

Abstract
AIM: To evaluate the effect of combined antisense oligonucleotides targeting midkine (MK-AS) and chemotherapeutic drugs [cisplatin(DDP), 5-fluorouracil (5-FU) and adriamycin (ADM)] on inhibition of HepG2 cell proliferation, and to analyze the efficacy of MK-AS used in combined ADM in in situ human hepatocellular carcinoma (HCC) model.
METHODS: HepG2 cells were treated with MK-AS and/or chemotherapeutic drugs mediated by Lipofectin, and cell growth activity was determined by MTS assay. An in situ HCC model was used in this experiment. MK-AS, ADM and MK-AS + ADM were given intravenously for 20 d, respectively. The animal body weight and their tumor weight were measured to assess the effect of the combined therapy in vivo.
RESULTS: Combined treatment with MK-AS reduced the IC50 of DDP, 5-FU and ADM in HepG2 cells. MK-AS significantly increased the inhibition rate of DDP, 5-FU and ADM. Additionally, synergism (Q 1.15) occurred at a lower concentration of ADM, 5-FU and DDP with combined MK-AS. Combined treatment with MK-AS and ADM resulted in the more growth inhibition on in situ human HCC model compared with treatment with chemotherapeutic drugs alone.
CONCLUSION: MK-AS increases the chemosensitivity in HepG2 cells and in situ human HCC model, and the combination of MK-AS and ADM has a much better in vitro and in vivo synergism.
Keywords: Antisense oligonucleotide, Midkine, Carci-noma, Hepatocellular, Combination therapy
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the malignant diseases with a high incidence and mortality in the world, which is often diagnosed at an advanced stage when most potentially curative therapies such as resection, transplantation or percutaneous and transarterial interventions are of limited efficacy[1-4]. It is accepted that there is no satisfactory treatment available for patients with HCC and chemotherapy has been extremely disappointing[5-7]. The poor prognosis of patients with HCC and the lack of satisfactory therapy for advanced cases indicate a need for more effective therapeutic options. Recent insights into the biology of HCC suggest that certain pathways and molecular alterations are likely to play essential roles in HCC development by promoting cell growth and survival. Dysregulation of growth factors, receptors and their downstream signaling pathway components represent a central pro-tumorigenic principle in human hepatocarcinogenesis.
Growth factors and the downstream signaling system are often overexpressed in tumors, and become the target of their treatment. It has been reported that MK, a heparin-binding growth factor or cytokine, is usually overexpressed in various malignant tumors, such as lung, breast, esophageal, gastric, colorectal, liver, pancreatic, ovarian, urinary bladder, prostatic, cerebral and renal malignancies[8-16], whereas in normal adult tissues, MK is low or undetectable[2,16]. Midkine (MK) can promote the growth, survival, and migration of various target cells[17]. The level of midkine expression correlates negatively with the patients’ prognosis. It is reported that antisense oligo (DNA) targeting MK suppresses the growth of tumors in nude mice[18,19]. Addtionally, siRNA or antisense oligo (DNA) targeting against MK inhibits neointima formation[20] and renal injury after ischemia[21]. All of these studies suggested that midkine may play an important role in carcinogenesis, development and metastasis of tumors, and that it could serve as a novel tumor marker.
A novel approach to cancer therapy is the integration of new cytotoxic agents with multiple selective inhibitors of relevant signaling pathways. Such a strategy indicates that it may be possible to use agents that cooperate at low doses and/or through simple administration modalities to increase tumor targeting and patient compliance and reduce toxicity. We intend to explore the potential of the antisense targeting against MK (MK-AS) combined with chemotherapeutic drugs (ADM, DDP and 5-FU) in inhibition of HCC growth.
MATERIALS AND METHODS
Oligonucleotides synthesis and drugs
Antisense phosphorothioate oligonucleotide (5’- CCCCGGGCCGCCCTTCTTCA-3’) were synthesized by an Applied Biosystems Model 391 DNA synthesizer on solid supports using Oligo Pilot II DNA (Amersham-Pharmacia, Piscataway, NJ, USA) and purified by HPLC Prep 4000 (Waters Delta, USA) with SOURCE 15Q (Amersham-Pharmacia, USA). Chemotherapeutic drugs (DDP, 5-FU and ADM) were purchased from the Shanghai Donghaipu Pharmaceuticals Company (Shanghai, China).
Cell culture conditions
Human HCC cell (HepG2) was obtained from the Cancer Institute, Chinese Academy of Medical Sciences. HepG2 cells were cultured in DMEM (GIBCO BRL, Grand Island, NY, USA), supplemented with 10% FCS (GIBCO BRL), 100 U/mL penicillin and 100 U/mL streptomycin. Tumor cells were kept at 37°C in a humidified atmosphere containing 50 mL/L CO2.
MK-AS and chemotherapeutic treatment
3 × 103 cells were seeded in 96-well microtiter plate and allowed to attach overnight. MK-AS was then tranfected into these cells mediated by Lipofectin (Invitrogen, USA). After incubation for 6 h, the cultural medium was replaced with 100 μL of cell culture medium with different chemotherapeutic drugs. The concentrations were used in this experiment around the IC50 of each chemotherapeutic drug (DDP: 0.25, 0.5, 1, 2, 4 mg/L; 5-FU: 1.0, 2.5, 5, 10, 20 mg/L; and ADM: 0.025, 0.05, 0.1, 0.2, 0.4 mg/L). Additionally, the concentrations of each anticancer drug were combined with three concentrations of MK-AS (0.1, 0.2, 0.4 μmol/L) respectively. After 48 h incubation, 20 μL of MTS (Promega, USA) was added into each well, followed by a 90 min incubation at 37°C. MTS assay was then used to assess the cell proliferation with a Victor 1420 Multilable Counter (WALLAC, USA). Zheng-Jun Jin’s methods[22] was used to analyze the combined effect (the antagonistic effects: Q < 0.85; the additive effect: 0.85 ≤ Q < 1.15; and the synergistic effects: Q ≥ 1.15).
Analysis of combined effects
The Balb/c nude mice (virgin female) used in these experiments were obtained from the Academy of Military Medical Sciences (Beijing, China). The human HCC model used in this experiment was described previously[23]. Briefly, the HCM-Y89 tumor was cut into 1 mm × 1 mm× 1 mm tissues, and implanted into the liver of mice. Two days after development of in situ HCC models, mice were injected intravenously with saline (saline alone used as control), MK-AS of 50 mg/kg per day and/or ADM of 10 mg/kg per day for 20 d. The body weight and general physical status of the animals were recorded daily. Mice were killed at different time points by cervical dislocation and the tumors were removed and weighed.
Western blotting and RT-PCR
The total RNA was extracted and RT-PCR reaction was performed using an RT-PCR kit (Promega, Madison, WI, USA). PCR products were analyzed using 2.0% agarose gel and visualized by ethidium bromide staining. For Western-blotting, the tumor tissues were lysed with lysis buffer (50 mmol/L Tris-HCl, pH 7.4, 0.5 mmol/L EDTA, 0.5% NP40, and 150 mmol/L NaCl) in the presence of protease inhibitors. The lysates were then centrifuged at 15 000 ×g for 15 min to remove debris. Protein samples (60 μg) were separated by 12% SDS-PAGE gel and transferred onto PVDF membranes (Hybond-polyvinylidene difluoride membranes, Amersham Biosciences). The reactive band was visualized with an ECL-plus Detection Kit (Amersham Biosciences, Piscataway, NJ) and scanned by Gel Doc 1000 (Bio-Rad CA, USA). β-actin was used as a control.
Statistical analysis
Data were expressed as means ± SD, statistical analysis was carried out using Student’s t test (two tailed), and P < 0.05 indicates statistical significance.
RESULTS
MK-AS transfer increases the cytotoxicity of DDP, 5-Fu and ADM in HepG2
After transfection with MK-AS, cells were treated with 5-FU, ADM or DDP at different concentrations. Transfection of MK-AS was found to enhance the cytotoxicity of 5-FU, ADM and DDP significantly. As shown in Figure 1A, the IC50 of ADM alone is 0.109 mg/L. However, combined ADM and MK-AS (0.1 μmol/L) decreased the IC50 from 0.109 mg/L to 0.0517 mg/L. Meanwhile, we also observed that 0.1 μmol/L MK-AS decreased the IC50 of 5-FU from 5.6147 mg/L to 2.61 mg/L (Figure 1B), and the IC50 of DDP from 1.048 mg/L to 0.594 mg/L. All these results indicated that MK-AS transfer increased the chemosensitivity in HepG2 cells.
Figure 1.
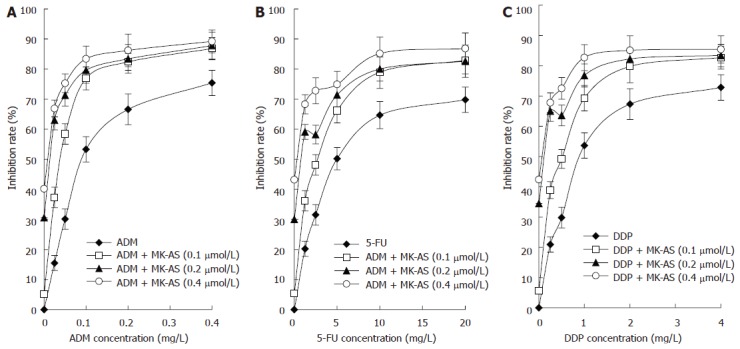
Analysis of combined effect of MK-AS and ADM (A), 5-FU (B) and DDP (C) in HepG2 cells. Each value represents the mean ± SD from triplicate determinations.
MK-AS synergistically interacts with chemotherapeutic drugs in HepG2
Furthermore, we used Zheng-Jun Jin’s method to analyze the antagonism, additivity or synergy of the interaction between MK-AS and the anticancer drugs in HepG2 cells. The Q values is presented in Figure 2. The synergistic effects (Q ≥ 1.15)of chemotherapeutic drugs with MK-AS only occurred at lower concentrations of anticancer drugs. With the increase of chemotherapeutic drug concentration, the additive effect(0.85 ≤ Q < 1.15) occurred. It should to be noted that there are no antagonistic effects(Q < 0.85) using the combined MK-AS with all these chemotherapeutic drugs. Meanwhile, the combined treatment of ADM with MK-AS showed better synergistic effects than that of the combined treatment with other anti-drugs. The highest Q value for the treatment of ADM and MK-AS was 1.87.
Figure 2.
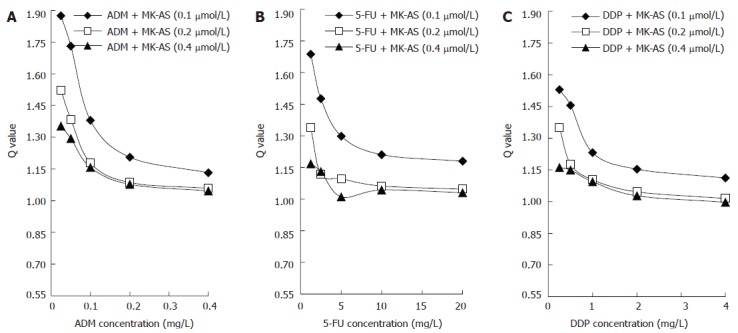
Q values for combined treatment of MK-AS and ADM (A), 5- FU (B) and DDP (C) in HepG2 cells. Q values were calculated from the dose-response curves shown in Figure 1 and analyzed by Zheng-Jun Jin’s method.
Combination of MK-AS and ADM on in situ HCC xenograft growth
In the present study, we used an in situ HCC model in mice to evaluate the antitumor activity of MK-AS in vivo. In this experiment, MK-AS (50 mg/kg per day), ADM (0.2 mg/kg per day )and saline were administered intravenously for 20 d. Tumors were removed after the mice were killed after treatment for 0 d, 4 d, 8 d,12 d, 16 d or 20 d. The tumors were then weighed. Results showed that both ADM and MK-AS treatment resulted in a significant inhibition of tumor weight compared to saline-treated mice (Figure 3). However, the combination of ADM with MK-AS showed a more marked inhibition effect than MK-AS or ADM treatment alone. Additionally, the mean body weight of the mice was not significantly different between the groups from the beginning to the end (Figure 4), suggesting that all the treatment groups showed no remarkable toxicity in animal.
Figure 3.
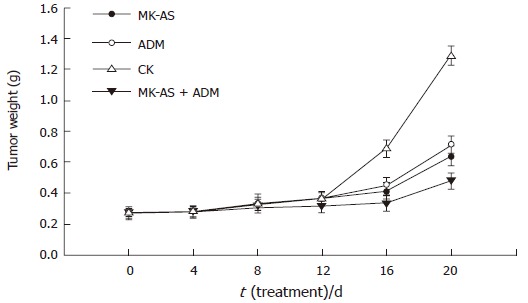
Tumor growth in situ human HCC model after treatment with MK-AS and/or ADM. CK: Treatment with saline; ADM: Treatment with ADM; MK-AS: Treatment with MK-AS; ADM + MK-AS: Combination of ADM with MK-AS. Each value represents the mean ± SD from 7 determinations.
Figure 4.
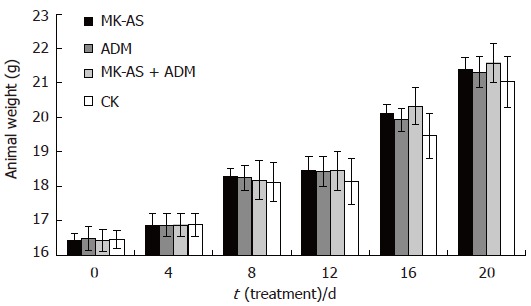
The human HCC model animal weight after treatment with MK-AS and/or ADM. CK: Treatment with saline; ADM: Treatment with ADM; MK-AS: Treatment with MK-AS; ADM + MK-AS: Combination of ADM with MK-AS. Each value represents the mean ± SD from 7 determinations.
MK-AS treatment decreased MK mRNA and protein level
We analyzed the effect of ADM or MK-AS treatment in MK mRNA and protein in HepG2 cells (Figure 5). Results showed that MK treatment alone or combination with ADM significantly downregulated MK expression including the protein and mRNA level in in situ HCC model in mice. However, ADM treatment alone has little effect on MK expression.
Figure 5.
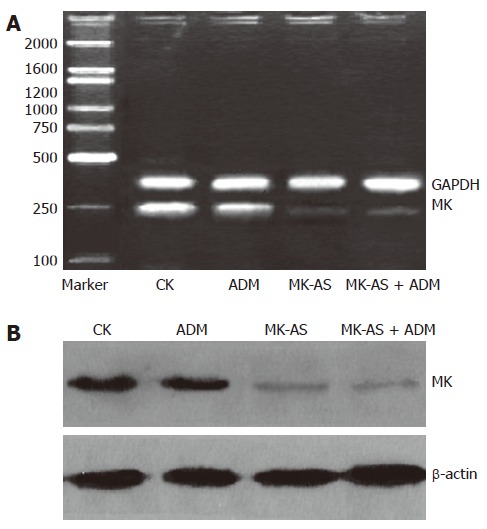
Effects of MK-AS and ADM on MK expression in in situ human hepatocellular carcinoma (HCC) model. A: Electrophoresis of RT-PCR products of MK gene and GAPDH gene, GAPDH is used as control; B: The total protein were separated by SDS gel electrophoresis, transferred to PVDF membranes, and blotted with anti-MK body. β-actin rabbit polyclonal antibodies was used as a loading control. CK: Treatment with saline; ADM: Treatment with ADM; MK-AS: Treatment with MK-AS; ADM + MK-AS: Combination of ADM with MK-AS.
DISCUSSION
MK is a heparin-binding growth factor identified as a product of a retinoic acid response gene[24,25]. The pathophysiological effects of MK include an enhanced plasminogen activity[26], oncogenic transformation of fibroblasts[27], antiapoptotic activity[28], and angiogenic activity[29]. MK and pleiotrophin (also known as the heparin-binding growth-associated molecule) comprise a family of heparin-binding growth/differentiation factors, different from other heparin-binding growth factors such as fibroblast growth factor and hepatocyte growth factor[2-4]. It was reported that MK activates mitogen-activated protein kinase pathways through inducing the phosphorylation of p44/42 MAPK, subsequently promoting cell growth[30]. Moreover, MK also activates extracellular signal-regulated kinases 1 and 2, which are well known as signal transducers[28]. The activated MAPK pathway was believed to downregulate caspase-3 activity in neurons[30]. In addition, MK can induce phosphorylation of protein kinase B. The phosphorylated AKT can promote a series of anti-apoptosis pathways in cells. Recently, HCC tumor cells were found to overexpress MK[31]. These findings suggest that MK could be a potential target for HCC therapy.
At present, although surgery and chemotherapy are effective in patients with localized tumors, the prognosis of patients with advanced or metastatic tumors is not ideal. Therefore, novel treatment approaches to the cancer are urgently needed. We analyzed the combination of MK-AS with chemotherapeutic drugs. The results showed that MK-AS transfer increased the anti-cancer effect of chemotherapeutic drugs. Furthermore, we also observed the synergism at a lower concentration of all these chemotherapeutic drugs. This result implied that combination of MK-AS with chemotherapeutic drugs will possibly provide a more marked therapeutic effect. ADM is a reagent that leads to DNA break in cells. However, MK-AS can inhibit MK growth factor expression. Taken together, we supposed that this good synergism might be due to the different antitumor functions.
Considering the in vitro and in vivo difference, we then analyzed the combination of MK-AS with ADM in in situ human HCC model. Both the MK-AS and ADM administration showed a remarkable inhibition of tumor weight. Interestingly, the combination decreased the tumor weight more than MK-AS or ADM treatment alone. Our experimental model of HCC has some advantages compared with the inoculated tumor model[23]. The tumor originated from human HCC maintained a series of characteristics of human HCC tissues, such as AFP secretion and drug sensitivity. Additionally, the pathological evidence also suggests that this model shows various features in clinical HCC patients. Therefore, the results obtained from this model can reflect the true clinical picture of patients to some degree. We also observed that MK-AS suppressed MK expression, including the mRNA and protein. However, ADM treatment showed little effect on MK expression, suggesting that the suppressed tumor effect of MK-AS is due to the inhibition of MK expression.
In summary, our results suggest that MK-AS can increase the therapeutic effect of chemotherapeutic drugs both in vivo and in vitro. The combined ADM and MK-AS showed a better synergism at a low concentration, which will provide another possible strategy for cancer treatment.
Footnotes
Supported by grants from the Zhejiang Province Medicine and Health Research Fund, No. 2003A077 and Huzhou Natural Science Foundation, No. 2004SZX07-11, China
S- Editor Liu Y L- Editor Ma JY E- Editor Ma WH
References
- 1.Kadomatsu K, Tomomura M, Muramatsu T. cDNA cloning and sequencing of a new gene intensely expressed in early differentiation stages of embryonal carcinoma cells and in mid-gestation period of mouse embryogenesis. Biochem Biophys Res Commun. 1988;151:1312–1318. doi: 10.1016/s0006-291x(88)80505-9. [DOI] [PubMed] [Google Scholar]
- 2.Li YS, Milner PG, Chauhan AK, Watson MA, Hoffman RM, Kodner CM, Milbrandt J, Deuel TF. Cloning and expression of a developmentally regulated protein that induces mitogenic and neurite outgrowth activity. Science. 1990;250:1690–1694. doi: 10.1126/science.2270483. [DOI] [PubMed] [Google Scholar]
- 3.Merenmies J, Rauvala H. Molecular cloning of the 18-kDa growth-associated protein of developing brain. J Biol Chem. 1990;265:16721–16724. [PubMed] [Google Scholar]
- 4.Muramatsu T. Midkine (MK), the product of a retinoic acid responsive gene, and pleiotrophin constitute a new protein family regulating growth and differentiation. Int J Dev Biol. 1993;37:183–188. [PubMed] [Google Scholar]
- 5.Minemura M, Tanimura H, Tabor E. Overexpression of multidrug resistance genes MDR1 and cMOAT in human hepatocellular carcinoma and hepatoblastoma cell lines. Int J Oncol. 1999;15:559–563. doi: 10.3892/ijo.15.3.559. [DOI] [PubMed] [Google Scholar]
- 6.Huang M, Liu G. The study of innate drug resistance of human hepatocellular carcinoma Bel7402 cell line. Cancer Lett. 1999;135:97–105. doi: 10.1016/s0304-3835(98)00280-8. [DOI] [PubMed] [Google Scholar]
- 7.Nies AT, König J, Pfannschmidt M, Klar E, Hofmann WJ, Keppler D. Expression of the multidrug resistance proteins MRP2 and MRP3 in human hepatocellular carcinoma. Int J Cancer. 2001;94:492–499. doi: 10.1002/ijc.1498. [DOI] [PubMed] [Google Scholar]
- 8.Garver RI, Chan CS, Milner PG. Reciprocal expression of pleiotrophin and midkine in normal versus malignant lung tissues. Am J Respir Cell Mol Biol. 1993;9:463–466. doi: 10.1165/ajrcmb/9.5.463. [DOI] [PubMed] [Google Scholar]
- 9.Garver RI, Radford DM, Donis-Keller H, Wick MR, Milner PG. Midkine and pleiotrophin expression in normal and malignant breast tissue. Cancer. 1994;74:1584–1590. doi: 10.1002/1097-0142(19940901)74:5<1584::aid-cncr2820740514>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 10.Aridome K, Tsutsui J, Takao S, Kadomatsu K, Ozawa M, Aikou T, Muramatsu T. Increased midkine gene expression in human gastrointestinal cancers. Jpn J Cancer Res. 1995;86:655–661. doi: 10.1111/j.1349-7006.1995.tb02449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakanishi T, Kadomatsu K, Okamoto T, Tomoda Y, Muramatsu T. Expression of midkine and pleiotropin in ovarian tumors. Obstet Gynecol. 1997;90:285–290. doi: 10.1016/S0029-7844(97)00237-8. [DOI] [PubMed] [Google Scholar]
- 12.O'Brien T, Cranston D, Fuggle S, Bicknell R, Harris AL. The angiogenic factor midkine is expressed in bladder cancer, and overexpression correlates with a poor outcome in patients with invasive cancers. Cancer Res. 1996;56:2515–2518. [PubMed] [Google Scholar]
- 13.Konishi N, Nakamura M, Nakaoka S, Hiasa Y, Cho M, Uemura H, Hirao Y, Muramatsu T, Kadomatsu K. Immunohistochemical analysis of midkine expression in human prostate carcinoma. Oncology. 1999;57:253–257. doi: 10.1159/000012039. [DOI] [PubMed] [Google Scholar]
- 14.Mishima K, Asai A, Kadomatsu K, Ino Y, Nomura K, Narita Y, Muramatsu T, Kirino T. Increased expression of midkine during the progression of human astrocytomas. Neurosci Lett. 1997;233:29–32. doi: 10.1016/s0304-3940(97)00619-8. [DOI] [PubMed] [Google Scholar]
- 15.Nakagawara A, Milbrandt J, Muramatsu T, Deuel TF, Zhao H, Cnaan A, Brodeur GM. Differential expression of pleiotrophin and midkine in advanced neuroblastomas. Cancer Res. 1995;55:1792–1797. [PubMed] [Google Scholar]
- 16.Tsutsui J, Kadomatsu K, Matsubara S, Nakagawara A, Hamanoue M, Takao S, Shimazu H, Ohi Y, Muramatsu T. A new family of heparin-binding growth/differentiation factors: increased midkine expression in Wilms' tumor and other human carcinomas. Cancer Res. 1993;53:1281–1285. [PubMed] [Google Scholar]
- 17.Muramatsu T. Midkine and pleiotrophin: two related proteins involved in development, survival, inflammation and tumorigenesis. J Biochem. 2002;132:359–371. doi: 10.1093/oxfordjournals.jbchem.a003231. [DOI] [PubMed] [Google Scholar]
- 18.Takei Y, Kadomatsu K, Matsuo S, Itoh H, Nakazawa K, Kubota S, Muramatsu T. Antisense oligodeoxynucleotide targeted to Midkine, a heparin-binding growth factor, suppresses tumorigenicity of mouse rectal carcinoma cells. Cancer Res. 2001;61:8486–8491. [PubMed] [Google Scholar]
- 19.Takei Y, Kadomatsu K, Goto T, Muramatsu T. Combinational antitumor effect of siRNA against midkine and paclitaxel on growth of human prostate cancer xenografts. Cancer. 2006;107:864–873. doi: 10.1002/cncr.22068. [DOI] [PubMed] [Google Scholar]
- 20.Banno H, Takei Y, Muramatsu T, Komori K, Kadomatsu K. Controlled release of small interfering RNA targeting midkine attenuates intimal hyperplasia in vein grafts. J Vasc Surg. 2006;44:633–641. doi: 10.1016/j.jvs.2006.04.044. [DOI] [PubMed] [Google Scholar]
- 21.Sato W, Takei Y, Yuzawa Y, Matsuo S, Kadomatsu K, Muramatsu T. Midkine antisense oligodeoxyribonucleotide inhibits renal damage induced by ischemic reperfusion. Kidney Int. 2005;67:1330–1339. doi: 10.1111/j.1523-1755.2005.00210.x. [DOI] [PubMed] [Google Scholar]
- 22.Jin ZJ. Addition in drug combination (author's transl) Zhongguo Yao Li Xue Bao. 1980;1:70–76. [PubMed] [Google Scholar]
- 23.Lin RX, Tuo CW, Lü QJ, Zhang W, Wang SQ. Inhibition of tumor growth and metastasis with antisense oligonucleotides (Cantide) targeting hTERT in an in situ human hepatocellular carcinoma model. Acta Pharmacol Sin. 2005;26:762–768. doi: 10.1111/j.1745-7254.2005.00762.x. [DOI] [PubMed] [Google Scholar]
- 24.Tomomura M, Kadomatsu K, Matsubara S, Muramatsu T. A retinoic acid-responsive gene, MK, found in the teratocarcinoma system. Heterogeneity of the transcript and the nature of the translation product. J Biol Chem. 1990;265:10765–10770. [PubMed] [Google Scholar]
- 25.Kadomatsu K, Huang RP, Suganuma T, Murata F, Muramatsu T. A retinoic acid responsive gene MK found in the teratocarcinoma system is expressed in spatially and temporally controlled manner during mouse embryogenesis. J Cell Biol. 1990;110:607–616. doi: 10.1083/jcb.110.3.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kojima S, Inui T, Muramatsu H, Kimura T, Sakakibara S, Muramatsu T. Midkine is a heat and acid stable polypeptide capable of enhancing plasminogen activator activity and neurite outgrowth extension. Biochem Biophys Res Commun. 1995;216:574–581. doi: 10.1006/bbrc.1995.2661. [DOI] [PubMed] [Google Scholar]
- 27.Kadomatsu K, Hagihara M, Akhter S, Fan QW, Muramatsu H, Muramatsu T. Midkine induces the transformation of NIH3T3 cells. Br J Cancer. 1997;75:354–359. doi: 10.1038/bjc.1997.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Owada K, Sanjo N, Kobayashi T, Mizusawa H, Muramatsu H, Muramatsu T, Michikawa M. Midkine inhibits caspase-dependent apoptosis via the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase in cultured neurons. J Neurochem. 1999;73:2084–2092. [PubMed] [Google Scholar]
- 29.Choudhuri R, Zhang HT, Donnini S, Ziche M, Bicknell R. An angiogenic role for the neurokines midkine and pleiotrophin in tumorigenesis. Cancer Res. 1997;57:1814–1819. [PubMed] [Google Scholar]
- 30.Sandra F, Harada H, Nakamura N, Ohishi M. Midkine induced growth of ameloblastoma through MAPK and Akt pathways. Oral Oncol. 2004;40:274–280. doi: 10.1016/j.oraloncology.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 31.Kato M, Shinozawa T, Kato S, Awaya A, Terada T. Increased midkine expression in hepatocellular carcinoma. Arch Pathol Lab Med. 2000;124:848–852. doi: 10.5858/2000-124-0848-IMEIHC. [DOI] [PubMed] [Google Scholar]