

Abstract
AIM: To describe the diagnostic criteria for acute liver failure due to Wilson disease (WD), which is an uncommon cause of acute liver failure (ALF).
METHODS: We compared findings of patients presenting with ALF due to WD to those with ALF of other etiologies.
RESULTS: Previously described criteria, such as low alkaline phosphatase activity, ratio of low alkaline phosphatase to total bilirubin or ratio of high aspartate aminotransferase (AST) to alanine aminotransferase (ALT), failed to identify patients with ALF due to WD. There were significant differences in low ALT and AST activities (53 ± 43 vs 1982 ± 938, P < 0.0001 and 87 ± 44 vs 2756 ± 2941, P = 0.037, respectively), low choline esterase activity (1.79 ± 1.2 vs 4.30 ± 1.2, P = 0.009), high urine copper concentrations (93.4 ± 144.0 vs 3.5 ± 1.8, P = 0.001) and low hemoglobin (7.0 ± 2.2 vs 12.6 ± 1.8, P < 0.0001) in patients with ALF caused by WD as compared with other etiologies. Interestingly, 4 of 7 patients with ALF due to WD survived without liver transplantation.
CONCLUSION: In ALF, these criteria can help establish a diagnosis of WD. Where applicable, slit-lamp examination for presence of Kayser-Fleischer rings and liver biopsy for determination of hepatic copper concentration still remain important for the diagnosis of ALF due to WD. The need for liver transplantation should be evaluated carefully as the prognosis is not necessarily fatal.
Keywords: Acute liver failure, Copper metabolism, Fulminant hepatic failure, Wilson disease, Liver transplantation
INTRODUCTION
Wilson disease (WD) is an autosomal recessively inherited disorder of copper metabolism with a prevalence of approximately 1:30 000. Mutations of the copper transporting ATPase ATP7B can lead to decreased biliary copper excretion and accumulation of copper predominantly in the liver and in extrahepatic organs such as the brain and the cornea[1]. The disease typically begins with an asymptomatic period with subclinical hepatitis and progresses to liver cirrhosis and neuropsychiatric symptoms[2,3]. A small proportion of patients, however, present with fulminant Wilson disease (FWD) without clinical signs of pre-existing liver disease fulfilling the criteria of acute liver failure (ALF).
Typically, diagnosis of WD is based on the finding of a low serum-ceruloplasmin level (< 20 mg/dL), presence of Kayser-Fleischer rings, increased hepatic copper content (> 250 μg/g dry weight) and increased urinary copper excretion [> 100 μg/24 h (= 1.6 μmol/24 h)][4]. However, there are limitations to all these criteria, especially in patients with acute liver failure. A ratio of alkaline phosphatase (AP) to bilirubin < 2 has been suggested to be diagnostic for FWD[5].
Clinically FWD patients appear as cases of acute liver failure. In this patient group, WD is a possible etiology for ALF amongst others like toxic liver damage, acute hepatitis or ischemic liver damage[6]. The mortality of patients is high, and orthotopic liver transplantation (OLT) often remains the only therapeutic option, which has been shown to be an effective treatment for patients with FWD[7].
The purpose of this study is to outline differences in clinical and biochemical findings between FWD and ALF of other etiologies that might identify cases of FWD.
MATERIALS AND METHODS
Acute liver failure was defined as new onset liver disease characterized by coagulopathy [international normalized ratio (INR) ≥ 1.5] and any grade of hepatic encephalopathy (HE) in absence of any pre-diagnosed liver disease[6,8]. For patients with FWD the prognostic score developed by Nazer et al[9] was calculated.
Seven patients with FWD were analyzed. Diagnosis of WD was confirmed by the presence of Kayser-Fleischer rings, hepatic copper content (> 250 μg/g dry weight) and/or genomic mutation analysis.
A control group of 8 patients with ALF due to other reasons rather than WD was analyzed. Initial diagnostic procedures in all patients included serological analysis for hepatitis A-E, cytomegalovirus (CMV), herpes simplex virus (HSV), varizella zoster virus (VZV), Ebstein-Barr virus (EBV), leptospirosis, screening for autoantibodies and ferritin[6]. Hepatic blood flow was analyzed by Doppler ultrasound and/or computed tomography. Liver biopsy was performed percutaneously. Hepatic copper content was measured in liver biopsies or explanted livers.
Mutational analysis
DNA analysis for presence of ATP7B mutations was performed by Ferenci et al[10] as part of an ongoing protocol to study genotype-phenotype correlations in patients with established diagnosis of WD. Mutation analysis was done by a stepwise procedure. First, a rapid, semi-nested PCR technique was used to detect the H1069Q mutation as described previously[11]. Patients not being homozygous for this mutation were further analysed exon by exon by denaturating HPLC (WAVE mutation detection system model 4000, Transgenomics, Crewe, UK). So far, the analysis has been completed for exons 3 to 20. Exons were amplified with published primers[12]. Samples with potential mutations identified by this approach were sequenced by the ABI Prism 310 Genetic Analyser (Perkin Elmer; Norwalk, CT, USA).
Statistical analysis
Statistical analyses were performed with SPSS for Windows, release 10.05 (SPSS, Chicago, IL). Comparisons of quantitative variables were performed by the unpaired t test. For urinary copper excretion, log values were used. Data are expresssed as means ± SD. A P value < 0.05 was considered statistically significant.
RESULTS
Patients’ characteristics
We have analysed the clinical and biochemical findings of 15 patients meeting diagnostic criteria for ALF. Of these 15 patients, seven had presented with ALF due to WD and eight with ALF of other etiologies.
Patients with FWD were diagnosed on the basis of a liver copper content of > 250 μg/g dry weight, presence of Kayser-Fleischer rings at split-lamp examination and/or presence of ATP7B-mutations (Table 1). Potential competing causes of ALF were excluded. Three patients received high-urgent OLT, and 4 patients survived on supportive care and initiation of medical therapy with D-penicillamine (n = 3) or trientine (n = 1). As controls, 8 patients with ALF of other causes were analyzed. One patient died from acetaminophen intoxication, all other patients survived on supportive care or on lamivudine therapy[13] in the cases of fulminant hepatitis B (Table 1).
Table 1.
Baseline characteristics at initial presentation
| Sex | Age (yr) | ALT (U/L) | AP (U/L) | Bilirubin (mg/dL) | INR | Creatinine (mg/dL) | U-Copper (μmol/24 h) | Grade of HE | Bx-Copper (μg/g) | KFR | Mutation | NS | Outcome | |
| FWD | F | 46 | 19 | 118 | 8.5 | 2.7 | 2.20 | 411.00 | 2 | ND | Yes | ND | 4 | OLT, alive |
| F | 16 | 45 | 122 | 10.6 | 2.0 | 0.60 | 104.00 | 1 | 1276 | No | ND | 5 | Alive | |
| F | 18 | 31 | 56 | 48.0 | 2.4 | 4.30 | 38.00 | 2 | 1500 | ND | H1069Q/H1069Q | 8 | OLT, alive | |
| F | 17 | 143 | n.d. | 15.0 | 1.6 | 1.00 | 60.00 | 2 | ND | No | H1069Q/IVS4-1:g > a | 7 | Alive | |
| F | 16 | 44 | 262 | 11.0 | 1.7 | 1.70 | 3.89 | 1 | 400 | Yes | H1069Q/2299Ins | 4 | Alive | |
| F | 18 | 64 | 193 | 52.0 | 2.6 | 0.77 | 25.00 | 1 | 1581 | ND | T977M/T977M | 7 | OLT, alive | |
| F | 10 | 22 | 19 | 13.0 | 1.9 | 0.60 | 11.93 | 1 | ND | Yes | ND | 5 | Alive | |
| ALF etiology | ||||||||||||||
| ALF | F | 52 | 3188 | 181 | 15.5 | 1.5 | 0.95 | 3.12 | 2 | Fulminant hepatitis B | Alive | |||
| F | 85 | 386 | 99 | 3.8 | 2.6 | 1.18 | 5.56 | 3 | Toxic (tuberculostatics) | Alive | ||||
| F | 35 | 1442 | 111 | 23.2 | 1.5 | 0.50 | 2.23 | 2 | Toxic (venlafaxin) | Alive | ||||
| F | 34 | 1782 | 266 | 1.5 | 1.7 | 0.55 | 1.72 | 1 | Unknown | Alive | ||||
| F | 59 | 2301 | 59 | 4.0 | 1.8 | 1.25 | 0.80 | 4 | Toxic (acetaminophen) | Death | ||||
| F | 43 | 1285 | 142 | 9.4 | 1.8 | 0.58 | 4.22 | 2 | Toxic (opipramol) | Alive | ||||
| M | 18 | 2953 | 179 | 24.5 | 2.4 | 0.64 | 4.39 | 2 | Fulminant hepatitis B | Alive | ||||
| M | 32 | 2522 | 79 | 38.0 | 3.2 | 0.89 | 5.80 | 1 | Unknown | OLT, alive |
FWD: acute liver failure secondary to WD; ALF: acute liver failure; F: female; M: male; ALT: alanine aminotransferase, normal range below 34 and 50 for female and male, respectively; AP: alkaline phosphatase, normal range below 126 and 130 for female and male, respectively; HE: hepatic encephalopathy; Bx-Copper: hepatic copper content; KFR: Kayser-Fleischer ring; NS: Nazer-Score; OLT: orthotopic liver transplantation; ND: Not determined.
Patients with ALF due to other causes rather than WD were significantly older than patients with FWD (44.8 ± 20.9 vs 20.1 ± 11.7 years of age, P < 0.05) (Tables 1 and 2). On admission, all patients had some degree of hepatic encephalopathy, with no significant difference between patients with FWD and patients with ALF duo to other causes (Tables 1 and 2).
Table 2.
Comparison of clinical findings between the groups
| FWD | ALF | P | |
| Age (yr) | 20.1 ± 11.7 | 44.8 ± 20.6 | 0.016 |
| MELD score | 27.6 ± 9.5 | 20.3 ± 7.0 | 0.108 |
| HE (grade 1-4) | 1.4 ± 0.5 | 2.13 ± 1.0 | 0.122 |
| Serum ALT (U/L) | 53 ± 43 | 1982 ± 938 | < 0.0001 |
| Serum AST (U/L) | 87 ± 44 | 2756 ± 2941 | 0.037 |
| AST-ALT ratio | 2.3 ± 1.5 | 1.5 ± 1.2 | 0.266 |
| Serum AP (U/L) | 128 ± 89 | 142 ± 66 | 0.749 |
| Serum LDH (U/L) | 535 ± 638 | 1567 ± 2707 | 0.382 |
| Serum bilirubin (mg/dL) | 23 ± 19 | 15 ± 13 | 0.372 |
| AP-bilirubin ratio | 9.3 ± 8.9 | 32.5 ± 59.0 | 0.363 |
| INR | 2.1 ± 0.4 | 2.1 ± 0.6 | 0.816 |
| Albumin (g/L) | 29.8 ± 4.7 | 32.0 ± 3.8 | 0.379 |
| CHE (kU/L) | 1.8 ± 1.2 | 4.3 ± 1.2 | 0.005 |
| Hemoglobin (g/L) | 7.0 ± 2.2 | 12.6 ± 1.8 | < 0.0001 |
| Serum copper (μmol/L) | 28.1 ± 29.4 | 17.8 ± 2.5 | 0.376 |
| Serum ceruloplasmin (g/L) | 0.12 ± 0.08 | 0.19 ± 0.03 | 0.07 |
| Urinary copper (μmol/24 h) | 93.4 ± 144.0 | 3.5 ± 1.8 | 0.001 |
FWD: acute liver failure secondary to WD; ALF: acute liver failure; HE: hepatic encephalopathy; ALT: alanine aminotransferase; AP: alkaline phosphatase; INR: international normalized ratio; CHE: choline esterase.
Parameters of copper metabolism
Compared with patients who had ALF due to other causes rather than WD, patients with FWD had a higher urinary copper (93.4 ± 144.0 μmol/24 h vs 3.5 ± 1.8 μmol/24 h, P < 0.01). Differences in serum ceruloplasmin and serum copper were not statistically significant between the two groups, although a trend towards lower serum ceruloplasmin levels was observed in FWD (Table 2).
Standard laboratory tests
Higher serum alanine aminotransferase (ALT) (53 ± 43 vs 1982 ± 938, P < 0.0001) and serum aspartate aminotransferase (AST) (87 ± 44 vs 2756 ± 2941, P < 0.05) were found in the control group than in the FWD group (Table 2). However, bilirubin, alkaline phosphatase (AP), albumin, INR, the AP to bilirubin ratio, and the AST to ALT ratio did not differ significantly between the two groups (Table 2).
Notably, serum cholinesterase (CHE) activity was significantly lower in patients with FWD than in patients with ALF due to other etiologies (1.8 ± 1.2 vs 4.3 ± 1.2, P < 0.01) (Table 2).
A low hemoglobin value, as the correlate of hemolysis, was present in all the patients with FWD, but only in 4 of the seven patients with ALF due to other causes. The mean hemoglobin value was significantly lower in the FWD group than in the Non-WD-ALF group (7.0 ± 2.2 vs 12.6 ± 1.8, P < 0.0001). However, the serum LDH showed no significant difference between the two groups.
Outcome
Four of seven patients with FWD were treated with chelating agents and survived without liver transplantation. Three patients with FWD underwent successful liver transplantation. Two of the three FWD patients with liver transplantation had a prognostic index ≥ 7 based on the score developed by Nazer et al[9]. All but one FWD patient surviving without liver transplantation had a score ≤ 7 (Table 1).
DISCUSSION
The lack of sensitive and specific criteria for the rapid diagnosis of FWD has made this disease quite difficult to identify. This complicates the potential decisions concerning liver transplantation and the initiation of medical therapy with chelating agents for WD.
In our study, we addressed clinical and biochemical differences between patients with FWD and those with ALF of other etiologies rather than WD to characterize variables for a rapid diagnosis.
Biochemical diagnosis of FWD usually is suggested by the association of a mild increase of serum transaminase concentrations compared with the degree of hepatic insufficiency by high serum bilirubin concentration, high serum and urinary copper concentration, and the presence of Coombs-negative hemolytic anemia[14].
Although hemolytic anemia associated with ALF is strongly suggestive of FWD, it is not pathognomonic. In our series, a low hemoglobin value, as the correlate of hemolysis, was present in all the patients with FWD and in four of the seven patients with ALF due to other causes. This is in concordance with Hillenbrand et al[15], who reported that anemia is present in 50% of adult patients with ALF.
Multiple indices have been used for FWD diagnosis. Berman et al[5] identified a ratio of AP to total bilirubin < 2.0 and an AST to ALT ratio > 4.0 for diagnosis of FWD. In our series, these two ratios showed no significant differences between the two groups. This is in concordance with other reports that doubt the validity of the two scores for diagnosis of FWD[16,17].
In our series, INR, grade of HE, and MELD score failed to discriminate FWD from ALF due to other causes. And, the previously reported low serum alkaline phosphatase activity in FWD[18] could not be observed in our FWD patients. However, FWD was characterized by statistically significant lower hemoglobin values, serum transaminases, and serum CHE. Low transaminases and CHE can be explained by the presence of an underlying chronic subclinical liver disease in FWD patients who may already have evidence of cirrhosis and impaired organ function. Typically, the presence of WD is not recognized before rapid deterioration and onset of FWD. Therefore, FWD is considered one of the special circumstances where ALF can still be diagnosed in patients with pre-existing liver disease[6,19].
An elevation in urinary copper excretion has been reported in other forms of ALF and is explained by the release of copper from necrotic hepatocytes. The urinary copper excretion was significantly elevated in patients with FWD. Other parameters of copper metabolism, serum copper and serum ceruloplasmin, showed no significant difference between the two groups. However, as shown in Figure 1, the overlap in serum ceruloplasmin concentration between FWD group and control group only concerned two patients. The absence of statistically significant difference between the two groups might be related to the small size of the samples. Based on the results in this series, it seems that by using a threshold value of about 1.5 g/L, serum ceruloplasmin may remain a relatively good marker. None of the patients in the control group had serum ceruloplasmin lower than 1.5 g/L as compared with 5 of 7 in the FWD group.
Figure 1.
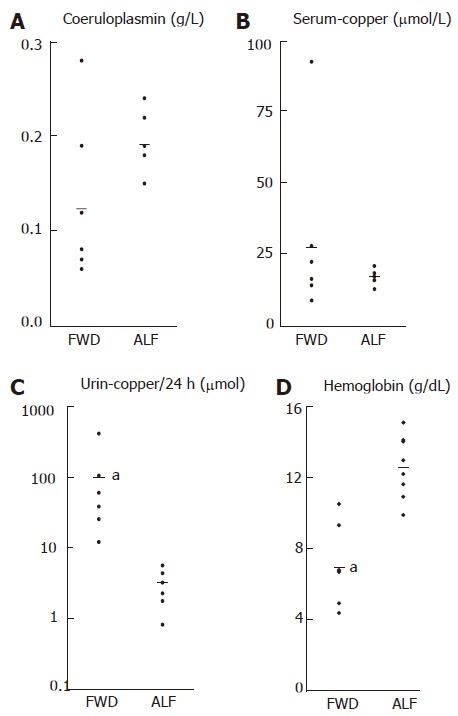
Comparison of copper metabolism parameters and hemoglobin. The linear scales in A, B, D and the logarithmic scale in C: Ceruloplasmin (g/L), serum-copper (μmol/L), urine-copper (μmol/24 h), hemoglobin (g/dL). FWD: acute liver failure secondary to Wilson disease; ALF: acute liver failure. aP ≤ 0.001).
Although copper metabolism parameters remain the most accurate means for WD diagnosis, it may be of limited value in the acute setting of FWD. Early FWD diagnosis could be achieved by identification of Kayser-Fleischer rings, a strikingly high urinary copper excretion, only mildly elevated transaminases and a low hemoglobin value.
Although it has been previously stated that FWD has a uniformly fatal outcome if liver transplantation is not performed timely[20,21], four out of our seven patients with FWD survived without liver transplantation. Nazer et al[9] have proposed a prognostic score for identification of patients with FWD who will not survive without liver transplantation (score ≥ 7). In our series only one FWD patient surviving without liver transplantation had a Nazer-score of 7, the other patients had lower scores.
In conclusion, the prognosis of FWD is not necessarily fatal. Patients with a Nazer-score of six or less can survive FWD without liver transplantation. Early consideration of the diagnosis of WD in patients with ALF is of paramount importance. Prompt initiation of appropriate treatment (chelation therapy vs liver transplantation) may be able to lower the high mortality from FWD.
ACKNOWLEDGMENTS
We are grateful to all physicians and nurses for taking care of the patients, and to Professor Peter Ferenci for performing the ATP7B H1069Q mutation analysis.
Footnotes
S- Editor Liu Y L- Editor Ma JY E- Editor Lu W
References
- 1.Scheinberg IH. Wilson's disease. J Rheumatol Suppl. 1981;7:90–93. [PubMed] [Google Scholar]
- 2.Gitlin JD. Wilson disease. Gastroenterology. 2003;125:1868–1877. doi: 10.1053/j.gastro.2003.05.010. [DOI] [PubMed] [Google Scholar]
- 3.Stremmel W, Meyerrose KW, Niederau C, Hefter H, Kreuzpaintner G, Strohmeyer G. Wilson disease: clinical presentation, treatment, and survival. Ann Intern Med. 1991;115:720–726. doi: 10.7326/0003-4819-115-9-720. [DOI] [PubMed] [Google Scholar]
- 4.Gitlin N. Wilson's disease: the scourge of copper. J Hepatol. 1998;28:734–739. doi: 10.1016/s0168-8278(98)80302-4. [DOI] [PubMed] [Google Scholar]
- 5.Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology. 1991;100:1129–1134. doi: 10.1016/0016-5085(91)90294-u. [DOI] [PubMed] [Google Scholar]
- 6.Polson J, Lee WM. AASLD position paper: the management of acute liver failure. Hepatology. 2005;41:1179–1197. doi: 10.1002/hep.20703. [DOI] [PubMed] [Google Scholar]
- 7.Bellary S, Hassanein T, Van Thiel DH. Liver transplantation for Wilson's disease. J Hepatol. 1995;23:373–381. doi: 10.1016/0168-8278(95)80194-4. [DOI] [PubMed] [Google Scholar]
- 8.Trey C, Davidson CS. The management of fulminant hepatic failure. Prog Liver Dis. 1970;3:282–298. [PubMed] [Google Scholar]
- 9.Nazer H, Ede RJ, Mowat AP, Williams R. Wilson's disease: clinical presentation and use of prognostic index. Gut. 1986;27:1377–1381. doi: 10.1136/gut.27.11.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, Schilsky M, Cox D, Berr F. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23:139–142. doi: 10.1034/j.1600-0676.2003.00824.x. [DOI] [PubMed] [Google Scholar]
- 11.Maier-Dobersberger T, Ferenci P, Polli C, Balać P, Dienes HP, Kaserer K, Datz C, Vogel W, Gangl A. Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Ann Intern Med. 1997;127:21–26. doi: 10.7326/0003-4819-127-1-199707010-00004. [DOI] [PubMed] [Google Scholar]
- 12.Thomas GR, Forbes JR, Roberts EA, Walshe JM, Cox DW. The Wilson disease gene: spectrum of mutations and their consequences. Nat Genet. 1995;9:210–217. doi: 10.1038/ng0295-210. [DOI] [PubMed] [Google Scholar]
- 13.Tillmann HL, Hadem J, Leifeld L, Zachou K, Canbay A, Eisenbach C, Graziadei I, Encke J, Schmidt H, Vogel W, et al. Safety and efficacy of lamivudine in patients with severe acute or fulminant hepatitis B, a multicenter experience. J Viral Hepat. 2006;13:256–263. doi: 10.1111/j.1365-2893.2005.00695.x. [DOI] [PubMed] [Google Scholar]
- 14.McCullough AJ, Fleming CR, Thistle JL, Baldus WP, Ludwig J, McCall JT, Dickson ER. Diagnosis of Wilson's disease presenting as fulminant hepatic failure. Gastroenterology. 1983;84:161–167. [PubMed] [Google Scholar]
- 15.Hillenbrand P, Parbhoo SP, Jedrychowski A, Sherlock S. Significance of intravascular coagulation and fibrinolysis in acute hepatic failure. Scand J Gastroenterol Suppl. 1973;19:133–134. [PubMed] [Google Scholar]
- 16.Sallie R, Katsiyiannakis L, Baldwin D, Davies S, O'Grady J, Mowat A, Mieli-Vergani G, Williams R. Failure of simple biochemical indexes to reliably differentiate fulminant Wilson's disease from other causes of fulminant liver failure. Hepatology. 1992;16:1206–1211. [PubMed] [Google Scholar]
- 17.Emre S, Atillasoy EO, Ozdemir S, Schilsky M, Rathna Varma CV, Thung SN, Sternlieb I, Guy SR, Sheiner PA, Schwartz ME, et al. Orthotopic liver transplantation for Wilson's disease: a single-center experience. Transplantation. 2001;72:1232–1236. doi: 10.1097/00007890-200110150-00008. [DOI] [PubMed] [Google Scholar]
- 18.Shaver WA, Bhatt H, Combes B. Low serum alkaline phosphatase activity in Wilson's disease. Hepatology. 1986;6:859–863. doi: 10.1002/hep.1840060509. [DOI] [PubMed] [Google Scholar]
- 19.Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology. 2003;37:1475–1492. doi: 10.1053/jhep.2003.50252. [DOI] [PubMed] [Google Scholar]
- 20.Sternlieb I. Wilson's disease: indications for liver transplants. Hepatology. 1984;4:15S–17S. doi: 10.1002/hep.1840040706. [DOI] [PubMed] [Google Scholar]
- 21.Stampfl DA, Muñoz SJ, Moritz MJ, Rubin R, Armenti VT, Jarrell BE, Maddrey WC. Heterotopic liver transplantation for fulminant Wilson's disease. Gastroenterology. 1990;99:1834–1836. doi: 10.1016/0016-5085(90)90497-o. [DOI] [PubMed] [Google Scholar]