

Abstract
Caroli’s syndrome is characterized by multiple segmental cystic or saccular dilatations of intrahepatic bile ducts associated with congenital hepatic fibrosis. The clinical features of this syndrome reflect both the characteristics of congenital hepatic fibrosis such as portal hypertension and that of Caroli’s disease named as recurrent cholangitis and cholelithiasis. The diagnosis depends on both histology and imaging methods which can show the communication between the sacculi and the bile ducts. Treatment consists of symptomatic treatment of cholangitis attacks by antibiotics, some endoscopic, radiological and surgical drainage procedures and surgery. Liver transplantation seems the ultimate treatment for this disease. Prognosis is fairly good unless recurrent cholangitis and renal failure develops.
Keywords: Caroli's syndrome, Liver transplantation, Endoscopic retrograde cholangiopancreatography
INTRODUCTION
Cystic lesions of the liver and bile ducts are increasingly being diagnosed. It is now clear that fibropolycystic diseases do not exist as single entities but as members of a family. The members are found in various combinations and are usually inherited. They consist of polycystic liver disease, microhamartoma, congenital hepatic fibrosis, Caroli’s disease and choledochal cysts[1].
Caroli’s syndrome consists of Caroli’s disease and congenital hepatic fibrosis. The term congenital hepatic fibrosis refers to a unique congenital liver histology characterized by bland portal fibrosis, hyperproliferation of interlobular bile ducts within the portal areas with variable shapes and sizes of bile ducts, and preservation of normal lobular architecture[2]. Caroli’s disease is however characterized by multiple segmental cystic dilatations of the intrahepatic bile ducts[3]. Many authors believe that the two conditions are actually different stages of the same disease characterized by periportal fibrosis and ductal dilatation[4].
EPIDEMIOLOGY
The major published reports concerning bile duct cysts have come from Great Britain, France, Japan and the United States[5]. More than 200 cases of Caroli’s disease have been reported in the literature[3] and the incidence of Caroli’s syndrome is more than the pure form of Caroli’s disease[6].
PATHOGENESIS
Caroli’s syndrome is a developmental anomaly and there are many theories explaining its pathogenesis. The most acceptable theory is related to ductal plate malformation at different levels of the intrahepatic biliary tree. Intrahepatic bile ducts develop from bipotential liver progenitor cells in contact with the mesenchyme of the portal vein which form from the ductal plates. The ductal plates are then remodeled into mature tubular ducts[7]. This process is regulated by factors influencing epithelial proliferation and apoptosis, the surrounding mesenchymal tissue, as well as the portal veins and various adhesion molecules[8]. Just as the formation of ductal plates follows the branching growth of portal vein from the hilus of the liver to its periphery, so does the remodelling process of the ductal plates beginning from the larger ducts to the smaller ducts. So the hereditary factors causing Caroli's syndrome seem to exert their influence not only during the early embryologic period of large intrahepatic bile duct formation, but also later during the development of the more proximal intralobular ducts related with congenital hepatic fibrosis[9]. It appears to be inherited in an autosomal recessive manner because mutations in PKHD1, the gene linked to adult recessive polycystic kidney disease (ARPKD) have also been identified in patients with Caroli's syndrome.
CLINICAL FEATURES
Caroli's syndrome presents a clinical syndrome which is a combination of Caroli's disease (bouts of cholangitis, hepatolithiasis, and gallbladder stones) and those of congenital hepatic fibrosis (portal hypertension). Clinical progression and presentation of Caroli's syndrome is highly variable and symptoms may appear early or late during life. The main consequences of congenital hepatic fibrosis are portal hypertension and the development of oesophageal varices. Portal hypertension may result in hematemesis or melena. In the majority of patients, portal hypertension will not be present or will appear only later in the disease evolution[10]. The late appearance of these symptoms in patients with Caroli's disease suggests that congenital hepatic fibrosis in Caroli's disease is dynamic and progressive[11]. Bile stagnation and hepatolithiasis explain the recurrent cholangitis which dominates the clinical course and which is the principal cause of morbidity and mortality. Chronic abdominal pain, pancreatitis resulting from biliary stones and liver abscess are other disease complications[10].
DIAGNOSIS
The laboratory findings are non-specific. Transaminase levels may be slightly elevated. The complete blood count may reveal thrombocytopenia and leukopenia if portal hypertension and hypersplenism are present. Elevated white blood cell count or erythrocyte sedimentation rate may indicate cholangitis. BUN and creatinine values should be obtained to detect associated renal disease.
Demonstration of communication between sacculi and bile ducts is important in showing the Caroli's disease component of Caroli's syndrome which can be achieved by USG, CT, MRCP or ERCP. The sonographic appearance in Caroli's disease is intrahepatic cystic anechoic areas in which fibrovascular bundles, stones and linear bridging or septum may be present. The fibrovascular bundles are composed of portal vein and hepatic arteries, which can be demonstrated by Doppler ultrasonography. Gorka mentioned that Doppler sonographic monitoring of portal vein in the fibrovascular bundle was useful in following the progression of congenital hepatic fibrosis component of Caroli's disease[3]. MR imaging is a non-invasive technique which can confirm the diagnosis especially in the large or small cystic patterns. The use of gadolinium enhanced sequences may allow the visualization of the dot sign which seems to be very specific of the malformation of the ductal plate. MR imaging can also suggest accompanying abnormalities such as portal hypertension, cirrhosis and renal involvement[12]. The diagnosis is more difficult to establish in the case of fusiform dilatations of the biliary tracts and ERCP is the gold standard in this situation. ERCP shows communication between the sacculi and bile ducts and diverticulum-like sacculi of the intrahepatic biliary tree[3] (Figure 1). Intra or extra hepatic cysts can easily be detected by these methods; whereas congenital hepatic fibrosis is a histopathological diagnosis. Histopathological intrahepatic bile duct ectasia and proliferation are associated with severe periportal fibrosis and confirm the congenital hepatic fibrosis component of Caroli's syndrome (Figure 2).
Figure 1.
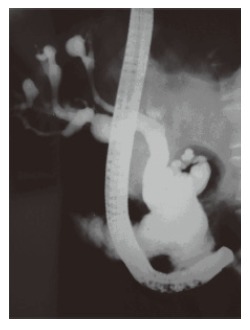
The ERCP image shows multiple cystic dilatations throughout the biliary tract.
Figure 2.
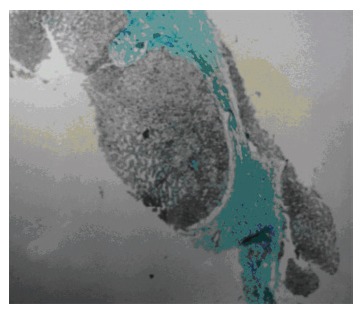
Severe peri-portal fibrotic bands upon histological exa-mination.
DIFFERENTIAL DIAGNOSIS
The appearance of ductal dilatation related to the Caroli’s disease component of Caroli's syndrome on CT/MRI can be confused with polycystic liver disease or obstructive bile duct dilatation. Whereas the cystic spaces in Caroli’s are irregular in shape and communicate with biliary tree, cysts in polycystic liver disease are rounder and smoother and they deform but do not communicate with bile ducts[13]. Portal hypertension is rare in polycystic liver disease[2].
In Caroli’s disease dilated bile ducts have a random bizarre pattern and there are focal areas of cystic ectasia. This differs from appearance in obstructive bile duct dilatation where dilatation is most marked centrally, tapers towards periphery in an organized pattern and lacks focal areas of cystic dilatation. Other diseases that should be considered in the differential diagnosis are primary sclerosing cholangitis, biliary papillomatosis and a choledochal cyst. The cholangiographic features of Caroli’s disease are well established as saccular or fusiform dilatation of the intrahepatic bile ducts. The association of Caroli's disease with extrahepatic bile duct dilatation is mentioned throughout the literature, as is the coexistence of Caroli's disease with choledochal cysts. The exact incidence of extrahepatic duct involvement in Caroli's disease is not known. A literature review of 46 cases found extrahepatic dilatation in 21% of patients. In the series of Levy including 17 patients (5 with Caroli's syndrome) this ratio is even higher with 53% of patients having extrahepatic duct dilatation. Recognition of this feature is important because Caroli's disease should be considered in the differential diagnosis for patients with intra- and extrahepatic biliary dilatation. Repeated bouts of cholangitis, stone formation and stone passage may explain extrahepatic duct dilatation in some patients with Caroli's disease or Caroli's syndrome. In the series of Levy, the presence of diffuse fusiform dilatation of the extrahepatic duct measuring 3 cm or less in diameter combined with the characteristic intrahepatic ductal findings has been suggested to be useful in differentiating patients with Caroli's disease from patients with a choledochal cyst and biliary dilatation[13].
ASSOCIATED CONDITIONS
As Caroli's syndrome is a member of the fibropolycystic disease family and its members are usually found in various combinations, presence of an association with any member should be sought.
Caroli's syndrome is associated with renal involvement in up to 60% of patients and implies a dilatation of the collecting renal tubules. Kidney lesions include renal tubular ectasia (medullary sponge kidney, cortical cyst) lesions of adult recessive polycystic kidney disease or rarely autosomal dominant polycystic kidney disease. The association between congenital hepatic fibrosis and ARKPD is caused by mutations in PKHD1 gene that encodes fibrocystin, a protein of primary cilia. Genetic defects in fibrocystin cause ciliary dysfunction, presently considered a major pathogenic event in cystogenesis[10]. In our study; between the time period 1971-2003 we followed-up 6 cases of Caroli’s syndrome in whom, 2 patients had cavernomatous transformation of portal vein and 2 had polycystic kidney disease as an associated condition. One of these patients died due to sepsis and two of them underwent liver transplantataion because of the development of secondary biliary cirrhosis[14].
Over the past years there has been an increasing recognition of the association of congenital hepatic fibrosis with not only fibropolycystic disease family related diseases but also with abnormalities in other organs such as pulmonary fibrosis, congenital heart disease, pulmonary hypertension with arteriovenous fistula, cavernomatous transformation of the portal vein and with Joubert’s syndrome[15-17].
COMPLICATIONS
Complications from Caroli’s syndrome are cholangitis, sepsis, choledocholithiasis, hepatic abscess, cholangio-carcinoma and portal hypertension[18]. After cholangitis occurs, a large number of patients die within 5-10 years. Caroli's syndrome may progress to cholangiocarcinoma[3].The occurence of hepatobiliary malignant transformation explained by chronic inflammation of the biliary tree, has been reported in 7%-14% of patients. Death is related to liver failure or complications of portal hypertension[18,19].
TREATMENT
Medical treatment of Caroli's syndrome using antibiotics may stabilize the acute cholangitis. Drainage procedures with ERCP or PTC are important and sphincterotomy can aid biliary drainage and stone removal or subsequent passage and may decrease bouts of cholangitis. Radiological, endoscopical and surgical biliary drainage procedures are however palliative treatments that may improve temporarily bile drainage but that become finally inefficient in relation to disease progression. Repeated palliative treatments should be avoided because of the prolongation of the symptomatic period and the increased risk of malignant transformation.
Imaging is essential in planning the surgical treatment which can consist of enterostomy, segmental or lobar hepatic resection, or liver transplantation[12] if the process is confined to one lobe, lobectomy completely relieves the symptoms. Internal surgical bypass procedures (choledochojejunostomy, Roux-en-Y hepaticojejunostomy) is helpful in diffuse forms of the disease. Liver transplantation represents the only curative treatment for symptomatic Caroli's disease or Caroli's syndrome. LT should be offered early in case of recurrent cholangitis and (suspicion of) early malignant transformation of the bilary tract. In case of associated renal disease, both renal and liver diseases should be documented in order to optimize the decision to perform sequential or combined hepatorenal transplantation[10].
As for portal hypertension, therapy for acute variceal haemorrhage includes stabilization with blood and blood products, vasopressin or somatostatin, early endoscopy sclerotherapy, and band ligation. Portocaval shunts are usually received as salvage treatment when sclerotherapy/banding ligation fails[20]. In cases when progression to biliary fibrosis might occur, transplantation has also to be considered. Early surgery with splenorenal or portocaval shunting may be required in recurrent bleeding episodes not available to sclerotherapy, oesophageal tamponade together with supportive medical treatment[15]. Transjugular intrahepatic portosystemic shunts are considered for patients not available to sclerotherapy. The inheritance of the disease seems to be autosomally recessive; therefore genetic counselling for the family may be important[21].
CONCLUSION
As a result, Caroli’s syndrome is an autosomal recessive disorder having diverse manifestations related to its association with the family of fibropolycystic diseases. Cholestasis can be observed in patients with Caroli's syndrome without cholangitis. This is in part due to the anatomical abnormality in small bile ducts related with Caroli's disease and also due to the small bile duct proliferation in the portal area related with congenital hepatic fibrosis which are both components of this syndrome. Another explanation for this situation is that stones can remain silent without causing cholangitis.
In conclusion, because of the slow and usually silent progress of Caroli’s syndrome along with its rarity and fatal complications, it should be considered in the differential diagnosis of recurrent cholangitis of unknown cause.
Footnotes
S- Editor Wang J L- Editor Zhu LH E- Editor Che YB
References
- 1.Sherlock S, Dooley J. Diseases of the liver and billiary system. 11th ed. Milano: Blackwell Sci Pub; 2002. p. 583. [Google Scholar]
- 2.Lu SC, Debian KA. Cystic diseases of the biliary tract. In: Yamada T, Alpers DH, Kaplowitz N, Laine L, Owyang C, et al., editors. Textbook of Gastroenterology. Philadelphia: Lippincott Williams and Wilkins; 2003. pp. 2225–2233. [Google Scholar]
- 3.Wu KL, Changchien CS, Kuo CM, Chuah SK, Chiu YC, Kuo CH. Caroli's disease - a report of two siblings. Eur J Gastroenterol Hepatol. 2002;14:1397–1399. doi: 10.1097/00042737-200212000-00019. [DOI] [PubMed] [Google Scholar]
- 4.Keramidas DC, Kapouleas GP, Sakellaris G. Isolated Caroli's disease presenting as an exophytic mass in the liver. Pediatr Surg Int. 1998;13:177–179. doi: 10.1007/s003830050281. [DOI] [PubMed] [Google Scholar]
- 5.Madjov R, Chervenkov P, Madjova V, Balev B. Caroli's disease. Report of 5 cases and review of literature. Hepatogastroenterology. 2005;52:606–609. [PubMed] [Google Scholar]
- 6.Gupta AK, Gupta A, Bhardwaj VK, Chansoria M. Caroli's disease. Indian J Pediatr. 2006;73:233–235. doi: 10.1007/BF02825490. [DOI] [PubMed] [Google Scholar]
- 7.Kim JT, Hur YJ, Park JM, Kim MJ, Park YN, Lee JS. Caroli's syndrome with autosomal recessive polycystic kidney disease in a two month old infant. Yonsei Med J. 2006;47:131–134. doi: 10.3349/ymj.2006.47.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Awasthi A, Das A, Srinivasan R, Joshi K. Morphological and immunohistochemical analysis of ductal plate malformation: correlation with fetal liver. Histopathology. 2004;45:260–267. doi: 10.1111/j.1365-2559.2004.01945.x. [DOI] [PubMed] [Google Scholar]
- 9.Desmet VJ. Pathogenesis of ductal plate malformation. J Gastroenterol Hepatol. 2004;19:S356–S360. [Google Scholar]
- 10.De Kerckhove L, De Meyer M, Verbaandert C, Mourad M, Sokal E, Goffette P, Geubel A, Karam V, Adam R, Lerut J. The place of liver transplantation in Caroli's disease and syndrome. Transpl Int. 2006;19:381–388. doi: 10.1111/j.1432-2277.2006.00292.x. [DOI] [PubMed] [Google Scholar]
- 11.Gorka W, Lewall DB. Value of Doppler sonography in the assessment of patients with Caroli's disease. J Clin Ultrasound. 1998;26:283–287. doi: 10.1002/(sici)1097-0096(199807/08)26:6<283::aid-jcu1>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 12.Guy F, Cognet F, Dranssart M, Cercueil JP, Conciatori L, Krausé D. Caroli's disease: magnetic resonance imaging features. Eur Radiol. 2002;12:2730–2736. doi: 10.1007/s00330-002-1471-6. [DOI] [PubMed] [Google Scholar]
- 13.Levy AD, Rohrmann CA, Murakata LA, Lonergan GJ. Caroli's disease: radiologic spectrum with pathologic correlation. AJR Am J Roentgenol. 2002;179:1053–1057. doi: 10.2214/ajr.179.4.1791053. [DOI] [PubMed] [Google Scholar]
- 14.Yönem O, Ozkayar N, Balkanci F, Harmanci O, Sökmensüer C, Ersoy O, Bayraktar Y. Is congenital hepatic fibrosis a pure liver disease. Am J Gastroenterol. 2006;101:1253–1259. doi: 10.1111/j.1572-0241.2006.00642.x. [DOI] [PubMed] [Google Scholar]
- 15.Abdullah AM, Nazer H, Atiyeh M, Ali MA. Congenital hepatic fibrosis in Saudi Arabia. J Trop Pediatr. 1991;37:240–243. doi: 10.1093/tropej/37.5.240. [DOI] [PubMed] [Google Scholar]
- 16.Bayraktar Y, Balkanci F, Kayhan B, Uzunalimoglu B, Ozenc A, Ozdemir A, Dündar S, Arslan S, Sivri B, Telatar H. Congenital hepatic fibrosis associated with cavernous transformation of the portal vein. Hepatogastroenterology. 1997;44:1588–1594. [PubMed] [Google Scholar]
- 17.Dahlstrom JE, Cookman J, Jain S. Joubert syndrome: an affected female with bilateral colobomata. Pathology. 2000;32:283–285. [PubMed] [Google Scholar]
- 18.Patil S, Das HS, Desai N, Manjunath SM, Thakur BS, Sawant P. Caroli's syndrome--a rare cause of portal hypertension. J Assoc Physicians India. 2004;52:261. [PubMed] [Google Scholar]
- 19.Kassahun WT, Kahn T, Wittekind C, Mössner J, Caca K, Hauss J, Lamesch P. Caroli's disease: liver resection and liver transplantation. Experience in 33 patients. Surgery. 2005;138:888–898. doi: 10.1016/j.surg.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 20.Shun A, Delaney DP, Martin HC, Henry GM, Stephen M. Portosystemic shunting for paediatric portal hypertension. J Pediatr Surg. 1997;32:489–493. doi: 10.1016/s0022-3468(97)90613-8. [DOI] [PubMed] [Google Scholar]
- 21.Yüce A, Koçak N, Akhan O, Gürakan F, Ozen H. Caroli's syndrome in two siblings. Am J Gastroenterol. 2002;97:1855–1856. doi: 10.1111/j.1572-0241.2002.05875.x. [DOI] [PubMed] [Google Scholar]