula (8 cm long) inserted through the rectum. Thereafter, animals were allowed access to standard chow and water ad libitum. Clinical manifestations such as diarrhea, bleeding and weight loss were observed during this period.
Experimental design
After an initial acclimation period of 1 wk, animals were assigned randomly to one of five groups of ten animals each, and followed during 1 wk. The colitis group was submitted to colitis-induction but did not receive any treatment and animals were sacrificed on experimental d 8. The therapeutic-thalidomide group was submitted to colitis-induction followed by treatment with thalidomide by gavage (200 mg/kg per day) during 7 d, whereas the placebo-treated group received just the solvent used for thalidomide administration (olive oil). The two groups were sacrificed on experimental d 15. The prophylactic-thalidomide group received a single dose of thalidomide by gavage (200 mg/kg per day) 6 h before colitis-induction, and animals were killed on experimental d 8. Normal rats not submitted to any intervention constituted the control group, being sacrificed after the acclimation period.
For the surgical procedure, animals were anesthetized by ether inhalation and submitted to a mid-laparotomy under sterile technique. The distal colon was removed, opened longitudinally and rinsed with sterile saline. The colon was immediately examined and any visible damage was scored. After scoring, three tissue samples were excised from the colon for histological assessment, immunohistochemical analysis and organ culture. A quick death procedure by cervical dislocation was uniformly performed in all animals.
Macroscopic assessment of the colon
Colon injury was evaluated and scored by two independent observers according to the following macroscopic parameters: adhesion, strictures, ulcer and thickness of colon wall. For adhesions grading was considered: 0, absent; 1, light; 2, multiple; for strictures 0, absent; 1, light; 3 accentuated with proximal dilatation. For ulcer grading the scoring was 0, absent; 1, linear ulcer < 1 cm; 2, two linear ulcers < 1 cm; 3, numerous ulcers > 1 cm; and for thickness of the colon wall, 0, < 1 mm; 1, 1 to 3 mm; 2, > 3 mm; total score ranging from 0 to 10[26].
Histological inflammatory scores of the colon
Specimens were fixed in 40 g/L formadehyde saline, embedded in paraffin, cut into 5 μm sections, stained with hematoxylin-eosin stain, and examined microscopically by two independent observers. The following histologic parameters were studied: ulceration, hyperplasia and inflammatory infiltrate. For both inflammatory infiltrate and hyperplasia, grading was considered: 3, severe; 2, moderate; 1, mild; 0, absent. For ulcers, grading was: 4, diffuse glandular disruption or extensive deep ulceration; 3, glandular disruption or focal deep ulceration; 2, diffuse superficial ulceration; 1, focal superficial ulceration; and 0, absent[27].
Immunohistochemical analyses
Tissue samples were immediately embedded in Tissue-Tek O.C.T. compound (Miles Scientific Laboratories Ltd, Naperville, IL) and snap frozen in isopentane in a liquid nitrogen bath. Samples were then stored at -80°C until processing, and cut into 6 μm section in a cryostat maintained at -20°C. Tissue sections were air-dried and fixed for 10 min in a 1:1 solution of chloroform-acetone. Immunologic assessment of the intestinal mucosa was made using indirect immunoperoxidase technique using the following antibodies: mouse monoclonal anti-rat CD68, to macrophages, mouse monoclonal anti-rat CD4, to CD4+ T cells (Serotec Ltd., Oxford, UK), and rabbit polyclonal anti-rat VEGF (Santa Cruz Biotechnology, Inc., CA). Briefly, tissue sections were incubated at room temperature with normal horse serum for 30 min and then with 50 μL of each monoclonal antibody for 1 h. One section from each animal was incubated with phosphate-buffered saline (PBS) alone or 50 μL of mouse IgG1 (Dakopatts A/S) and served as a negative control. After rinsing in PBS for 10 min, all sections were incubated for 45 min with 50 μL of either a goat anti-mouse or a goat anti-rabbit peroxidase conjugate IgG (Zymed Laboratories In.). Additional rinsing was followed by development with a solution containing hydrogen peroxide and diaminobenzidine, dehydrated and mounted in histological mounting medium. Sections were examined by two independent observers using light microscopy at 400X magnification. Lamina propria cells exhibiting identifiable reactivity on their surface distinct from background were regarded as positive. All cells within the lamina propria were assessed by a quantitative grading system in at least five distinct high power fields. The results were expressed as positive cells (immunoreactive) numbers per mm2 lamina propria.
Organ culture and cytokine measurements
Colonic mucosal explants were cultured for 24 h at 37°C with 95% CO2 by the established procedure. All culture media consisted of RPMI 1640 medium and 10% fetal calf serum (Life Technologies, Great Island, USA) with penicillin (100 KU/L) and streptomycin (100 mg/L). After culture for 24 h, the organ culture medium was collected and used for TNFα and IL-12 assays. Samples were centrifuged and the supernatants used for measurement of the concentration of TNFα by a comercial sensitive enzyme-linked immunosorbent assay (ELISA) method for rat TNFα (R&D System, Mineapolis, MN). In our data, biopsy specimens’ wet weight was shown to correlate closely with protein content of tissue homogenates. The minimum detectable concentration of rat TNFα. was typically less than 5.0 ng/L.
IL-12 was measured in supernatants samples using an ELISA assay (BIOSOURCE, Camarillo, CA). According to the manufacturer directions, the minimum detectable IL-12 concentration was 3.0 ng/L. All samples were analyzed in duplicate. Cytokine concentrations in organ culture supernatants were expressed as pg/μg protein. The total protein content of the biopsy specimens was estimated by the Lowry’s method.
Statistical analysis
Statistical analyses were performed using the statistical software SPSS for Windows (Version 10.0.1, SPSS Inc., 1989-1999, USA). Statistical differences among the experimental groups were evaluated with the one-way ANOVA test in which pairwise multiple comparisons were carried out using the Dunnett’s T3 test. Correlations between inflammatory scores, the densities of positive cells measured by immunohistochemistry, and cytokines levels were assessed using the Spearman rank correlation coefficient. Values are expressed as medians (1st quartile, 3rd quartile). The level of significance was set at P < 0.05.
RESULTS
Assessment of macroscopic and microscopic changes in the colon
TNBS-induced colitis manifested predominantly as patchy inflammatory lesions in the distal colon that gradually worsened throughout d 1-7. Involved areas of the colon demonstrated mucosal edema, ulceration, and evidence of transmural inflammation. Animals treated with either therapeutic or prophylactic thalidomide showed less inflammation compared to placebo-treated animals (data not shown).
Significantly higher microscopic inflammatory scores were found in the intestinal mucosa of all animals submitted to colitis-induction compared to the normal mucosa of controls. Scores in the thalidomide-treated group were significantly lower compared to those of the colitis group (Figures 1 and 2). Among histological parameters analyzed ulceration was the most affected by thalidomide treatment.
Figure 1.
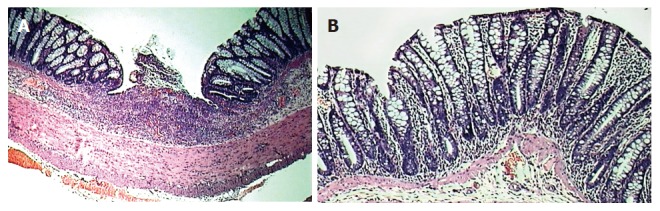
HE staining of tissue obtained from the left colon of animals: untreated colitis (A), therapeutic-thalidomide (B), respectively. Sample from untreated colitis group shows gross ulceration and intense cellular infiltration, congestion and edema, while thalidomide-treated animal presents less inflammation and no ulceration (Original magnification × 100).
Figure 2.
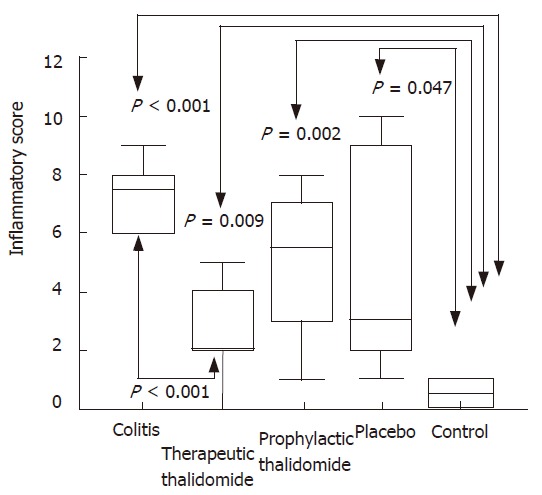
Histological inflammatory scores of the colon. Total score of histological parameters analyzed in all groups: untreated colitis, therapeutic- and prophylactic-thalidomide, placebo-treated, and normal controls, respectively (n = 10 in each group). Horizontal bars represent medians, boxes represent the 25th and 75th percentiles, and vertical bars represent ranges. Differences were analysed using one-way ANOVA with the Dunnett’s test for multiple comparisons. Significant differences are presented.
Number of CD4+ T-cells and macrophages in the colonic lamina propria
The numbers of lamina propria CD4+ T-cells were not significantly different among the study groups. The number of macrophages was significantly higher in colitis, placebo-treated, and also in the prophylactic-thalidomide group compared with that of the control group. In the therapeutic-thalidomide group the number of macrophages was similar to that of normal controls (Figures 3 and 4). A significant correlation was detected between the number of macrophages in the colon and the histological inflammatory scores (r = 0.56, P < 0.001).
Figure 3.
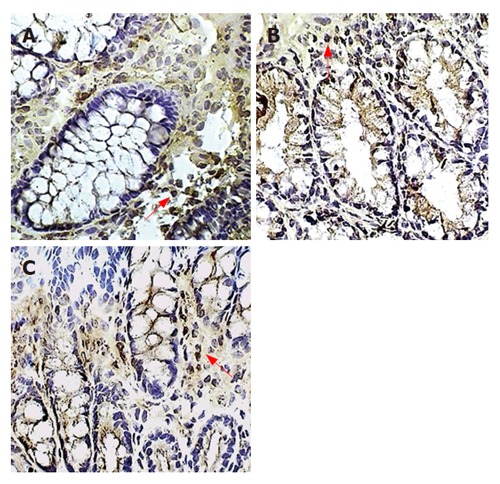
Macrophages in the colonic lamina propria. Immunoperoxidase staining for macrophages is shown in placebo-treated colitis (A), therapeutic-thalidomide (B), and in a normal control (C), respectively. Arrowheads show immunoreactive cells (Original magnification × 400).
Figure 4.
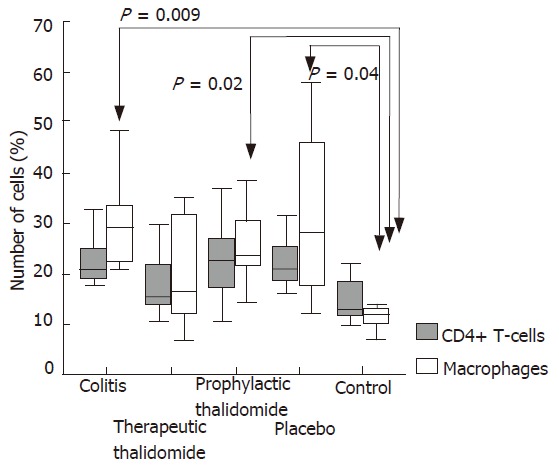
Quantitative analysis of CD4+ T-cells and macrophages in the colonic lamina propria of animals with untreated colitis, animals receiving therapeutic or prophylactic thalidomide, placebo-treated, and normal controls, respectively (n = 10 in each group). Horizontal bars represent medians, boxes represent the 25th and 75th percentiles, and vertical bars represent ranges. Significant values are presented. Differences were analysed using one-way ANOVA with the Dunnett’s test for multiple comparisons. Significant differences are presented.
Number of VEGF+ cells in the colonic lamina propria.
The number of lamina propria VEGF+ cells was significantly higher in colitis, placebo-treated, and also in the prophylactic-thalidomide group compared to the control group. However, therapeutic-thalidomide significantly reduced the number of VEGF+ cells in TNBS-induced colitis compared to colitis group and placebo-treated (Figures 5 and 6). A significant correlation was detected between the number of VEGF+ cells in the colon and the histological inflammatory scores (r = 0.63, P < 0.001).
Figure 5.
Quantitative analysis of CD4+ T-cells and macrophages in the colonic lamina propria of animals with untreated colitis, animals receiving therapeutic or prophylactic thalidomide, placebo-treated, and normal controls, respectively (n = 10 in each group). Horizontal bars represent medians, boxes represent the 25th and 75th percentiles, and vertical bars represent ranges. Significant values are presented. Differences were analysed using one-way ANOVA with the Dunnett’s test for multiple comparisons. Significant differences are presented.
Figure 6.
Quantitative analysis of VEGF+ cells in the colonic mucosa of the groups: untreated colitis, therapeutic- and prophylactic-thalidomide, placebo-treated, and normal controls, respectively (n = 10 in each group). Horizontal bars represent medians, boxes represent the 25th and 75th percentiles, and vertical bars represent ranges. Differences were analysed using one-way ANOVA with the Dunnett’s test for multiple comparisons. Significant differences are presented.
Cytokine production by colonic mucosa
Significantly higher levels of TNF-α were found in the supernatants from colitis, placebo-treated and therapeutic-thalidomide, compared to normal controls. However, levels were significantly lower in both therapeutic- and prophylactic-thalidomide groups compared with those obtained from colitis and placebo-treated groups (Figure 7). A significant correlation was found between the levels of TNF-α, and the histological inflammatory scores (r = 0.60, P < 0.001).
Figure 7.
TNF-α production by the colonic mucosa. Levels of TNF-α are measured in supernatants of organ cultures and presented as pg/μg of tissue protein, in the groups: untreated colitis, therapeutic- and prophylactic-thalidomide, placebo-treated, and normal controls, respectively (n = 10 in each group). Horizontal bars represent medians, boxes represent the 25th and 75th percentiles, and vertical bars represent ranges. Differences were analysed using one-way ANOVA with the Dunnett’s test for multiple comparisons. Significant differences are presented.
Significantly higher levels of IL-12 were detected in the supernatants from colitis and placebo-treated colitis compared to controls. Placebo-treated group also showed significantly increased levels compared with those of the therapeutic-thalidomide group (Figure 8). A significant correlation was found between the levels of IL-12 and both the histological inflammatory scores (r = 0.50, P < 0.001), and the number of lamina propria VEGF+ cells (r = 0.62, P = 0.001).
Figure 8.
IL-12 production by the colonic mucosa. Levels of IL-12 are measured in supernatants of organ cultures and presented as pg/μg of tissue protein, in the groups: untreated colitis, therapeutic- and prophylactic-thalidomide, placebo-treated, and normal controls, respectively (n = 10 in each group). Horizontal bars represent medians, boxes represent the 25th and 75th percentiles, and vertical bars represent ranges. Differences were analysed using one-way ANOVA with the Dunnett’s test for multiple comparisons. Significant differences are presented.
DISCUSSION
In the present study we evaluated both the therapeutic and prophylactic effect of thalidomide on TNBS-induced colitis. Experimental colitis showed a gradual worsening during the first week after TNBS administration. The pathologic process consisted basically of patchy inflammatory lesions and evidence of transmural inflammation in the distal colon, similar to lesions that can be seen in human Crohn’s disease (CD). A significant reduction in the inflammatory score was observed in the group treated with thalidomide compared with TNBS-induced colitis animals not receiving thalidomide. In particular, we demonstrated that thalidomide treatment reduces the percentages of VEGF+ cells in the colonic lamina propria, and the local production of TNF-α and IL-12.
Animal models have been extremely helpful not only to study mechanisms of inflammation underlying IBD but also providing information on which cytokines could be potential targets for therapeutic blockade. TNBS-induced colitis is one of the most common models of experimental colitis in which the colonic mucosa is exposed to the contact-sensitizing agent, TNBS, a covalently reactive compound that attaches to autologous proteins and stimulates a delayed-type hypersensitivity response to the hapten-modified self antigens. This TNBS-induced animal model of chronic intestinal inflammation exhibits a Th1-like inflammatory response and displays several features consistent with those observed in human CD[28].
In the most common animal models of colitis, the cytokines predominantly involved in mucosal inflammation are TNF-α, IL-12, and IFN-γ, and their neutralization with specific antibodies were shown to ameliorate the inflammatory process[29,30]. Interestingly, treatment with the anti-TNF-α antibody infliximab has been demonstrated to be very effective in active CD, whereas etanercept, a fusion protein containing the TNF receptor p75 did not show good results[31]. Although both have high affinity for TNF-α, it seems that etanercept has less specificity and avidity and, moreover, it does not induce lymphocyte apoptosis[32]. In fact, induction of T-cell apoptosis has been associated with clinical remission in CD[33,34]. Hence, it appears that the beneficial effect of infliximab in CD could be, at least in part, related to the elimination of inflammatory cells in the intestinal mucosa. In this study, although colitic animals showed elevated percentages of macrophages in the intestinal lamina propria, it appeared that their numbers were not significantly affected by thalidomide. Moreover, no significant difference was found between groups concerning the percentage of CD4+ T-cells. Therefore, colitis attenuation observed after thalidomide cannot be attributed to a change in the number of inflammatory cells in the colon. Consequently, it seems unlikely that apoptosis would constitute a primary mechanism of action of thalidomide in TNBS-induced colitis.
In vitro studies demonstrated that thalidomide is capable of inhibiting TNF-α production by human monocytes in culture, without affecting levels of interleukin 1 beta, interleukin 6, and granulocyte/macrophage colony-stimulating factor[35]. The mechanism of TNF-α inhibition by thalidomide is thought to be associated with the enhanced degradation of the TNF-α messenger RNA[36]. Thus, the selective blockade of TNF-α production makes thalidomide an interesting candidate for the treatment of inflammatory diseases in which TNF-α plays an important role such as CD. In fact, clinical efficacy of thalidomide on refractory CD has been reported in recent open label trials[22,23]. In this study, TNF-α levels in the colonic mucosa were significantly reduced among both therapeutically and prophylactically treated animals, and correlated with inflammatory scores. In keeping with our results, an other experimental study also demonstrated amelioration of colitis in association with the reduction of TNF-α tissue levels[37].
Nevertheless, exclusive inhibition of TNF-α may not be sufficient to explain the beneficial effect of thalidomide in IBD and experimental colitis. For example, oxpentifylline is capable of inhibiting the secretion of TNF-α by peripheral blood mononuclear cells and intestinal mucosa cultures from IBD patients[38]. However, in an open label study using oxpentifylline the resulting suppression of TNF-α in patients with corticosteroid-dependent CD did not improve clinical disease activity[24]. In our study, the finding that prophylactic abrogation of TNF-α did not prevent inflammation supports the idea that other cytokines or inflammatory pathways might be involved in the development of TNBS-induced colitis, and that additional effects beyond TNF-α inhibition are necessary to explain the efficacy of therapeutic thalidomide.
In addition to TNF-α neutralization, some preliminary clinical data also suggest there are beneficial effects of blockade of different cytokines including IL-12, IL-6, and IFN-γ[39,40]. IL-12 has a critical role in the development of cellular immune responses that operate largely by stimulating the production of IFN-γ, and it has been shown to be secreted by intestinal lamina propria mononuclear cells of CD patients[11]. In addition to the increased production of IL-12, the IL-12 receptor β2 subunit was found to be up-regulated at inflammatory sites in CD intestine, further supporting the importance of a signalling pathway used by IL-12 to induce the aberrant mucosal Th1 activation seen in CD[41]. The prevention of experimental colitis obtained following the administration of an antibody targeting IL-12 also contributes to the notion of the involvement of IL-12 in the development of intestinal inflammation[12]. In an open label trial in refractory CD, clinical efficacy of thalidomide was associated with a strong effect on both the TNF-α and IL-12 production by lamina propria mononuclear cells and peripheral blood monocytes[42]. Intestinal levels of both TNF-α and IL-12 in our study were significantly correlated with the inflammatory scores of the colon and decreased after thalidomide treatment. Hence, the suppression of IL-12 production by thalidomide seems to constitute an alternative mechanism by which the drug exerts its therapeutic activities, independent of, or synergistic with its anti-TNF-α effect. The combined inhibition of IL-12 and TNF-α by thalidomide may also explain the absence of response in studies using oxpentifylline.
Increased vascular density and angiogenic activity have been reported in different inflammatory diseases[43]. In IBD, an increased vessel density detected with Doppler ultrasonography has been reported during active CD[44]. Vascular endothelial cell growth factor (VEGF) is a potent angiogenic cytokine capable of increasing the proliferation, permeability and cell motility of endothelial cells[45]. Increased serum and tissue levels of VEGF have been shown in patients with active IBD[46,47]. In addition, VEGF was recently found to induce the expression of adhesion molecules such as intercellular adhesion molecule-1[47], and also to increase the adhesion of immune cells to the colonic endothelium[48]. On the other hand, thalidomide and its analogue supidimide, significantly attenuated TNBS-induced colitis, impairing adhesion of leukocytes to vascular endothelial cells and reducing the vascular cell adhesion molecule-1 mRNA expression[26]. Therefore, it is likely that VEGF may play a role as intermediary molecule acting in the interface between angiogenic and chronic inflammation in IBD. Consistent with this hypothesis, our results demonstrate that the percentage of colonic VEGF+ cells positively correlated to the inflammatory scores, and reduced significantly in response to therapeutic thalidomide.
In conclusion, our data suggest that thalidomide significantly attenuates TNBS-induced colitis by mechanisms including the inhibition of the intestinal production of TNF-α, IL-12, and VEGF. The thalidomide immunoregulatory mechanism acts on the cytokine imbalance present in inflammatory bowel disease mucosa. Although currently less effective than other novel therapies such as anti-TNF-α, thalidomide and its analogues with enhanced activity and hopefully less side effects, and the development of new methods for drug delivery in the gut, may possibly support the use of thalidomide as an alternate approach for selected patients with CD.
COMMENTS
Background
Thalidomide has been reported to downregulate the expression of tumor necrosis factor α (TNF-α), IL-12, and vascular endothelial growth factor (VEGF), a hallmark of intestinal inflammation in Crohn’s disease (CD). The aim of this study was evaluated the therapeutic and prophylactic effect of thalidomide on TNBS-induced colitis.
Research frontiers
Inflammatory bowel disease, etiopathogenesis of chronic mucosa inflammation, experimental colitis, cytokines.
Innovations and breakthroughs
We studied the thalidomide effect on the experimental colitis induced with 2, 4, 6-trinitrobenzene sulfonic acid (TNBS). Thalidomide significantly attenuates TNBS-induced colitis by mechanisms including the inhibition of the intestinal production of TNF-α, IL-12, and VEGF. Other studies observed the thalidomide effects on the TNF-α action. In this paper we add the effect of thalidomide in the IL-12 and VEGF mucosal levels. This immunoregulatory mechanism acts in the cytokine imbalance present in inflammatory bowel disease mucosa.
Applications
Although currently less effective than other novel therapies such as anti-TNF-α, thalidomide and its analogues with enhanced activity and hopefully less side effects, and the development of new methods for drug delivery in the gut, may possibly support the use of thalidomide as an alternate approach for selected patients with CD.
Peer review
This study supports the anti-inflammatory role of thalidomide by inhibiting the intestinal production of TNF-α, IL-12, and VEGF.
Footnotes
S- Editor Liu Y L- Editor Anand BS E- Editor Liu Y
References
- 1.Fiocchi C. Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology. 1998;115:182–205. doi: 10.1016/s0016-5085(98)70381-6. [DOI] [PubMed] [Google Scholar]
- 2.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 3.Papadakis KA, Targan SR. The role of chemokines and chemokine receptors in mucosal inflammation. Inflamm Bowel Dis. 2000;6:303–313. doi: 10.1002/ibd.3780060408. [DOI] [PubMed] [Google Scholar]
- 4.Reinecker HC, Steffen M, Witthoeft T, Pflueger I, Schreiber S, MacDermott RP, Raedler A. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn's disease. Clin Exp Immunol. 1993;94:174–181. doi: 10.1111/j.1365-2249.1993.tb05997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fuss IJ, Heller F, Boirivant M, Leon F, Yoshida M, Fichtner-Feigl S, Yang Z, Exley M, Kitani A, Blumberg RS, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113:1490–1497. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murch SH, Lamkin VA, Savage MO, Walker-Smith JA, MacDonald TT. Serum concentrations of tumour necrosis factor alpha in childhood chronic inflammatory bowel disease. Gut. 1991;32:913–917. doi: 10.1136/gut.32.8.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braegger CP, Nicholls S, Murch SH, Stephens S, MacDonald TT. Tumour necrosis factor alpha in stool as a marker of intestinal inflammation. Lancet. 1992;339:89–91. doi: 10.1016/0140-6736(92)90999-j. [DOI] [PubMed] [Google Scholar]
- 8.Schreiber S, Nikolaus S, Hampe J, Hämling J, Koop I, Groessner B, Lochs H, Raedler A. Tumour necrosis factor alpha and interleukin 1beta in relapse of Crohn's disease. Lancet. 1999;353:459–461. doi: 10.1016/S0140-6736(98)03339-X. [DOI] [PubMed] [Google Scholar]
- 9.van Dullemen HM, van Deventer SJ, Hommes DW, Bijl HA, Jansen J, Tytgat GN, Woody J. Treatment of Crohn's disease with anti-tumor necrosis factor chimeric monoclonal antibody (cA2) Gastroenterology. 1995;109:129–135. doi: 10.1016/0016-5085(95)90277-5. [DOI] [PubMed] [Google Scholar]
- 10.Targan SR, Hanauer SB, van Deventer SJ, Mayer L, Present DH, Braakman T, DeWoody KL, Schaible TF, Rutgeerts PJ. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn's disease. Crohn's Disease cA2 Study Group. N Engl J Med. 1997;337:1029–1035. doi: 10.1056/NEJM199710093371502. [DOI] [PubMed] [Google Scholar]
- 11.Monteleone G, Biancone L, Marasco R, Morrone G, Marasco O, Luzza F, Pallone F. Interleukin 12 is expressed and actively released by Crohn's disease intestinal lamina propria mononuclear cells. Gastroenterology. 1997;112:1169–1178. doi: 10.1016/s0016-5085(97)70128-8. [DOI] [PubMed] [Google Scholar]
- 12.Neurath MF, Fuss I, Kelsall BL, Stüber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1281–1290. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griga T, Tromm A, Spranger J, May B. Increased serum levels of vascular endothelial growth factor in patients with inflammatory bowel disease. Scand J Gastroenterol. 1998;33:504–508. doi: 10.1080/00365529850172070. [DOI] [PubMed] [Google Scholar]
- 14.Kanazawa S, Tsunoda T, Onuma E, Majima T, Kagiyama M, Kikuchi K. VEGF, basic-FGF, and TGF-beta in Crohn's disease and ulcerative colitis: a novel mechanism of chronic intestinal inflammation. Am J Gastroenterol. 2001;96:822–828. doi: 10.1111/j.1572-0241.2001.03527.x. [DOI] [PubMed] [Google Scholar]
- 15.Griga T, Voigt E, Gretzer B, Brasch F, May B. Increased production of vascular endothelial growth factor by intestinal mucosa of patients with inflammatory bowel disease. Hepatogastroenterology. 1999;46:920–923. [PubMed] [Google Scholar]
- 16.Di Sabatino A, Ciccocioppo R, Benazzato L, Sturniolo GC, Corazza GR. Infliximab downregulates basic fibroblast growth factor and vascular endothelial growth factor in Crohn's disease patients. Aliment Pharmacol Ther. 2004;19:1019–1024. doi: 10.1111/j.1365-2036.2004.01927.x. [DOI] [PubMed] [Google Scholar]
- 17.Gutiérrez-Rodríguez O, Starusta-Bacal P, Gutiérrez-Montes O. Treatment of refractory rheumatoid arthritis--the thalidomide experience. J Rheumatol. 1989;16:158–163. [PubMed] [Google Scholar]
- 18.Vogelsang GB, Farmer ER, Hess AD, Altamonte V, Beschorner WE, Jabs DA, Corio RL, Levin LS, Colvin OM, Wingard JR. Thalidomide for the treatment of chronic graft-versus-host disease. N Engl J Med. 1992;326:1055–1058. doi: 10.1056/NEJM199204163261604. [DOI] [PubMed] [Google Scholar]
- 19.Holm AL, Bowers KE, McMeekin TO, Gaspari AA. Chronic cutaneous lupus erythematosus treated with thalidomide. Arch Dermatol. 1993;129:1548–1550. [PubMed] [Google Scholar]
- 20.Corral LG, Haslett PA, Muller GW, Chen R, Wong LM, Ocampo CJ, Patterson RT, Stirling DI, Kaplan G. Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-alpha. J Immunol. 1999;163:380–386. [PubMed] [Google Scholar]
- 21.Moller DR, Wysocka M, Greenlee BM, Ma X, Wahl L, Flockhart DA, Trinchieri G, Karp CL. Inhibition of IL-12 production by thalidomide. J Immunol. 1997;159:5157–5161. [PubMed] [Google Scholar]
- 22.Ehrenpreis ED, Kane SV, Cohen LB, Cohen RD, Hanauer SB. Thalidomide therapy for patients with refractory Crohn's disease: an open-label trial. Gastroenterology. 1999;117:1271–1277. doi: 10.1016/s0016-5085(99)70276-3. [DOI] [PubMed] [Google Scholar]
- 23.Vasiliauskas EA, Kam LY, Abreu-Martin MT, Hassard PV, Papadakis KA, Yang H, Zeldis JB, Targan SR. An open-label pilot study of low-dose thalidomide in chronically active, steroid-dependent Crohn's disease. Gastroenterology. 1999;117:1278–1287. doi: 10.1016/s0016-5085(99)70277-5. [DOI] [PubMed] [Google Scholar]
- 24.Bauditz J, Haemling J, Ortner M, Lochs H, Raedler A, Schreiber S. Treatment with tumour necrosis factor inhibitor oxpentifylline does not improve corticosteroid dependent chronic active Crohn's disease. Gut. 1997;40:470–474. doi: 10.1136/gut.40.4.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council. Guide for the Care and Use of Laboratory animals. Washington, DC: National Academy Press; 1996. [Google Scholar]
- 26.Lienenlüke B, Stojanovic T, Fiebig T, Fayyazi A, Germann T, Hecker M. Thalidomide impairment of trinitrobenzene sulphonic acid-induced colitis in the rat - role of endothelial cell-leukocyte interaction. Br J Pharmacol. 2001;133:1414–1423. doi: 10.1038/sj.bjp.0704193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hahm KB, Im YH, Parks TW, Park SH, Markowitz S, Jung HY, Green J, Kim SJ. Loss of transforming growth factor beta signalling in the intestine contributes to tissue injury in inflammatory bowel disease. Gut. 2001;49:190–198. doi: 10.1136/gut.49.2.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris GP, Beck PL, Herridge MS, Depew WT, Szewczuk MR, Wallace JL. Hapten-induced model of chronic inflammation and ulceration in the rat colon. Gastroenterology. 1989;96:795–803. [PubMed] [Google Scholar]
- 29.Marini M, Bamias G, Rivera-Nieves J, Moskaluk CA, Hoang SB, Ross WG, Pizarro TT, Cominelli F. TNF-alpha neutralization ameliorates the severity of murine Crohn's-like ileitis by abrogation of intestinal epithelial cell apoptosis. Proc Natl Acad Sci USA. 2003;100:8366–8371. doi: 10.1073/pnas.1432897100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Totsuka T, Kanai T, Uraushihara K, Iiyama R, Yamazaki M, Akiba H, Yagita H, Okumura K, Watanabe M. Therapeutic effect of anti-OX40L and anti-TNF-alpha MAbs in a murine model of chronic colitis. Am J Physiol Gastrointest Liver Physiol. 2003;284:G595–G603. doi: 10.1152/ajpgi.00450.2002. [DOI] [PubMed] [Google Scholar]
- 31.Van den Brande JM, Braat H, van den Brink GR, Versteeg HH, Bauer CA, Hoedemaeker I, van Montfrans C, Hommes DW, Peppelenbosch MP, van Deventer SJ. Infliximab but not etanercept induces apoptosis in lamina propria T-lymphocytes from patients with Crohn's disease. Gastroenterology. 2003;124:1774–1785. doi: 10.1016/s0016-5085(03)00382-2. [DOI] [PubMed] [Google Scholar]
- 32.van Deventer SJ. Transmembrane TNF-alpha, induction of apoptosis, and the efficacy of TNF-targeting therapies in Crohn's disease. Gastroenterology. 2001;121:1242–1246. doi: 10.1053/gast.2001.29035. [DOI] [PubMed] [Google Scholar]
- 33.ten Hove T, van Montfrans C, Peppelenbosch MP, van Deventer SJ. Infliximab treatment induces apoptosis of lamina propria T lymphocytes in Crohn's disease. Gut. 2002;50:206–211. doi: 10.1136/gut.50.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di Sabatino A, Ciccocioppo R, Cinque B, Millimaggi D, Morera R, Ricevuti L, Cifone MG, Corazza GR. Defective mucosal T cell death is sustainably reverted by infliximab in a caspase dependent pathway in Crohn's disease. Gut. 2004;53:70–77. doi: 10.1136/gut.53.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sampaio EP, Sarno EN, Galilly R, Cohn ZA, Kaplan G. Thalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytes. J Exp Med. 1991;173:699–703. doi: 10.1084/jem.173.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moreira AL, Sampaio EP, Zmuidzinas A, Frindt P, Smith KA, Kaplan G. Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mRNA degradation. J Exp Med. 1993;177:1675–1680. doi: 10.1084/jem.177.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mazzon E, Muià C, Di Paola R, Genovese T, De Sarro A, Cuzzocrea S. Thalidomide treatment reduces colon injury induced by experimental colitis. Shock. 2005;23:556–564. [PubMed] [Google Scholar]
- 38.Reimund JM, Dumont S, Muller CD, Kenney JS, Kedinger M, Baumann R, Poindron P, Duclos B. In vitro effects of oxpentifylline on inflammatory cytokine release in patients with inflammatory bowel disease. Gut. 1997;40:475–480. doi: 10.1136/gut.40.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, Dolin B, Goodman N, Groden C, Hornung RL, et al. Anti-interleukin-12 antibody for active Crohn's disease. N Engl J Med. 2004;351:2069–2079. doi: 10.1056/NEJMoa033402. [DOI] [PubMed] [Google Scholar]
- 40.Ito H, Takazoe M, Fukuda Y, Hibi T, Kusugami K, Andoh A, Matsumoto T, Yamamura T, Azuma J, Nishimoto N, et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn's disease. Gastroenterology. 2004;126:989–996; discussion 947. doi: 10.1053/j.gastro.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 41.Parrello T, Monteleone G, Cucchiara S, Monteleone I, Sebkova L, Doldo P, Luzza F, Pallone F. Up-regulation of the IL-12 receptor beta 2 chain in Crohn's disease. J Immunol. 2000;165:7234–7239. doi: 10.4049/jimmunol.165.12.7234. [DOI] [PubMed] [Google Scholar]
- 42.Bauditz J, Wedel S, Lochs H. Thalidomide reduces tumour necrosis factor alpha and interleukin 12 production in patients with chronic active Crohn's disease. Gut. 2002;50:196–200. doi: 10.1136/gut.50.2.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carmeliet P, Collen D. Molecular analysis of blood vessel formation and disease. Am J Physiol. 1997;273:H2091–H2104. doi: 10.1152/ajpheart.1997.273.5.H2091. [DOI] [PubMed] [Google Scholar]
- 44.Spalinger J, Patriquin H, Miron MC, Marx G, Herzog D, Dubois J, Dubinsky M, Seidman EG. Doppler US in patients with crohn disease: vessel density in the diseased bowel reflects disease activity. Radiology. 2000;217:787–791. doi: 10.1148/radiology.217.3.r00dc19787. [DOI] [PubMed] [Google Scholar]
- 45.Kevil CG, Payne DK, Mire E, Alexander JS. Vascular permeability factor/vascular endothelial cell growth factor-mediated permeability occurs through disorganization of endothelial junctional proteins. J Biol Chem. 1998;273:15099–15103. doi: 10.1074/jbc.273.24.15099. [DOI] [PubMed] [Google Scholar]
- 46.Bousvaros A, Leichtner A, Zurakowski D, Kwon J, Law T, Keough K, Fishman S. Elevated serum vascular endothelial growth factor in children and young adults with Crohn's disease. Dig Dis Sci. 1999;44:424–430. doi: 10.1023/a:1026635308127. [DOI] [PubMed] [Google Scholar]
- 47.Kim I, Moon SO, Kim SH, Kim HJ, Koh YS, Koh GY. Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J Biol Chem. 2001;276:7614–7620. doi: 10.1074/jbc.M009705200. [DOI] [PubMed] [Google Scholar]
- 48.Goebel S, Huang M, Davis WC, Jennings M, Siahaan TJ, Alexander JS, Kevil CG. VEGF-A stimulation of leukocyte adhesion to colonic microvascular endothelium: implications for inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2006;290:G648–G654. doi: 10.1152/ajpgi.00466.2005. [DOI] [PubMed] [Google Scholar]