

Abstract
AIM: To investigate the effect of ceramide on the cell cycle in human hepatocarcinoma Bel7402 cells. Possible molecular mechanisms were explored.
METHODS: [3- (4, 5)-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide (MTT) assay, plasmid transfection, reporter assay, FACS and Western blotting analyses were employed to investigate the effect and the related molecular mechanisms of C2-ceramide on the cell cycle of Bel7402 cells.
RESULTS: C2-ceramide was found to inhibit the growth of Bel7402 cells by inducing cell cycle arrest. During the process, the expression of p21 protein increased, while that of cyclinD1, phospho-ERK1/2 and c-myc decreased. Furthermore, the level of CDK7 was downregulated, while the transcriptional activity of PPARγ was upregulated. Addition of GW9662, which is a PPARγ specific antagonist, could reserve the modulation action on CDK7.
CONCLUSION: Our results support the hypothesis that cell cycle arrest induced by C2-ceramide may be mediated via accumulation of p21 and reduction of cyclinD1 and CDK7, at least partly, through PPARγ activation. The ERK signaling pathway was involved in this process.
Keywords: Ceramide, Cell cycle arrest, Human hepatocarcinoma cells, P21, CyclinD1, CDK7, PPARγ, ERK
INTRODUCTION
Ceramide has emerged as a novel lipid second messenger with specific roles in mediating cell growth, differentiation, stress responses and apoptosis[1-3]. Ceramide is generated through the hydrolysis of sphingomyelin by the activation of sphingomyelinase (SMase). A number of stimuli have been reported to activate SMase[4-6]. Exogenously administered synthetic ceramide mimicked the action of these inducers in the regulation of various cell functions. Ceramide mediates numerous cellular functions such as differentiation, growth arrest, apoptosis and proliferation[7,8]. Ceramide is thought to be involved in modulating ceramide-activated protein kinase (CAPK), mitogen-activated protein kinase (MAPK), ceramide-activated protein phosphatase (CAPP) and phospholipaseA2 (PLA2), etc[9]. Apoptosis induction by ceramide is associated with Bcl-2 phosphorylation, SAPK/JNK and caspase pathway activation[10]. On the other hand, activation of PKC and Bcl-2 expression can inhibit the ceramide signal pathway[11,12]. However, the link between ceramide signaling and the cell cycle is poorly understood.
To exploit the effect of ceramide on the cell cycle, human hepatocarcinoma Bel7402 cells were employed and treated with C2-ceramide. Hepatocarcinoma occurs with high incidence in southern China and southeast Asia. Radiation is one of the agents that activates ceramide signaling[13], and therefore, it is of interest in investigating the effect of ceramide on Bel7402 cells. In this study, we observed inhibition of cell proliferation and cell cycle arrest in the G1 phase following C2-ceramide treatment in Bel7402 cells. Subsequent studies suggested that modulation might be mediated via accumulation of p21 and reduction of cyclinD1 and CDK7, at least partly, through PPARγ activation. The ERK signaling pathway was also involved in this process.
MATERIALS AND METHODS
Materials
C2-ceramide was purchased from Sigma. Co (St Louis Mo, USA); mouse monoclonal anti-p21, anti-cyclinD1, anti-CDK7, anti-p-ERK and anti-c-myc were purchased from Santa Cruz Biotechnology, Inc (CA, USA); antibody of PE-E-cadherin was purchased from DAKO Co. Horseradish peroxidase-conjugated secondary antibodies were purchased from Jackson Immuno-Research Laboratories, Inc (West Grove, PA, USA). Lipofectamine and lipofectin reagents were from Gibco, Inc. RNaseA, MTT, propiudium iodide and other chemicals were all from Sigma.
Cell culture
Bel7402 cells were provided by the Institute of Zoology, Chinese Academy of Sciences, China. Cells were maintained in Dulbecco’s minimal essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS) in a humidified atmosphere of 95% air/5% CO2 at 37°C. A subculture of cells was processed by enzymatic digestion (trypsin/EDTA solution: 0.25/0.02%). C2-ceramide dissolved in ethanol was used without filtration. The final concentration of ethanol in culture medium was < 0.3%.
MTT assay
Bel7402 cells were seeded onto 96-well plates at a concentration of 2.5 × 103 cells/well in DMEM plus 5% FBS. The stock of C2-ceramide was diluted with medium, and then added to wells for desired final concentrations. After exposure to C2-ceramide for the desired time, 10 μL of 5 mg/mL MTT was added to each well and incubated for 4 h, and the liquid in wells was evaporated. To dissolve the formazam, 100 μL of DMSO was added. The absorbance was determined with a microplate reader model 550 at the wavelength of 570 nm.
Cell cycle analysis
The proportions of cells in G0-G1, S, and G2-M were determined by flow cytometric analysis of DNA content. Briefly, cells were obtained by trypsinization following treatment with C2-ceramide, and then washed twice with PBS. Cells were then incubated with RNase at a concentration of 0.25 mg/mL at 37°C for 1 h following incubation with PI (50 μg/mL in PBS) for 30 min at 4°C in the dark. Before flow cytometry, samples were syringed through a 25-gauge needle to prevent nuclear clumping. PI was excited at 488 nm, and fluorescence was analyzed at 620 nm. All measurements were carried out under the same instrumental settings.
PE-E-cadherin labeling
Cells were washed once with cold PBS, and centrifuged to collect the cell pellet (350 g × 5 min) following treatment with C2-ceramide.The cell pellet was resuspended in cold PBS (4°C) and PE-E-cadherin and the corresponding isotype antibodies were added to the cell suspension and mixed gently. The tube was then incubated for 30-60 min in the dark at room temperature prior to flow cytometry. PE-E-cadherin binding was analyzed by flow cytometry collecting the fluorescence of 10 000 cells using a FACScan (Becton Dickinson) according to the manufacturer’s instructions. All experiments were replicated three times.
Western blot analysis
To determine the expression levels of E-cadherin, p21, cyclinD1, CDK7 and c-myc, cells were lysed in buffer (150 mmol/L NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mmoL/L Tris, pH 8.0, 1 mmoL/L phenylmethylsulfonyl fluoride (PMSF), and 10 μg/mL aprotinin). To determine the level and phosphorylation state of ERK, cells were harvested in lysis buffer containing 50 mmol/LTES (N-tris [hydroxymethyl] methyl-2-aminoethane sulfonic acid) (pH 7.4), 2 mmol/L EGTA, 1 mmol/L EDTA, 250 mmol/L sucrose, 40 mmol/L phenylphosphate, 1 mmol/L MgCl2, 2 mmoL/L Na3VO4, 10 mmol/L Na4P2O7, 100 mmol/L NaF, 5 μg/mL aprotinin, 1 mmol/L PMSF, 1 μg/mL leupeptin, 5 mmol/L benzamidine, and 10 mmol/L dithiothreitol. Protein (40-80 μg) was separated by 12%-15% SDS-polyacrylamide gel in the separation buffer (25 mmol/L Tris, 250 mmol/L Glycine, 0.1% SDS). Total proteins were transferred onto a PVDF membrane after electrophoresis. Western blot analyses using anti-p21, anti-cyclinD1, anti-CDK7, anti-c-myc, anti-phospho-p42/p44ERK and anti-totalERK antibodies were performed. As an internal control, mouse monoclonal anti-β-actin antibody was used.
Transient transfection and luciferase assay for PPARγ activity
Bel7402 cells were seeded at a concentration of 1 × 105 cells/35 mm dish. After 12 h, the medium was changed from complete medium to DMEM without antibiotic. Transfection was done using LipofectAMINE reagent mixed with 2 μg of acyl-CoA oxidase promoter-luciferase plasmid pAOXPPREluc and the control pAOXBluc basic vector (kindly donated by Dr. Osumi) for 8 h. After the transfection mixture was replaced by a medium containing 10% FBS, cells were then incubated with or without different concentrations of ceramide for a desired time. Luciferase activity was measured according to the manufacturer’s protocol (Promega).
Statistical analysis
All statistical analyses were performed with SPSS 10.0 statistical package for Microsoft Windows. Data were expressed as mean ± SE for all measurements. P < 0.05 was considered statistically significant.
RESULTS
Ceramide inhibited proliferation by halting the cell cycle in Bel7402 cells
Treatment with different concentrations of C2-ceramide for 24 h exhibited significant inhibition of cell proliferation of human hepatocarcinoma Bel7402 cells as suggested by MTT assay. Under concentrations of 0, 5, 10, 15, 30 and 60 μmmo/L, inhibitory rates were 0, 21.5% ± 1.3%, 52.7% ± 0.9%, 69.3% ± 1.2%, 77.2% ± 0.8% and 83.8% ± 1.2%, respectively (Figure 1A). Cytotoxicity was further indicated by determination of E-cadherin, which is a marker for many tumor cells with high expression. PE-E-cadherin antibodies were stained with cells to determine the expression of E-cadherin on the cell surface. Flow cytometry analysis results indicated that E-cadherin was significantly down-regulated by C2-ceramide (Figure 1B), which was also suggested by blot assay (Figure 1C). To test whether the cytotoxicity was derived from the effect on the cell cycle, flow cytometry analysis was applied following treatment with different concentrations of C2-ceramide at 0, 5, 15, 30 and 60 μmol/L. As shown in Table 1, the cell cycle was halted in the G1 phase, and the percentage of cells in the G1 phase was 35.3% ± 0.7%, 36.8% ± 1.2%, 43.9% ± 1.2%, 57.2% ± 0.6% and 76.2% ± 1.3%, respectively.
Figure 1.
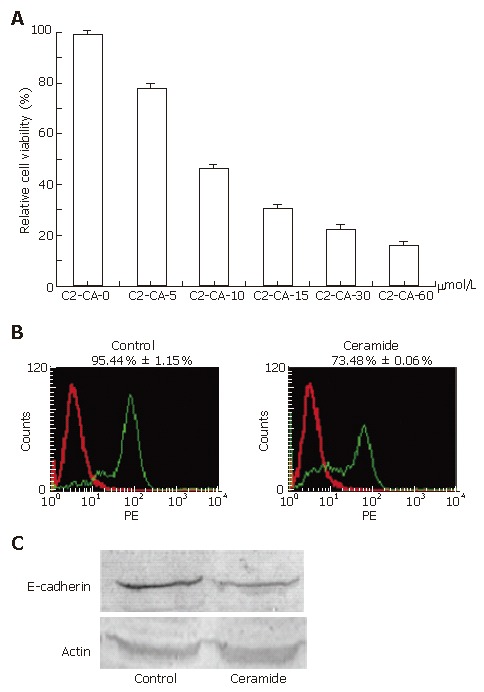
Ceramide inhibited proliferation by halt of cell cycle in Bel7402 cells. A: ceramide inhibited cell proliferation in Bel7402 cells. Cells were cultured in the medium with different concentrations of C2-ceramide for 24 h. Cell viability was analyzed by MTT assay and presented as cell proliferative rates. The results show the mean ± SE (n = 3-4); B and C: ceramide down-regulated the expression of E-cadherin. Bel7402 cells were incubated with or without 30 μmmo/L of ceramide for 24 h. Cells were then harvested and stained with anti-E-cadherin directly labeled by PE. The protein levels of E-cadherin were determined by flow cytometry. Data was represented by the mean ± SE of 3 or 4 separate experiments (B). Bel7402 cells were incubated with or without 30 μmmo/L of ceramide for 24 h. Cells were then harvested, lysed and resolved in 12% SDS-PAGE. The expression of E-cadherin was determined by Western blot. Actin was used as a control. Data represent the results of three separate experiments (C).
Table 1.
Ceramide halted cell cycle in Bel7402 cells (mean ± SE, n = 3)
| Ceramide (μmol/L) | G0/G1 (%) | S (%) | G2/M |
| 0 | 35.3 ± 0.7 | 44.3 ± 0.3 | 20.4 ± 0.1 |
| 5 | 36.8 ± 1.2 | 43.9 ± 0.4 | 19.3 ± 1.8a |
| 15 | 43.9 ± 1.2a | 41.9 ± 0.7a | 14.2 ± 0.8a |
| 30 | 57.2 ± 0.6a | 28.8 ± 0.5a | 14.0 ± 0.5a |
| 60 | 76.2 ± 1.3a | 8.2 ± 0.2a | 15.6 ± 1.3a |
P < 0.05 vs control.
Ceramide up-regulated the expression of p21WAF1/BIP1 and down-regulated that of cyclinD1
In order to elucidate whether molecules were involved in the G1/S transition following C2-ceramide treatment, cell extracts were prepared from Bel7402 cells treated with different concentrations of C2-ceramide. Protein expression was indicated by blot using anti-p21 and anti-cyclinD1 antibodies, respectively. As shown in Figure 2, C2-ceramide increased the expression of p21 protein, but decreased that of cyclinD1 protein.
Figure 2.
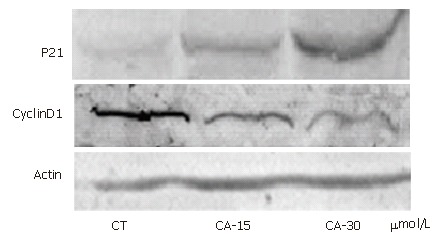
Ceramide up-regulated the expression of p21WAF1/BIP1 and down-regulated that of cyclinD1. Bel7402 cells were incubated with different concentrations of ceramide as indicated for 24 h. Cells were harvested, lysed and resolved in 15% SDS-PAGE. The expression of p21 and cyclinD1 were determined by Western blot. Actin was used as a control. Data represent the results of three separate experiments.
CDK7 and PPARγ pathways involved in cell cycle arrest induced by C2-ceramide
In our previous study, we reported that C2-ceramide could activate PPARγ transcription activity in human colon cancer HT29 cells[14]. In this study, as shown in Figure 3A, C2-ceramide could also markedly activate the transcriptional activity of PPARγ in hepatocarcinoma Bel7402 cells.
Figure 3.
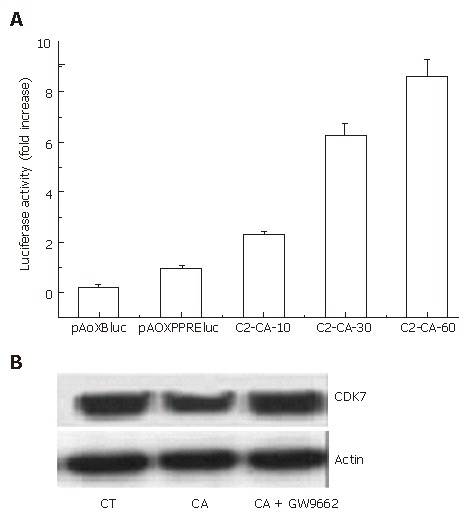
CDK7 and PPARγ pathways involved in cell cycle arrest induced by C2-ceramide. A: activation of PPARγ transcriptional activity induced by C2-ceramide. Bel7402 cells were transiently transfected with the PPAR responsive element (PPRE) reporter construct (pAOXPPREluc) or the promoter-less control vector pAOXBluc following the treatment of different concentrations of C2-ceramide as indicated, PPARγ transcriptional activity was measured as described. The bar represents the relative fold increase of luciferase activity. The results show means ± SE (n = 3); B: C2-ceramide down-regulated the expression of CDK7, which was blocked by GW9662. Bel7402 cells were cultured with 30 μmol/L C2-ceramide in the presence or absence of GW9662, which was claimed as a specific PPARγ antagonist for 24 h. Total proteins were extracted and resolved on SDS-PAGE followed by Western blot assay using anti-CDK7 antibody. Data represents the results of three separate experiments.
Cyclin-dependent kinase 7 (CDK7) is critical for cell cycle and transcriptional programs[15]. Therefore, we investigated the expression of CDK7 by blotting. It was observed that CDK7 expression decreased following treatment with 30 μmmo/L C2-ceramide for 24 h. However, addition of the PPARγ specific antagonist GW9662 markedly reversed the inhibition (Figure 3B).
Ceramide inhibited the activation of ERK1/2 in Bel7402 cells
ERK plays a key role in cell survival in many cells. To examine whether ceramide inhibited the activation of ERK, Bel7402 cells were treated with different concentrations of C2-ceramide and phospho-ERK1/2 was determined. As shown in Figure 4, the expression of p-ERK decreased significantly with the treatment of C2-ceramide, while total ERK protein expression was unaffected.
Figure 4.
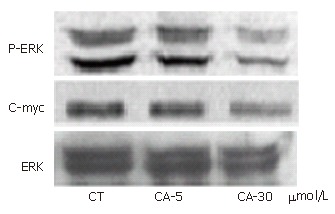
Ceramide inhibited activation of ERK pathway in Bel7402. Bel7402 cells were incubated with different concentrations of ceramide as indicated. Total proteins were extracted and resolved on SDS-PAGE. Phospho-ERK1/2 and c-myc were normalized to total Erk1/2 and internal standard actin was determined by Western blot. Data represents the results of three separate experiments.
To complete the study of the MAPK pathway, c-myc content was also indicated by blot using anti-c-myc antibodies. As shown in Figure 4, the modulation pattern on the expression of c-myc induced by C2-ceramide was similar to that of p-ERK.
DISCUSSION
Since ceramide was affirmed as an important lipid second messenger in 1994, the importance of ceramide in cell metabolism has been broadly investigated. Ceramide has shown inhibition of cell growth via apoptosis in a variety of cancers. However, the link between ceramide signaling and the cell cycle is poorly understood. The present work demonstrates that C2-ceramide halted the distribution of cell cycles in G1 phase, and the mechanisms involved were explored.
As displayed in Table 1, C2-ceramide induced cell cycle arrest in the G1 phase. Accordingly, down-regulation of E-cadherin, a marker for many tumor cells with high expression, suggested that C2-ceramide inhibited cancer cell growth (Figure 1B).
In order to elucidate the mechanisms involved in cell cycle arrest following treatment with C2-ceramide in Bel7402 cells, we firstly investigated whether the cell cycle control genes; i.e., cyclin D1 and the cyclin-dependent kinase inhibitor p21, are involved in ceramide-mediated cell cycle regulation in Bel7402 cells.
Cell cycle progression is regulated by interactions between cyclins and cyclin-dependent kinases, which are modulated by a family of negative cell cycle regulators; i.e., cyclin-dependent kinase inhibitors, which are especially involved in controlling the transition from G1 to S-phase[16,17]. The latter includes two families, the CIP/KIP family and the INK4 family. p21 is a member of the CIP/KIP family and plays a crucial role in growth arrest by a variety of mechanisms[18]. Consistent with this idea, we showed that C2-ceramide up-regulated the expression of p21 protein concomitant with inhibition of cyclin D1 protein (Figure 2).
PPARγ is clearly involved in lipid metabolism and is essential for cell differentiation[19,20]. Activation of PPARγ by its ligands can induce growth inhibition and cytotoxicity in human prostate cancer cells, colon cancer cells and liposarcoma cells, and their biological activities are attributed to inhibition of proliferation and induction of apoptosis by PPARγ[21,22]. We have previously shown C2-ceramide could induce apoptosis via a PPARγ dependent pathway in human colon cancer HT29 cells[14].To examine whether the PPARγ pathway was involved in the modulation of cell cycle arrest induced by ceramide in human hepatocarcinoma Bel7402 cells, the luciferase reporter of PPRE3x-tk-luc was transfected and luciferase activity was assayed. The result shown in Figure 3A suggested that ceramide activated the transcriptional activity of PPARγ in a dose-dependent manner.
CDK7 distributes normally throughout the nucleus and cytoplasm, and in the nucleus, it attaches to a DNA template with other TFIIH subunits initiating gene transcription[23], which is very important in modulating cell proliferation. Participating in basal transcription by phosphorylating the carboxy-terminal domain of the largest subunit of RNA polymerase II, CDK7 is critical for the cell cycle and transcriptional programmes, which also phosphorylate other CDKs as an essential step for their activation[24,25]. Often, phosphorylation of NRs by kinases that are associated with general transcription factors (e.g. CDK7 within TFIIH), or activated in response to a variety of signals (MAPKs, Akt, PKA, PKC), facilitates the recruitment of coactivators or components of the transcription machinery and, therefore, cooperates with the ligand to enhance transcription activation.
Though there is much to be explored, data have shown that C2-ceramide inhibited cell proliferation through attenuation of CDK7 (Figure 3B). However, GW9662, which is a PPARγ specific antagonist, could markedly block this action. These data suggested that CDK7 was related to PPARγ, which was consistent with the opinion described above.
ERK signaling plays a key role in cell survival, and Erk1/2 are proteins belonging to the MAPK pathway, whose members are active when phosphorylated. Consequently, dephosphorylation of these proteins inhibits their activity and the transcription factor c-myc. The increase of PP2A, which is a serine/threonine phosphatase causing dephosphorylation of MAPKs[26], is one of the causes of Erk1/2 dephosphorylation. Indeed, the ERK1/2 pathway, that regulates cellular growth and proliferation, has been shown to be pro[27-30] or anti-apoptotic[31-33], depending on experimental conditions and/or cell types. To examine whether C2-ceramide can inhibit activation of ERK, phospho-ERK was determined. It was found that phospho-ERK decreased with the treatment of C2-ceramide in Bel7402 cells, however, the expression level of total ERK protein was unaffected. Accordingly, the protein expression of c-myc also decreased (Figure 4).
In summary, with consideration that there is much to be explored, it was concluded that C2-ceramide plays an important role in the inhibition of cell growth in Bel7402 cells. Our results showed that there is p21 accumulation in accordance with decreased cyclinD1, inactivation of the ERK pathway, downregulation of CDK7 and stimulation of PPARγ transcriptional activity. As such, our results support the hypothesis that suppression of hepatocarcinoma cell growth through cell cycle arrest induced by C2-ceramide may be mediated via accumulation of p21 and reduction of cyclinD1 and CDK7, at least partly, through PPARγ activation. The ERK signaling pathway was involved in this process.
Footnotes
S- Editor Liu Y L- Editor Lutze M E- Editor Lu W
References
- 1.Obeid LM, Linardic CM, Karolak LA, Hannun YA. Programmed cell death induced by ceramide. Science. 1993;259:1769–1771. doi: 10.1126/science.8456305. [DOI] [PubMed] [Google Scholar]
- 2.Hannun YA, Obeid LM. The Ceramide-centric universe of lipid-mediated cell regulation: stress encounters of the lipid kind. J Biol Chem. 2002;277:25847–25850. doi: 10.1074/jbc.R200008200. [DOI] [PubMed] [Google Scholar]
- 3.Mathias S, Peña LA, Kolesnick RN. Signal transduction of stress via ceramide. Biochem J. 1998;335(Pt 3):465–480. doi: 10.1042/bj3350465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tavarini S, Colombaioni L, Garcia-Gil M. Sphingomyelinase metabolites control survival and apoptotic death in SH-SY5Y neuroblastoma cells. Neurosci Lett. 2000;285:185–188. doi: 10.1016/s0304-3940(00)01054-5. [DOI] [PubMed] [Google Scholar]
- 5.Tomassini B, Testi R. Mitochondria as sensors of sphingolipids. Biochimie. 2002;84:123–129. doi: 10.1016/s0300-9084(02)01377-9. [DOI] [PubMed] [Google Scholar]
- 6.Jaffrézou JP, Levade T, Bettaïeb A, Andrieu N, Bezombes C, Maestre N, Vermeersch S, Rousse A, Laurent G. Daunorubicin-induced apoptosis: triggering of ceramide generation through sphingomyelin hydrolysis. EMBO J. 1996;15:2417–2424. [PMC free article] [PubMed] [Google Scholar]
- 7.Levade T, Malagarie-Cazenave S, Gouazé V, Ségui B, Tardy C, Betito S, Andrieu-Abadie N, Cuvillier O. Ceramide in apoptosis: a revisited role. Neurochem Res. 2002;27:601–607. doi: 10.1023/a:1020215815013. [DOI] [PubMed] [Google Scholar]
- 8.Haimovitz-Friedman A, Kolesnick RN, Fuks Z. Ceramide signaling in apoptosis. Br Med Bull. 1997;53:539–553. doi: 10.1093/oxfordjournals.bmb.a011629. [DOI] [PubMed] [Google Scholar]
- 9.Hannun YA. Functions of ceramide in coordinating cellular responses to stress. Science. 1996;274:1855–1859. doi: 10.1126/science.274.5294.1855. [DOI] [PubMed] [Google Scholar]
- 10.Raisova M, Goltz G, Bektas M, Bielawska A, Riebeling C, Hossini AM, Eberle J, Hannun YA, Orfanos CE, Geilen CC. Bcl-2 overexpression prevents apoptosis induced by ceramidase inhibitors in malignant melanoma and HaCaT keratinocytes. FEBS Lett. 2002;516:47–52. doi: 10.1016/s0014-5793(02)02472-9. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J, Alter N, Reed JC, Borner C, Obeid LM, Hannun YA. Bcl-2 interrupts the ceramide-mediated pathway of cell death. Proc Natl Acad Sci USA. 1996;93:5325–5328. doi: 10.1073/pnas.93.11.5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sawai H, Okazaki T, Takeda Y, Tashima M, Sawada H, Okuma M, Kishi S, Umehara H, Domae N. Ceramide-induced translocation of protein kinase C-delta and -epsilon to the cytosol. Implications in apoptosis. J Biol Chem. 1997;272:2452–2458. doi: 10.1074/jbc.272.4.2452. [DOI] [PubMed] [Google Scholar]
- 13.Haimovitz-Friedman A, Kan CC, Ehleiter D, Persaud RS, McLoughlin M, Fuks Z, Kolesnick RN. Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J Exp Med. 1994;180:525–535. doi: 10.1084/jem.180.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Lv X, Shi J, Hu X. Ceramide induces apoptosis via a peroxisome proliferator-activated receptor gamma-dependent pathway. Apoptosis. 2006;11:2043–2052. doi: 10.1007/s10495-006-0191-9. [DOI] [PubMed] [Google Scholar]
- 15.Xiao D, Singh SV. Phenethyl isothiocyanate-induced apoptosis in p53-deficient PC-3 human prostate cancer cell line is mediated by extracellular signal-regulated kinases. Cancer Res. 2002;62:3615–3619. [PubMed] [Google Scholar]
- 16.Fang JY, Lu YY. Effects of histone acetylation and DNA methylation on p21( WAF1) regulation. World J Gastroenterol. 2002;8:400–405. doi: 10.3748/wjg.v8.i3.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chellappan SP, Giordano A, Fisher PB. Role of cyclin-dependent kinases and their inhibitors in cellular differentiation and development. Curr Top Microbiol Immunol. 1998;227:57–103. doi: 10.1007/978-3-642-71941-7_4. [DOI] [PubMed] [Google Scholar]
- 18.Kim JS, Lee S, Lee T, Lee YW, Trepel JB. Transcriptional activation of p21(WAF1/CIP1) by apicidin, a novel histone deacetylase inhibitor. Biochem Biophys Res Commun. 2001;281:866–871. doi: 10.1006/bbrc.2001.4434. [DOI] [PubMed] [Google Scholar]
- 19.Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, Spiegelman BM, Mortensen RM. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell. 1999;4:611–617. doi: 10.1016/s1097-2765(00)80211-7. [DOI] [PubMed] [Google Scholar]
- 20.Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, Evans RM. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell. 1999;4:585–595. doi: 10.1016/s1097-2765(00)80209-9. [DOI] [PubMed] [Google Scholar]
- 21.Chinetti G, Griglio S, Antonucci M, Torra IP, Delerive P, Majd Z, Fruchart JC, Chapman J, Najib J, Staels B. Activation of proliferator-activated receptors alpha and gamma induces apoptosis of human monocyte-derived macrophages. J Biol Chem. 1998;273:25573–25580. doi: 10.1074/jbc.273.40.25573. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi N, Okumura T, Motomura W, Fujimoto Y, Kawabata I, Kohgo Y. Activation of PPARgamma inhibits cell growth and induces apoptosis in human gastric cancer cells. FEBS Lett. 1999;455:135–139. doi: 10.1016/s0014-5793(99)00871-6. [DOI] [PubMed] [Google Scholar]
- 23.Chen J, Larochelle S, Li X, Suter B. Xpd/Ercc2 regulates CAK activity and mitotic progression. Nature. 2003;424:228–232. doi: 10.1038/nature01746. [DOI] [PubMed] [Google Scholar]
- 24.Bushnell DA, Westover KD, Davis RE, Kornberg RD. Structural basis of transcription: an RNA polymerase II-TFIIB cocrystal at 4.5 Angstroms. Science. 2004;303:983–988. doi: 10.1126/science.1090838. [DOI] [PubMed] [Google Scholar]
- 25.Oelgeschläger T. Regulation of RNA polymerase II activity by CTD phosphorylation and cell cycle control. J Cell Physiol. 2002;190:160–169. doi: 10.1002/jcp.10058. [DOI] [PubMed] [Google Scholar]
- 26.Westermarck J, Li SP, Kallunki T, Han J, Kähäri VM. p38 mitogen-activated protein kinase-dependent activation of protein phosphatases 1 and 2A inhibits MEK1 and MEK2 activity and collagenase 1 (MMP-1) gene expression. Mol Cell Biol. 2001;21:2373–2383. doi: 10.1128/MCB.21.7.2373-2383.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garg TK, Chang JY. Oxidative stress causes ERK phosphorylation and cell death in cultured retinal pigment epithelium: prevention of cell death by AG126 and 15-deoxy-delta 12, 14-PGJ2. BMC Ophthalmol. 2003;3:5. doi: 10.1186/1471-2415-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi YJ, Lim SY, Woo JH, Kim YH, Kwon YK, Suh SI, Lee SH, Choi WY, Kim JG, Lee IS, et al. Sodium orthovanadate potentiates EGCG-induced apoptosis that is dependent on the ERK pathway. Biochem Biophys Res Commun. 2003;305:176–185. doi: 10.1016/s0006-291x(03)00719-8. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, Martindale JL, Holbrook NJ. Requirement for ERK activation in cisplatin-induced apoptosis. J Biol Chem. 2000;275:39435–39443. doi: 10.1074/jbc.M004583200. [DOI] [PubMed] [Google Scholar]
- 30.Calabrese C, Frank A, Maclean K, Gilbertson R. Medulloblastoma sensitivity to 17-allylamino-17-demethoxygeldanamycin requires MEK/ERKM. J Biol Chem. 2003;278:24951–24959. doi: 10.1074/jbc.M211600200. [DOI] [PubMed] [Google Scholar]
- 31.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 32.Smalley KS. A pivotal role for ERK in the oncogenic behaviour of malignant melanoma? Int J Cancer. 2003;104:527–532. doi: 10.1002/ijc.10978. [DOI] [PubMed] [Google Scholar]
- 33.Zheng B, Fiumara P, Li YV, Georgakis G, Snell V, Younes M, Vauthey JN, Carbone A, Younes A. MEK/ERK pathway is aberrantly active in Hodgkin disease: a signaling pathway shared by CD30, CD40, and RANK that regulates cell proliferation and survival. Blood. 2003;102:1019–1027. doi: 10.1182/blood-2002-11-3507. [DOI] [PubMed] [Google Scholar]