

Abstract
Congenital hepatic fibrosis (CHF) is an autosomal recessive disorder that belongs to the family of fibropolycystic liver diseases. This family includes a spectrum of disorders which are usually found in combination with each other and are usually inherited. Clinically fibropolycystic diseases have three effects being present in different proportions, those of a space occupying lesion, of portal hypertension and of cholangitis. In most patients, the first manifestations of CHF are signs and symptoms related to portal hypertension such as splenomegaly and varices. Portal hypertension in these patients has been attributed to the hypoplasia or compression of the portal vein radicles in the fibrous bands. Cavernous transformation of the portal vein (CTPV) is a relatively rare condition resulting from extrahepatic portal vein obstruction with recanalization or collateral vein formation to bypass the obstruction. It has been found that patients with CHF having an accompanying CTPV have relatively large splenomegaly and suffers more frequent episodes of bleeding from esophageal varices.We believe that CTPV is a congenital component of CHF and also one of the important causative factors of portal hypertension in these patients.
Keywords: Congenital hepatic fibrosis, Cavernous transformation of portal vein, Portal hypertension
INTRODUCTION
Liver diseases characterized by some degree of dilatation of segments of intrahepatic bile ducts with associated fibrosis are named as fibropolycystic liver diseases[1]. It is now clear with the help of improved methods of imaging that these diseases do not exist as single entities, but as members of a family. The members are found in various combinations[2]. They consist of congenital hepatic fibrosis, Caroli’s disease, autosomal dominant polycystic kidney disease, autosomal recessive polycystic kidney disease, Von Meyenburg complex and mesenchymal hamartoma[3]. Since their original recognition, many reports on the pathogenesis, genetics and combinations with other morphological alterations of these groups of diseases have been published[4]. Congenital hepatic fibrosis (CHF) belongs to this group, is an autosomal recessive inherited condition and is characterized by a destructive type of cholangiopathy of variable speed and duration associated with scarring fibrosis[5].
CLINICAL CHARACTERISTICS OF CHF WITH AN EMPHASIS ON PORTAL HYPERTENSION
Typically, patients with CHF do not have cirrhosis and maintain normal hepatic lobular architecture with normal hepatic function. Onset of symptoms and signs is highly variable and ranges from early childhood to the 5th or 6th decades of life, although this disorder is diagnosed in most patients during adolescence or young adulthood[6].
In most patients, the first manifestations of the disease are signs or symptoms related to portal hypertension especially spelenomegaly and varices- often with spontaneous gastrointestinal bleeding[7]. Portal hypertension, a very common clinical feature of CHF, has been attributed to the compression of portal vein radicles in the fibrous bands and to an anomaly in the branching pattern of the portal vein, giving rise to hypoplastic and involutive branches[8].
EMBRYOLOGICAL EXPLANATION OF THE LINK BETWEEN PORTAL VEIN AND CHF
In view of the embryology, it is not surprising that anomaly of the vessels and of the intrahepatic bile ducts are usually combined because a close relationship exists between vascular and bile ductal development[9]. The liver develops in the fourth week of gestation on the hepatic diverticulum. Intrahepatic ducts develop from primitive hepatocytes around branches of the portal vein. The layer of cells surrounding the portal vein transforms in to a double-walled cylinder with a slit like lumen and is termed the ductal plate. A progressive progress termed remodelling starts during the 12th wk of gestation. It is the lack of remodelling of the ductal plate that results in persistence of an excess of embryonic duct structures. This abnormality has been termed the ductal plate malformation (DPM) and consists of persistence of the ductal plate with an increase in ducts elements and an increase in portal fibroid tissue[1,9] DPM is frequently associated with abnormalities in the ramification pattern of the portal vein, referred to as a “pollard willow” pattern, resulting in too many, too small, and too closely spaced branches. A transverse section through such a pollard willow area will appear as an enlarged portal tract (in fact, a fusion of several portal tracts) containing several ductal plates (or remnants of incompletely remodelled ductal plates) surrounding several hypoplastic or even obliterated portal vein branches. The ductal plates appear around the mesenchyme surrounding the developing portal vein branches, whereas a similar phenomenon does not occur around the draining hepatic veins. The portal vein clearly has a dominant role in determining the three-dimensional configuration of the biliary tree, in as much as it has a critical role in the development of the liver as a whole[1]. Also Terada et al conducted a detailed study of the development of the human prebiliary capillary plexus and observed that the development and maturation of the prebiliary capillary plexus progress parallel to that of the intrahepatic bile ducts. They hypothesized that the development of both these structures is regulated by common substances on growth factors, which have yet to be determined[3].
CAVERNOUS TRANSFORMATION OF THE PORTAL VEIN AND CHF
Cavernous transformation of portal vein (CTPV) in adults is an uncommon finding, whose cause cannot usually be identified. Some researches consider this condition to be congenital, while others accept that it is a consequence of neonatal umbilical vein sepsis or a complication of an inflammatory neoplastic process occurring later in life[10]. CTPV is frequently associated with congenital anomalies (Figure 1). Among these abnormalities the most frequent are atrial septal defects or malformations of the biliary tract or of the inferior vena cava. Much less common is the association of prehepatic portal hypertension with intestinal telangiectasia, cutaneous hemangioma, valvular aortic stenosis, left renal agenesis, oesophageal atresia or pyloric stenosis. In one study Bayraktar et al[11]. have found that almost 50% of the cases of congenital hepatic fibrosis had CTPV. In this study also; it was found that congenital hepatic fibrosis patients having CTPV had relatively larger splenomegaly than those who had no CTPV and they also suffered more frequent bleeding episodes from esophageal varices.
Figure 1.
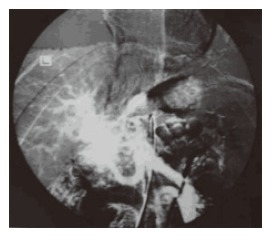
Arterial portography shows the cavernous transformation in portal vein.
CONCLUSION
In conclusion, CHF is an uncommon but important cause of portal hypertension[12]. As a close relationship exists between the development of portal vein and bile ducts; we think that this association between CHF and CTPV is not just a coincidence but rather one of the congenital components of this clinical condition. For this reason we believe that when CTPV is found in a patient with CHF, serious complications of portal hypertension such as bleeding should be expected.
Footnotes
S- Editor Wang J L- Editor Zhu LH E- Editor Che YB
References
- 1.Desmet VJ. Ludwig symposium on biliary disorders-part I. Pathogenesis of ductal plate abnormalities. Mayo Clin Proc. 1998;73:80–89. doi: 10.4065/73.1.80. [DOI] [PubMed] [Google Scholar]
- 2.Sherlock S, Dooley J. Diseases of the liver and billiary system. 11th ed. Milano: Blackwell Sci Pub; 2002. p. 583. [Google Scholar]
- 3.Awasthi A, Das A, Srinivasan R, Joshi K. Morphological and immunohistochemical analysis of ductal plate malformation: correlation with fetal liver. Histopathology. 2004;45:260–267. doi: 10.1111/j.1365-2559.2004.01945.x. [DOI] [PubMed] [Google Scholar]
- 4.Teufel J, Farack UM. Hepatobiliary fibropolycystic diseases. Two cases of Caroli's disease. Scand J Gastroenterol Suppl. 1987;139:76–80. doi: 10.3109/00365528709089778. [DOI] [PubMed] [Google Scholar]
- 5.Giouleme O, Nikolaidis N, Tziomalos K, Patsiaoura K, Vassiliadis T, Grammatikos N, Papanikolaou V, Eugenidis N. Ductal plate malformation and congenital hepatic fibrosis Clinical and histological findings in four patients. Hepatol Res. 2006;35:147–150. doi: 10.1016/j.hepres.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 6.Zeitoun D, Brancatelli G, Colombat M, Federle MP, Valla D, Wu T, Degott C, Vilgrain V. Congenital hepatic fibrosis: CT findings in 18 adults. Radiology. 2004;231:109–116. doi: 10.1148/radiol.2311030108. [DOI] [PubMed] [Google Scholar]
- 7.Mindikoglu AL, Regev A, O'Sullivan MJ, Schiff ER. Multiple normal deliveries in a woman with severe portal hypertension due to congenital hepatic fibrosis: the importance of preserved hepatocellular function. Am J Gastroenterol. 2005;100:2359–2361. doi: 10.1111/j.1572-0241.2005.00255.x. [DOI] [PubMed] [Google Scholar]
- 8.El-Youssef M, Mu Y, Huang L, Stellmach V, Crawford SE. Increased expression of transforming growth factor-beta1 and thrombospondin-1 in congenital hepatic fibrosis: possible role of the hepatic stellate cell. J Pediatr Gastroenterol Nutr. 1999;28:386–392. doi: 10.1097/00005176-199904000-00008. [DOI] [PubMed] [Google Scholar]
- 9.Desmet VJ. Pathogenesis of ductal plate malformation. J Gastroenterol Hepatol. 2004;19:S356–S360. [Google Scholar]
- 10.Bayraktar Y, Tuncer ZS, Kabukçu A, Uzunalimoğlu B, Ayhan A. Pregnancy complicated by congenital hepatic fibrosis with cavernous transformation of the portal vein: a case report. Am J Obstet Gynecol. 1997;177:459–461. doi: 10.1016/s0002-9378(97)70216-3. [DOI] [PubMed] [Google Scholar]
- 11.Bayraktar Y, Balkanci F, Kayhan B, Uzunalimoglu B, Ozenc A, Ozdemir A, Dündar S, Arslan S, Sivri B, Telatar H. Congenital hepatic fibrosis associated with cavernous transformation of the portal vein. Hepatogastroenterology. 1997;44:1588–1594. [PubMed] [Google Scholar]
- 12.Poddar U, Thapa BR, Vashishta RK, Girish CS, Singh K. Congenital hepatic fibrosis in Indian children. J Gastroenterol Hepatol. 1999;14:1192–1196. doi: 10.1046/j.1440-1746.1999.02028.x. [DOI] [PubMed] [Google Scholar]