

Abstract
AIM: To investigate the reversal effect of LY980503, a benflumetol derivative, on multidrug resistance in vincristine (VCR) -resistant human gastric carcinoma cell line SGC7901/VCR.
METHODS: Cells of a human gastric cancer cell line, SGC7901, and its VCR-resistant variant, SGC7901/VCR, were cultivated with LY980503 and /or doxorubicin (DOX). The cytotoxicity of drugs in vitro was assayed by MTT method. Based on the flow cytometric technology, the uptake of DOX was detected in these cells by measuring DOX -associated mean fluorescence intensity (MFI).
RESULTS: SGC7901/VCR cells were 23.5 times more resistant to DOX in comparison with SGC7901 cells. LY980503 at the concentrations of 2.0 μmol/L -10 μmol/L had no obvious cytotoxicity to SGC7901 and SGC7901/VCR cells. After simultaneous treatment with LY980503 at the concentrations of 2.0, 4.0 and 10 μmol/L, the IC50 of DOX to SGC7901/VCR cells decreased from 1.6 ± 0.12 μmol/L to 0.55 ± 0.024, 0.25 ± 0.032 and 0.11 ± 0.015 μmol/L, respectively, thus, increasing the DOX sensitivity by 2.9-fold (P < 0. 05), 6.4-fold (P < 0. 01) and 14.5-fold (P < 0. 01), respectively. In the uptake study of DOX, simultaneous incubation of SGC7901/VCR cells with LY980503 significantly increased the DOX -associated MFI in SGC7901/VCR cells. No such results were found in parental SGC7901 cells.
CONCLUSION: LY980503 at non-cytotoxic concen-trations can effectively circumvent resistance of SGC7901/VCR cells to DOX by increasing intracellular DOX accumulation.
Keywords: Multidrug resistance, Benflumetol, Doxorubicin, Gastric cancer, Reversal
INTRODUCTION
Resistance of tumor cells to chemotherapeutic drugs is a major problem in clinical treatment of cancer. Multidrug resistance (MDR) is the most important form of drug resistance characterized by decreased cellular sensitivity to a broad range of chemotherapeutic drugs[1,2]. Classic MDR is mainly caused by over-expression of P-glycoprotein(Pgp), which is coded by the MDR1 gene and functions as an ATP-dependent drug-efflux membrane transporter that rapidly extrudes a variety of hydrophobic anticancer drugs from exerting cytotoxic effect[1-3]. Agents that inhibit the ATP-dependent transporter can suppress the efflux of hydrophobic drugs from resistant cells and increase their intracellular accumulation. A number of agents, including verapamil and cyclosporin A, bind to and inhibit Pgp to restore drug sensitivity to cancer cells in vitro. However, to date, no effective resistance modulators have been used in clinic due to their toxicity or unacceptable side effects[3,4]. Therefore, development of safe and effective MDR reversal agents is eagerly required.
Our previous study has proved that LY980503, a derivative of benflumetol (Figure 1), could effectively reverse Pgp-mediated MDR in doxorubicin (DOX) -resistant human breast cancer cell line MCF/DOX[5]. However, the effect of LY980503 on MDR in human gastric cancer cells remains unknown. The present study was therefore undertaken to explore the reversal effect of LY980503 on MDR in vincristine (VCR)-resistant human gastric cancer cell line SGC7901/VCR.
Figure 1.
Chemical structures of benflumetol and benflumetol derivative LY980503.
MATERIALS AND METHODS
Drugs and chemicals
LY980503 was synthesized in our laboratory, and dissolved in dimethyl sulfoxide (DMSO). MTT, DOX were purchased from Sigma, VCR from the Twelfth Shanghai Pharmaceutical Product Factory.
Cell culture
Human gastric cancer cell line SGC7901, and its VCR-resistant counterpart SGC7901/VCR selected by stepwise exposure of parental SGC7901 cells to increasing concentrations of VCR, were purchased from Wuhan University Type Culture Collection (Wuhan, China). Both cell lines were grown in RPMI 1640 medium supplemented with 10% fetal calf serum at 37°C in a humidified atmosphere of 5% CO2. The SGC7901/VCR cells were cultured in the presence of 0.8 μmol/L VCR and grown in drug-free medium 2 wk before the experiments.
Cytotoxicity assay
The ability of LY980503 to potentiate DOX cytotoxicity was evaluated in SGC7901/VCR and SGC7901 cells by MTT method[5]. SGC7901 and SGC7901/VCR cells in exponential growth were seeded in 96- well plates at a density of 4 × 104 cells per well and 24 h later graded DOX (at the concentrations of 0, 0.01, 0.03, 0.1, 0.3, 1.0, 3.0, 10 μmol/L) and LY980503 (at the concentration of 1.0, 2.0, 4.0, 10 μmol/L ) were added simultaneously, respectively. The total medium volume of each well was 200 μL. Three days after drug addition, 10 μL MTT (5.0 g/L in PBS) was added. After 4 h incubation, supernatants were removed and replaced by 150 μL DMSO. After formazan solubilization, the absorbance at 570 nm was recorded using an automated microplate reader. IC50 (concentration resulting in 50% inhibition of cell growth) values for DOX were calculated as 100% from plotted results using untreated cells. The reversal fold (RF) was calculated as follows:
RF = IC50 of DOX/IC50 of DOX in the presence of LY980503
Intrinsic cytotoxicity assay
SGC7901 and SGC7901/VCR cells were seeded into 96- well plates at 4 × 104/well. After 24 h incubation, 2.0, 4.0, 10, 20, 40 μmol/L of LY980503 were added, respectively. The cells were incubated for 72 h, and the intrinsic cytotoxicity of LY980503 was determined as above.
Intracellular DOX accumulation
The assay was performed as described by Broxterman et al[6]. Briefly, the cells in exponential growth were seeded in 96- well plates at a density of 4 × 104/mL. After 24 h incubation, the SGC7901 and SGC7901/VCR cells were exposed to 4.0 μmol/L DOX in the presence of graded LY980503 (at the concentration of 1.0, 2.0, 4.0, 10 μmol/L, respectively) for 60 min. Following incubation, the cells were washed twice with ice-cold PBS and respended in 100 μL of ice-cold PBS. DOX-associated MFI was measured by flow cytometry at an excitation wavelength of 488 nm and an emission wavelength of 550 nm.
Statistical analysis
Data were expressed as mean ± SD. Student’s t test was used to assess statistical significance of differences. P < 0.05 was considered statistically significant.
RESULTS
Modulating effect of LY980503 on MDR in SGC7901/VCR cells
Using the MTT assay, the in vitro cytotoxicity of DOX combined with LY980503 in SGC7901 and SGC7901/VCR cells was examined. As shown in Table 1, SGC7901/VCR cells were 23.5 times more resistant to DOX in comparison with SGC7901 cells. LY980503 gave a significant reversal of resistance of SGC7901/VCR cells to DOX at the concentration as low as 2.0 μmol/L. LY980503 at 10 μmol/L decreased the IC50 of DOX on SGC7901/VCR cells from 1.6 ± 0.12 μmol/L to 0.11 ± 0.015 μmol/L (the reversal fold was 14.5), indicating that LY980503 had a potent reversal effect on MDR in SGC7901/VCR cells. The IC50 of DOX on SGC7901 cells decreased from 0.068 ± 0.0081 μmol/L to 0.021 ± 0.0029 μmol/L in the presence of 10 μmol/L LY980503 (the reversal fold was 3.2), indicating that LY980503 could slightly enhance the sensitivity of SGC7901 cells to DOX.
Table 1.
Effect of LY980503 on the sensitivity of SGC7901 and SGC7901/VCR cells to DOX (mean ± SD)
Intrinsic cytotoxicity
As shown in Table 2, LY980503 up to 10 μmol/L had no obvious cytotoxicity to SGC7901 and SGC7901/VCR cells.
Table 2.
Effect of LY980503 on the growth of SGC7901 and SGC7901/VCR cells (mean ± SD)
| LY980503 |
Survival rate (%) |
|
| (μmol/L) | SGC7901 | SGC7901/VCR |
| 2 | 97 ± 8.6 | 103 ± 9.7 |
| 4 | 94 ± 7.8 | 98 ± 8.3 |
| 10 | 89 ± 6.5 | 87 ± 8.9 |
| 20 | 61 ± 4.6 | 67 ± 6.8 |
| 40 | 34 ± 4.5 | 39 ± 3.9 |
Effect of LY980503 on intracellular accumulation of DOX
The ability of LY980503 to inhibit the Pgp function was evaluated by determining the intracellular DOX-associated MFI in SGC7901/VCR and SGC7901 cells. As shown In Figure 2, in the uptake study, there was a high level of DOX accumulation in the drug-sensitive SGC7901 cells, that was unaffected by LY980503. There was a very low level of accumulation in SGC7901/VCR cells in the absence of LY980503. In contrast, LY980503 dose-dependently restored the level of accumulation of DOX in SGC7901/VCR cells. LY980503 at 2.0, 4.0, 10 μmol/L enhanced DOX-associated MFI in SGC7901/VCR cells by 1.4-, 2.3-, 3.4-fold, respectively.
Figure 2.
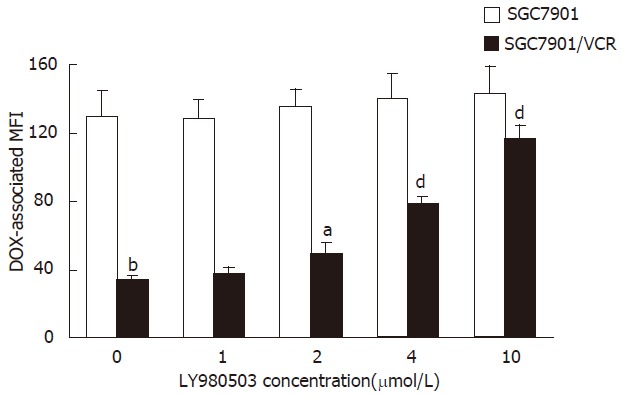
Effect of LY980503 at various concentrations on accumulation of intracellular DOX in SGC7901 and SGC7901/VCR cells. SGC7901 and SGC7901/VCR cells were incubated with 4.0 μmol/L DOX for 60 min, in the presence of 0.0, 1.0, 2.0, 4.0 and 10 μmol/L LY980503, respectively. Each point represents the mean ± SD from three experiments. bP < 0. 01 vs SGC7901 cells in the absence of LY980503; aP < 0.05, dP < 0.01 vs SGC7901/VCR cells in the absence of LY980503.
DISCUSSION
Many mechanisms are involved in resistance to cancer chemotherapy, including decreased drug accumulation, altered intracellular drug distribution, increased detoxification, diminished drug-target interaction, increased DNA repair, altered cell-cycle regulation, etc. Of these, over-expression of Pgp is the main mechanism of MDR[3,4].
SGC7901/VCR is an established VCR-resistant cell sub-line selected by stepwise exposure of parental SGC7901 cells to increasing concentrations of VCR. Several reports have proved that the VCR-resistant SGC7901/VCR cells possess the characteristics of MDR with over-expression of Pgp[7-12]. The SGC7901/VCR cell line has been successfully used as an in vitro MDR reversal model by several study groups[10-12].
Benflumetol is a newly developed antimalarial drug[13]. Our previous study found that 10 μmol/L benflumetol could decrease the IC50 values of DOX on MCF/DOX cells from 2.8 μmol/L to 0.55 μmol/L, indicating that benflumetol has a slight reversal effect on MDR[5]. We synthesized a series of benflumetol derivatives. Among the 22 benflumetol derivatives, twelve compounds have varied MDR reversal effects, and the effect of LY980503 was the most potent on MDR in MCF/DOX cells[5].
SGC7901/VCR cell line was 23.5-fold more resistant to DOX than the parental SGC7901 cell line. LY980503, at concentrations of 2.0, 4.0, 10 μmol/L, could increase the chemosensitivity of SGC7901/VCR cells to DOX by 2.9-, 6.4- and 14.5-fold, respectively. Restoration of DOX accumulation in the resistant SGC7901/VCR cells close to the level seen in the sensitive SGC7901 cells in the presence of 10 μmol/L LY980503 suggests that LY980503 can inhibit efflux of DOX by Pgp. Taken together, it seems reasonable to conclude that LY980503 can inhibit the function of Pgp and therefore enchance the sensitivity of the resistant cells to anticancer agent DOX. Furthermore, although LY980503 reverses Pgp-mediated MDR at the concentration of 2.0 μmol/L, the compound is non-cytotoxic by itself at the concentration up to 10 μmol/L in SGC7901/VCR and parental SGC7901 cells.
In conclusion, LY980503 at a non-cytotoxic concentration exerts a potent reversal effect on MDR in SGC7901/VCR cells by inhibiting Pgp-mediated drug transport, indicating that LY980503 may be a promising MDR chemosensitizer.
COMMENTS
Background
Multidrug resistance (MDR) was first observed in experimental oncology in 1970 by Biedler and Riehm, and has been extensively studied since then. MDR involves the simultaneous cross-resistance to several structurally unrelated natural products. Unfortunately, approximately 50% of all antineoplastic agents are derived from natural sources and fall under the umbrella of the MDR mechanism. The MDR phenotype confers resistance to anthracyclines, vinca alkaloids, epipodophyllotoxins, taxanes, antibiotics and some of the new topo-I inhibitors. The most important factor of the mechanisms leading to MDR of tumor cells is the increased activity of transporter proteins. The best-characterized transporter protein is P-glycoprotein (Pgp), and a number of clinical investigations have suggested that its intrinsic or acquired over-expression results in a poor clinical outcome of chemotherapy. An interesting feature of the MDR phenotype is its susceptibility to inhibition by certain non-natural product agents. Calcium channel blockers, calmodulin antagonists, immunosuppressants, hormonal drugs and other agents have been found to inhibit Pgp function and the MDR phenotype. Structural similarities in many of these reversal agents include a ternary nitrogen, aromatic ring and amphipathic characteristics. The mechanism of action is thought to be direct binding to the Pgp and displacing anticancer drugs, which increase intracellular drug accumulation. The design of nontoxic agents that would overcome the MDR of tumors has been a challenging area for pharmaceutical development.
Research frontiers
Many agents that modulate the Pgp transporter, including verapamil, cyclosporin, tamoxifen, and several calmodulin antagonists, were identified in the 1980s.These agents often produce disappointing results in vivo because their low binding affinities necessitate the use of high doses, resulting in unacceptable toxicity. Many of the first chemosensitizers identified are substrates for Pgp and thus work by competing with the cytotoxic compounds for efflux by the Pgp pump. However, many of these chemosensitizers are substrates for other transporters and enzyme systems, resulting in unpredictable pharmacokinetic interactions in the presence of chemotherapy agents. The second-generation Pgp modulators include dexverapamil, dexniguldipine, valspodar and biricodar. These agents are more potent and also less toxic than their predecessors. The best characterized of these agents is valspodar, a nonimmunosuppressive derivative of cyclosporin D. Valspodar has been studied in numerous clinical trials in combination with cytotoxic agents. A study reported that valspodar at 1 nmol/L had a modest effect (20% increase) on anthracycline accumulation in Pgp positive sarcoma samples. The co-administration of second-generation Pgp modulators and chemotherapy agents in clinical trials has resulted in the reversal of MDR and some limited success in treating refractory cancers. Second-generation Pgp modulators have a better pharmacologic profile than the first-generation compounds, but they also retain some characteristics that limit their clinical usefulness. In particular, these compounds significantly inhibit the metabolism and excretion of cytotoxic agents, thus leading to unacceptable toxicity that has necessitated chemotherapy dose reductions in clinical trials. Third-generation molecules that specifically and potently inhibit Pgp function have been developed by using structure-activity relationships and combinatorial chemistry to overcome the limitations of the second generation Pgp modulators. These agents do not affect cytochrome P450 3A4 at relevant concentrations, thus do not alter the plasma pharmacokinetics of anticancer drugs. Similarly, third-generation agents typically do not inhibit other ABC transporters. This specificity for the Pgp pump minimizes the possibility that the blockade of more than one pump might result in altered bioavailability or excretion of the chemotherapy agents. The third generation Pgp inhibitors currently in clinical development include tariquidar, zosuquidar and laniquidar.
Innovations and breakthroughs
As a benflumetol derivative, LY980503 was synthesized in our laboratory. In vitro assay showed that LY980503 effectively reversed MDR in MCF/DOX cells. It is a new MDR reversing compound, deserving of further study.
Applications
LY980503 has a good prospect of clinical application. However, it has to be further studied before clinical trial, including elucidation of the exact mechanisms of LY980503 and pharmacokinetic interaction between LY980503 and anticancer drugs in vivo.
Peer review
New synthetic compound, that inhibits P-glycoprotein-mediated efflux in a human gastric cell line SGC7901 and its vincistrin resistant variant. It was previously shown that the vincristin resistancy in this cell line is related to the inproper P-glycoprotein-mediated efflux. The authors show that their compound increase doxorubycin sensitivity in this cell line by promoting doxorubycin uptake.
Footnotes
Supported by National Natural Science Foundation of China, No. 39800181
S- Editor Wang J L- Editor Wang XL E- Editor Zhou T
References
- 1.Wu DL, Huang F, Lu HZ. Drug-resistant proteins in breast cancer: recent progress in multidrug resistance. Ai Zheng. 2003;22:441–444. [PubMed] [Google Scholar]
- 2.Wu DL, L i MJ, Gao HZ, Wu DZ. Daunorubicin-albumin conjugate reverses resistance in multidrug resistant KB/VCR cells to daunorubicin. Zhongguo Yaoli Duli Zazhi. 1997;11:54–58. [Google Scholar]
- 3.Nobili S, Landini I, Giglioni B, Mini E. Pharmacological strategies for overcoming multidrug resistance. Curr Drug Targets. 2006;7:861–879. doi: 10.2174/138945006777709593. [DOI] [PubMed] [Google Scholar]
- 4.Takara K, Sakaeda T, Okumura K. An update on overcoming MDR1-mediated multidrug resistance in cancer chemotherapy. Curr Pharm Des. 2006;12:273–286. doi: 10.2174/138161206775201965. [DOI] [PubMed] [Google Scholar]
- 5.Wu DL, Lu HZ, Huang F, Yin LX, Wan YL, Guo JH, Wu DZ. Reversal of resistance of multidrug resistant MCF/ Dox cell line to doxorubicin by a benflumetol derivative LY980503. Zhongguo Yaoli Duli Zazhi. 2002;16:211–215. [Google Scholar]
- 6.Feller N, Kuiper CM, Lankelma J, Ruhdal JK, Scheper RJ, Pinedo HM, Broxterman HJ. Functional detection of MDR1/P170 and MRP/P190-mediated multidrug resistance in tumour cells by flow cytometry. Br J Cancer. 1995;72:543–549. doi: 10.1038/bjc.1995.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang YX, Xiao ZQ, Chen ZC, Zhang GY, Yi H, Zhang PF, Li JL, Zhu G. Proteome analysis of multidrug resistance in vincristine-resistant human gastric cancer cell line SGC7901/VCR. Proteomics. 2006;6:2009–2021. doi: 10.1002/pmic.200402031. [DOI] [PubMed] [Google Scholar]
- 8.Zhao Y, Xiao B, Chen B, Qiao T, Fan D. Upregulation of drug sensitivity of multidrug-resistant SGC7901/VCR human gastric cancer cells by bax gene transduction. Chin Med J (Engl) 2000;113:977–980. [PubMed] [Google Scholar]
- 9.Han Y, Shi Y, Zhang H. Alteration of subcellular distribution of protein kinase C isoforms in swelling-activated multi-drug-resistant gastric cancer cells and its significance. Zhonghua YiXue ZaZhi. 2001;81:328–331. [PubMed] [Google Scholar]
- 10.Tang XQ, Bi H, Feng JQ, Cao JG. Effect of curcumin on multidrug resistance in resistant human gastric carcinoma cell line SGC7901/VCR. Acta Pharmacol Sin. 2005;26:1009–1016. doi: 10.1111/j.1745-7254.2005.00149.x. [DOI] [PubMed] [Google Scholar]
- 11.Li DQ, Wang ZB, Bai J, Zhao J, Wang Y, Hu K, Du YH. Reversal of multidrug resistance in drug-resistant human gastric cancer cell line SGC7901/VCR by antiprogestin drug mifepristone. World J Gastroenterol. 2004;10:1722–1725. doi: 10.3748/wjg.v10.i12.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao FL, Wang F, Wu JL, LE XP, Zhang QX. Screening effective sequences of small interfering RNAs targeting MDR1 gene in human gastric cancer SGC7901/VCR cells. Zhonghua ZhongLiu ZaZhi. 2006;28:178–182. [PubMed] [Google Scholar]
- 13.Wiesner J, Ortmann R, Jomaa H, Schlitzer M. New antimalarial drugs. Angew Chem Int Ed Engl. 2003;42:5274–5293. doi: 10.1002/anie.200200569. [DOI] [PubMed] [Google Scholar]