

Abstract
AIM: To investigate the significance of p16 and O6-methylguanine-DNA methyltransferase (MGMT) genes promoter hypermethylation and K-ras mutations on colorectal tumorigenesis and progression.
METHODS: p16 and MGMT methylation status was examined on 47 tumor samples, and K-ras mutational status was examined on 85 tumor samples. For methylation analysis, a methylation specific PCR (MS-PCR) method was used.
RESULTS: p16 and MGMT promoter methylation was found in 51% (24/47) and 43% (20/47) of CRCs, respectively, and the K-ras mutation was found in 44% (37/85) of CRCs. Comethylation of p16 and MGMT genes was significantly associated with lower aggressiveness of the disease within a two-year period of observation. Only 27% of patients with simultaneous p16 and MGMT methylation showed the detectible occurrence of metastasis and/or death, compared to 67% of patients without double methylation or with no methylation (3/11 vs 22/33, P < 0.05, χ2-test). In addition, p16 and MGMT comethylation showed a trend toward an association with longer survival in patients with CRCs (35.5 ± 6.0 mo vs 23.1 ± 3.2 mo, P = 0.072, Log-rank test). Progression of the disease within a two-year period was observed in 66% of patients carrying the K-ras mutation, compared to only 19% of patients with wild type K-ras (29/44 vs 7/37, P < 0.001, χ2-test). The presence of the K-ras mutation significantly correlated to shortened overall survival (20.0 ± 1.9 mo vs 37.0 ± 1.8 mo, P < 0.001, Log-rank test). The comethylation of p16 and MGMT genes was significantly associated with lower aggressiveness of the disease even when K-ras mutations were included in the analysis as an independent variable.
CONCLUSION: Our data suggest that comethylation of promoters of p16 and MGMT genes could have a prognostic value in patients with CRC. Specifically, concurrent methylation of both genes correlates with better prognosis.
Keywords: Colorectal carcinoma, DNA methylation, p16, MGMT, K-ras mutation
INTRODUCTION
Point mutations in the K-ras gene are among the most common genetic features of colorectal cancers (CRCs)[1]. In addition to mutational changes, epigenetic mechanisms also play an important role in the pathogenesis of this tumor type. The main epigenetic modification observed in the human genome is methylation of cytosine residues within CpG dinucleotides. Aberrant de novo methylation of CpG islands within the promoter region may lead to silencing of gene transcription through a complex process involving chromatin condensation and histone deacetylation[2,3]. Epigenetic silencing through DNA methylation can begin very early in tumor progression and may affect multiple genes involved in different cellular pathways, including cell cycle control, DNA repair and many others[4,5].
Inactivation of cell cycle regulatory genes, such as p16, confers a selective growth advantage to affected cells. This tumor suppressor gene encodes a cyclin-dependent kinase inhibitor, which is critical for maintaining the retinoblastoma (Rb) protein in its active, non-phosphorylated state in the cyclin D-Rb pathway[6]. Promoter hypermethylation may lead to transcriptional inactivation of p16 resulting in abnormal cell cycling and uncontrolled cell growth, which are the hallmarks of cancer cells[7].
In addition, epigenetic modification can cause inactivation of DNA repair genes, such as O6-methylguanine-DNA methyltransferase (MGMT)[8]. MGMT is a DNA repair protein that removes mutagenic and cytotoxic alkyl adducts from the O6 position of guanine[9]. In the absence of MGMT activity, the adducts are not removed allowing the O6-alkyl guanine to mispair with thymine during DNA replication, resulting in a G-to-A transition[10]. This kind of mutation may accumulate in the genes specifically associated with particular tumor types, such as K-ras in CRCs[9].
The literature suggests that hypermethylation of p16 and MGMT genes occurs as frequently in CRCs as previously reported for the K-ras gene[7,11,12]. Epigenetic changes usually begin very early in carcinogenesis, are potentially reversible, and can advance to gene alterations. For this reason, detection of aberrant methylation can be important for early diagnosis, prognosis and treatment of patients affected by this disease[5,13].
In the present study, we analyzed the methylation status of p16 and MGMT gene promoters in patients with primary CRC and compared the frequency of these changes with the occurrence of the K-ras gene mutations. In addition, we evaluated the correlation of these gene alterations with standard clinicopathological parameters, such as Dukes’ stage, differentiation, location, histological and macroscopical type of tumor, patient gender and age. We also examined the possible correlation between these gene alterations and some immunohistochemical parameters that are known to be indicators of cell transformation, migration and metastasis of tumor cells. These parameters include altered expression of adhesive molecules E-cadherin and CD44, decrease or loss of laminin, which is a major component of epithelial basal membrane, and increased proliferate activity of the neoplastic cell. The ultimate aim of this study was to evaluate the influence of these genetic and epigenetic changes on disease progression and patient survival.
MATERIAL AND METHODS
Patients and tumor specimens
Tumor material in the form of formalin-fixed and paraffin-embedded tissue was obtained from 85 patients who underwent radical surgical resection (R0) at the Institute for Pathology and Forensic Medicine, Military Medical Academy, Belgrade. All patients gave informed consent prior to specimen collection according to institutional guidelines. None of the patients had preoperative or postoperative chemotherapy. The progression of the disease (occurrence of metastasis or/and death) was monitored during the two-year period following surgery, and survival over a five-year period was estimated. Mean follow-up was 31, 4 mo (range, 2 to 66 mo).
Tumor tissue chosen for analysis was routinely processed and microscopically examined for the regions enriched in neoplastic cells. Several 10 μm sections with neoplastic cell content greater than 85% were taken from each specimen and used for direct in vitro amplification of the K-ras gene by polymerase chain reaction (PCR). For K-ras mutational analysis, the sample of 47 was augmented with 38 samples from our previous study[14]. For analysis of p16 and MGMT methylation status, DNA was isolated from 47 samples.
Analysis of mutation status of the K-ras gene
Sections (10 μm thick) of the formalin-fixed and paraffin-embedded tumor specimens were de-paraffined and kept under 70% ethanol at 4°C until PCR. In vitro amplification of 111-bp DNA fragments encompassing codons 12 and 13 of the K-ras gene was performed on tissue from sections as described by Almoguera et al[15]. Detection of K-ras mutations was performed on 85 patients with primary CRC by a highly selective oligonucleotide hybridization technique, as described earlier[14]. Two specimens were analyzed by single strand conformation polymorphism (SSCP) electrophoresis.
Analysis of methylation status of the p16 and MGMT genes
DNA methylation patterns in the promoter CpG islands of the p16 and MGMT genes were determined in 47 samples by methylation-specific PCR (MSP) following the bisulfite modification of isolated genomic DNA, as described by Herman et al[16], with some modifications[17]. Briefly, DNA was isolated from deparaffined tumor specimens using standard Proteinase K, phenol/chloroform/isoamyl alcohol extraction, and ethanol precipitation. Two μg of isolated DNA were denatured by NaOH (final 0.3 mmol/L) at 42°C for 30 min and modified by sodium bisulfite (5.20-5.69 mol/L, pH 5.0, Sigma, USA) for 4 hr at 55°C. After incubation, DNA was purified using the DNA extraction KIT (MBI Fermentas, Lithuania), again treated by NaOH (final 0.3 mol/L), at 37°C for 20 min, precipitated with ethanol/ammonium acetate and resuspended in 40 μL of 1 mmol/L Tris-HCl, pH 8.0. Aliquots of 4 μL of bisulfite modified DNA were used for MSP reactions. The PCR mixture contained 1 x PCR buffer (16 mmol/L ammonium sulfate, 67 mmol/L Tris-HCL, pH 8.8, 10 mmol/L 2-mercaptoethanol), 6.7 mmol/L MgCl2, dNTP (each at 1.25 mmol/L) and primers (300 ng each per reaction) in a final volume of 50 μL. Reactions were hot-started at 95°C for 5 min before the addition of 1.25 units of Taq polymerase (MBI Fermentas, Lithuania). Amplification was carried out in a Hybaid OmniGene temperature cycler for 35 cycles (30 s at 95°C, 30 s at the adequate annealing temperature, and 30 s at 72°C), followed by a final extension for 4 min at 72°C. Primers for a methylated and unmethylated promoter of the p16 gene were used from Herman [16] and for the MGMT gene from Esteller et al[8]. The annealing temperatures for unmethylated and methylated p16 amplification were 60°C and 65°C, respectively, whereas the annealing temperature for both unmethylated and methylated MGMT amplification was 57°C. Controls without DNA were performed for each set of PCRs. Modified DNA from the human lymphoma cell line Raji (ECACC No: 85011429) served as positive control for methylated alleles, and modified DNA from normal lymphocytes as negative control for methylated alleles of p16 and MGMT genes. 10 μL of each PCR reaction were directly loaded on to nondenaturing 8% polyacrylamide gels, stained with AgNO3 and visualized by Na2CO3, or 2% agarose gels, stained with SYBR GreenIand visualized under UV illumination.
Immunohistochemistry
Immunohistochemistry was performed using the labeled streptavidin-biotin method (LSAB Kit+, Dako, Denmark) with microwave pretreatment for antigen retrieval. Endogenous peroxidase activity was blocked by incubating with 3% H2O2. Sections were then incubated with primary monoclonal antibodies against E-cadherin (HECD-1, Zymed Laboratories, San Francisco, USA), CD44 (Clone DF 1485, Dakocytomation, Denmark) and Laminin (4C7, Dakocytomation, Denmark) and Proliferation Cell Nuclear Antigen-PCNA (PC-10, Dakocytomation, Denmark) in optimal concentrations. Amino-ethyl carbazole was used as chromogen and finally Mayer’s hematoxylin was used for counterstaining. The presence of a positive reaction in some normal tissue structures served as a positive control. Negative controls were prepared by replacing the primary antibody with Tris-buffered saline.
Immunostaining (membrane cytoplasmic immunoreac-tivity) for CD44 was calculated as the percentage of positive tumor cells in relation to the total number of tumor cells in representative fields. The presence of less then 10%, from 10% to 50%, and for more than 50% of positive tumor cells of CD44 were considered as low, moderate and extensive expression, respectively. Present, heterogenous and absent expression in the case of E-cadherin were distinguished. The loss of laminin immunoreactivity at the tumor-stroma borders was considered according to a three-point semiquantitive scale as follows: in more than 75%, between 25%-75% and in less than 25% of glandular structures. The PCNA index was considered to be low if less than 10% of tumor cell nuclei were positive, moderate for 10%-50%, and high for more than 50% of PCNA positive tumor cell nuclei.
Statistical analysis
Contingency tables were analyzed using Pearson′s χ2-test or Fisher′s exact two-tailed test (F-test), when expected frequencies were lower than five. Continuous variables were compared with the use of Student′s t-test. Overall survival distributions were estimated by the Kaplan-Meier method and differences were evaluated by the Log-rank test. The data was fitted to Cox′s proportional risks regression model. In all tests, a P value less than 0.05 were considered as statistically significant.
RESULTS
K-ras mutations, p16 and MGMT promoter methylation in primary colorectal carcinomas
We detected K-ras mutations in 44% (37/85) of patients with CRC. The distribution of mutations was 57% (21/37) of G-to-T transversions, 32% (12/37) of G-to-A transitions and 11% (4/37) of G-to-C transversions. In codons 12 and 13, we detected mutations in 73% (27/37) and 27% (10/37), respectively.
Aberrant methylation of p16 and MGMT genes was detected in 51% (24/47) and 43% (20/47) of patients, respectively. Representative examples of the methylation analysis are shown in Figure 1A and B.
Figure 1.
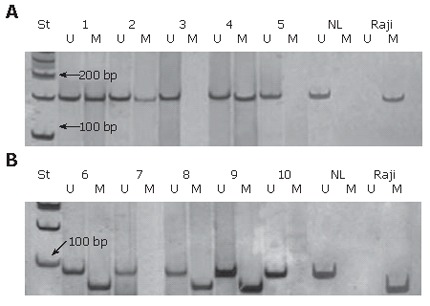
Analysis of p16 (A) and MGMT (B) gene methylation by MSP. The presence of a visible PCR product in lanes U indicates the presence of unmethylated genes of p16 (151 bp) and MGMT (93 bp); the presence of product in lanes M indicates the presence of methylated genes of p16 (150 bp) and MGMT (81 bp). The samples of colorectal carcinoma 1, 2 and 4 show p16 promoter hypermethylation, while 6, 8 and 9 show MGMT promoter hypermethylation. NL-normal lymphocytes as a positive control for unmethylated alleles; Raji-commercial cell line as a positive control for methylated alleles; St-molecular weight marker (50 bp).
Of the total number of cases analyzed for methylation, 45% (21/47) had a mutant K-ras gene. Simultaneous alterations of K-ras and p16 genes were detected in 26% (12/47) of tumors. These events seemed to occur independently, as estimated by a χ2-test (results not shown). The type of mutation at K-ras did not show a significant association with the p16 methylation by an F-test, although 55% (6/11) of samples with the hypermethylated p16 promoter carried G-to-T transversions in K-ras.
Simultaneous occurrence of K-ras mutation and MGMT methylation was detected in 21% (10/47) of the patients. Our results showed that MGMT methylation is significantly associated with a G-to-A transition in the K-ras gene (P = 0.01, F-test). This type of oncogene transition mutation occurs in 67% (6/9) of tumors carrying MGMT methylation, in contrast to only 10% (1/10) of tumors without MGMT methylation, but with the same type of K-ras mutation.
Concurrent methylation of p16 and MGMT genes was detected in 11 out of 47 (23%) cases, while simultaneous K-ras, p16 and MGMT alterations were present in 6 out of 47 cases (13%). Methylation of p16 and MGMT as well as K-ras mutation vs comethylation of p16 and MGMT genes seemed to occur independently as assessed by a χ2-test (results not shown).
Correlation of K-ras mutations and hypermethylation of p16 and MGMT genes with clinicopathological and immunohistochemical parameters
We further examined correlation between genetic and epigenetic alterations and some clinicopathological and immunohistochemical parameters that might be important for disease prognosis in CRC patients. The results of these analyses are summarized in Tables 1 and 2.
Table 1.
Association between p16 and MGMT methylation, K-ras mutations and clinicopathological features of colorectal cancer
| Methylation (%) | Mutations (%) | ||
| Variable | p16 (n = 46) | MGMT (n = 46) | K-ras (n = 84) |
| Gender | |||
| Female | 9/15 (60.0) | 7/15 (46.7) | 15/23 (65.2)a |
| Male | 15/31 (48.4) | 13/31 (41.9) | 22/61 (36.1) |
| Dukes’ stage | |||
| A | 5/9 (55.5) | 5/9 (55.5) | 8/21 (38.1) |
| B | 7/10 (70.0) | 4/10 (40.0) | 12/24 (50.0) |
| C | 8/15 (53.3) | 8/15 (53.3) | 17/39 (43.6)4 |
| D | 4/12 (33.3) | 3/12 (25.0) | |
| Differentiation | |||
| Poor | 15/25 (60.0) | 10/25 (40.0) | 18/44 (40.9) |
| Moderate | 8/18 (44.4) | 10/18 (55.5) | 16/26 (61.5) |
| Well | 1/3 (33.3) | 0/3 (0) | 3/14 (21.4) |
| Macrosopic type2 | |||
| Polypoid | 11/23 (47.8) | 14/23 (60.7)a | 25/48 (52.1) |
| Flat | 13/23 (56.5) | 6/23 (26.1) | 12/35 (34.3) |
| Histological type2 | |||
| Mucinous | 8/19 (42.1) | 6/19 (31.6) | 16/34 (47.1) |
| Tubular | 16/27 (59.3) | 14/27 (51.8) | 21/49 (42.8) |
| Location2 | |||
| Proximal | 3/3 (100) | 1/3 (33.3) | 5/9 (55.5) |
| Distal | 21/43 (48.8) | 19/43 (44.2) | 32/74 (43.2) |
| Progression (2 yr)1,3 | |||
| Metastasis or/and death | 11/25 (44.0) | 9/25 (36.0) | 29/44 (65.9)b |
| Without progression | 12/19 (63.1) | 11/19 (57.9) | 7/37 (18.9) |
Complete clinicopathological data were missing on one sample of colorectal cancer, and it was only included in analysis of genetic alterations;
P < 0.05;
P < 0.001 (all P values were revealed by χ2-test ); Data are missing on two1 subjects for p16 and MGMT methylation status analysis, and on one2 and three3 subjects for K-ras mutation analysis, respectively.
Data for C and D Dukes’ stages are considered together for K-ras mutation analysis.
Table 2.
Association between p16 and MGMT methylation, K-ras mutations and immunohistochemical features of colorectal cancer
| Methylation (%) | Mutations (%) | ||
| Variable | p16 (n = 46) | MGMT (n = 46) | K-ras (n = 84) |
| E-cadherin1,3 | |||
| Absent | 13/19 (68.4) | 9/19 (47.4) | 28/37 (75.7)b |
| Heterogeneous | 4/14 (28.6) | 7/14 (50.0) | 4/23 (17.4) |
| Present | 6/11 (54.5) | 4/11 (36.4) | 4/21 (19.0) |
| CD44 expression1,3 | |||
| Absent | 7/11 (63.6) | 6/11 (54.5) | 4/22 (18.2) |
| Low | 6/9 (66.7) | 4/9 (44.4) | 5/17 (29.4) |
| Moderate | 2/7 (28.6) | 4/7 (57.1) | 4/10 (40.0) |
| Extensive | 8/17 (47.0) | 6/17 (35.3) | 23/32 (71.9)b |
| CD44 type2,4 | |||
| Membranous | 9/17 (52.9) | 7/17 (41.2) | 9/36 (25.0) |
| Cytoplasmic | 13/26 (50.0) | 12/26 (46.1) | 27/44 (71.9)b |
| Laminin1,3 | |||
| 0-25% | 5/8 (62.5) | 5/8 (62.5) | 2/15 (13.3) |
| 25%-75% | 5/12 (41.7) | 5/12 (41.7) | 5/22 (22.7) |
| > 75% | 13/24 (54.2) | 10/24 (41.7) | 29/44 (65.9)b |
| PCNAi1,3 | |||
| 0 | 1/2 (50.0) | 2/2 (100) | 0/2 (0) |
| 0-10% | 5/9 (55.5) | 4/9 (44.4) | 3/17 (17.6) |
| 10%-50% | 6/9 (66.7) | 5/9 (55.5) | 3/20 (15.0) |
| > 50% | 11/24 (45.8) | 9/24 (37.5) | 30/42 (71.4)b |
Complete immunohistochemical data were missing on one sample of colorectal cancer, and it was only included in analysis of genetic alterations;
P < 0.001 (all P values were revealed by χ2-test ); Data are missing on two1 and three2 subjects for p16 and MGMT methylation status analysis, respectively, and on three3 and four4 subjects for K-ras mutation analysis, respectively.
The presence of K-ras mutations was significantly more frequent in female than in male patients (P < 0.05, χ2-test, Table 1). In addition, the average age of female patients carrying K-ras mutations was 53.3 ± 11.8 years, which was significantly lower (P < 0.01, t -test) than the average age for males with K-ras mutations (64.1 ± 8.7 years). As presented in Table 1, there was no significant association between the presence of the K-ras mutations and other clinicopathological features. However, the presence of K-ras mutations correlated significantly with the lack of E-cadherin expression, extensive expression and cytoplasmic type of CD44 antigen, decreased expression of laminin and the high PCNA index (P < 0.001 by χ2-test in all cases; Table 2). These correlations were independent of the type of K-ras mutation, except in the case of E-cadherin, where the lack of expression of this adhesive molecule showed a significant correlation with the G-to-T transversions (P < 0.05, χ2-test, results not shown).
p16 methylation was not associated with any of the variables considered, as presented in Table 1 and 2, though p16 methylation tended to be more prevalent in tumors not expressing E-cadherin (P < 0.1, χ2-test). Methylation of MGMT gene was significantly more prevalent in polyploid than in flat colorectal carcinomas (P < 0.05, χ2-test, Table 1). There was no significant association between MGMT methylation and any other clinicopathological features studied. In addition, comethylation of p16 and MGMT genes did not correlate with any clinicopathological or immunohistochemical parameters considered.
Survival analysis
In this study, we also monitored the progression of the disease in the two-year period following surgery. Methylation of p16 and MGMT genes, when they were considered separately, showed slight but insignificant correlation with slower progression of disease (Table 1). However, during that period, only 27% (3/11) of patients with simultaneous p16 and MGMT methylation showed the occurrence of metastasis and/or death, compared to 67% (22/33) of patients without this double or any gene methylation (P < 0.05, χ2-test, Figure 2A).
Figure 2.
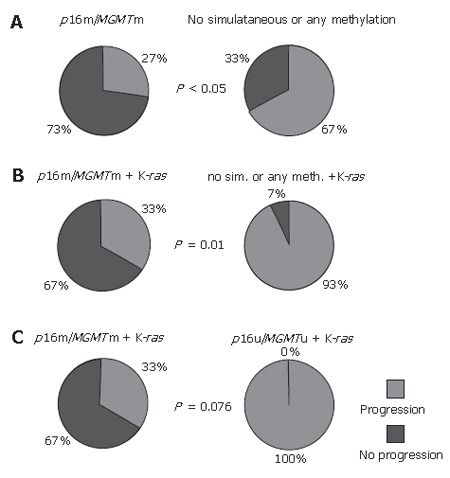
Graphical distribution of progression of the disease in the two-year period, as a function of simultaneous p16 and MGMT methylation status (A); simultaneous p16, MGMT methylation and mutated K-ras gene compared either to no simultaneous or any methylation of two genes (B) or to unmethylated both (C), respectively.
This association was preserved and was even higher (P < 0.01, F-test, Figure 2B) when K-ras mutations were included as an independent variable. Namely, the progression of the disease occurred in only 2 of 6 (33%) patients with p16 and MGMT comethylation and K-ras mutations, while disease recurred in 13 of 14 (93%) patients with the K-ras mutation, but with only one methylated or both unmethylated genes. However, when both genes were unmethylated in the presence of the mutant K-ras gene, it was observed that all 4 patients from this group exhibited the progression of the disease within two years, compared to only 2 of 6 patients with p16 and MGMT comethylation and K-ras gene mutations (P = 0.076, F-test, Figure 2C).
When the analyzed group was augmented with 38 patients from our previous study on CRC[14], we observed the progression of disease within a two-year period in 66% (29/44) of patients carrying the K-ras mutation, compared to only 19% (7/37) of patients with the wild type K-ras gene(P < 0.001, χ2 test, Table 1).
The Kaplan-Meier analysis of the few-year survival rate revealed a slight but insignificant influence of p16 and MGMT gene methylation on longer overall survival when the alterations of these genes were considered separately (results not shown). However, simultaneous p16 and MGMT methylation showed a trend toward association with longer survival in patients with CRCs who underwent curative surgery (P = 0.072, Log-rank-test, Figure 3A), as the median survival for these patients was 35.5 ± 6.0 mo in contrast to 23.1 ± 3.2 mo for patients without simultaneous or any methylation of examined genes. Multivariate analysis revealed that p16 and MGMT comethylation had no prognostic value without additional variables. By the same analysis, the presence of K-ras mutations significantly correlated to shortened overall survival (20.0 ± 1.9 mo vs 37.0 ± 1.8 mo) for 85 patients included in the analysis (P < 0.001, Log-rank test, Figure 3B). However, a Kaplan-Meier analysis considering simultaneous presence of K-ras mutations according to the methylated status of p16 and MGMT was not possible because there was a small number of samples.
Figure 3.
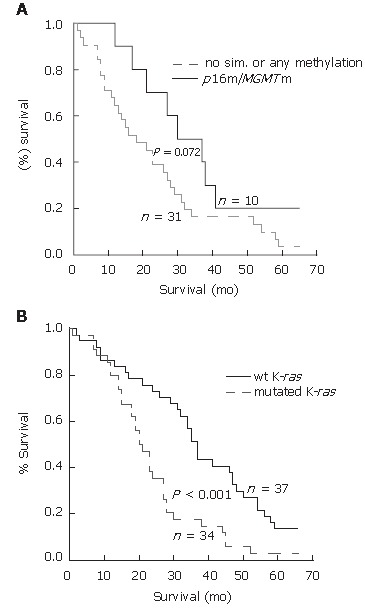
Overall survival among the patients with CRC according to the methylation status of p16 and MGMT genes (A), and mutational status of K-ras gene (B). Overall survival is longer in the group of the patients with simultaneous methylation of p16 and MGMT gene than in group with no simultaneous or any methylation (P = 0.072, Log-rank test). Overall survival is significantly lower in the group of patients with mutated K-ras than in group with unchanged, wild type K-ras gene (P < 0.001, Log-rank test). Survival curves were constructed by Kaplan-Meier method.
DISCUSSION
The significance of p16 and MGMT methylation for tumor formation and progression, as well as correlation with the occurrence of classical genetic changes in K-ras genes, was the major subject of this study. While the frequency of the genetic and epigenetic alterations in these genes was similar to those previously reported[7,8,11,18,19], this is the first study to assess their cumulative and individual effects on disease progression.
While there was no clear association between methylation and standard prognostic parameters in CRC, the data obtained from survival analysis suggest that the simultaneous methylation of p16 and MGMT genes could be a better predictive factor for the course of disease. The most important finding of the current study was that comethylation of p16 and MGMT genes was significantly associated with lower aggressiveness of the disease and tended to associate with longer overall survival in patients with CRC who underwent curative surgery. More importantly, the influence of the double gene alteration was significant in spite of the simultaneous presence of K-ras mutations, which is another well established molecular marker of unfavorable prognosis in CRCs. Consistent with our previous findings[14] and those of the multicentric RASCAL study[20], our present results confirm that the mutated K-ras gene is a molecular marker for the more aggressive course of disease and was connected to reduced overall survival. In addition, we demonstrated that K-ras mutation is strongly associated with parameters that are indicators of poor prognosis. These include the absence of E-cadherin expression[21], extensive expression of the cytoplasmic type of CD44 antigen[22], low expression of laminin[23] and high PCNA index[24].
The functional association between the effect of p16 and MGMT gene inactivation and K-ras mutation on the progression of disease is not clear. Yet, in the absence of methylation of both p16 and MGMT genes together with the mutated K-ras, a severe form of the disease is likely to commence. There is evidence that CRC does not evolve through a single sequence of molecular alterations, but evolves through different pathways. Each of them has distinct clinical, pathological and molecular characteristics[25]. One possible explanation for our findings could be that the colorectal tumors without comethylation of the two genes could arise through different mechanisms. These include large genetic rearrangements and chromosomal instability, with a significant degree of gene amplification and deletion[26,27] and such changes could be associated with a more aggressive course of the disease as proposed by Verma and Srivastava[13]. The other proposed pathway of colorectal carcinogenesis involves the occurrence of microsatellite instability (MSI), which is a phenotype resulting from alteration in the mismatch repair genes; e.g. hMLH1 and hMLH2[28,26]. Some authors reported a relationship between MSI and CpG island methylator phenotype (CIMP+), as hMLH1 is frequently inactivated by aberrant promoter methylation in sporadic MSI-H CRCs[29]. On the other hand, the CIMP+ phenotype is described as the occurrence of simultaneous methylation of a large number of genes, including p16 and hMLH1[29,30]. Though the biological and clinical properties of CIMP+ CRCs remain largely unknown, there is no longer doubt that those epigenetic changes mark a distinct group of tumors that have unique molecular profiles and etiology. More over, Wynter et al[31] demonstrated that there are at least two pathways for colorectal carcinogenesis, both implicating CIMP-high status. One group of tumors arises through BRAF mutations and hMLH1 methylation, and another with K-ras instead of BRAF mutations and with MGMT instead of hMLH1 promoter hypermethylation. Whitehall et al[32] demonstrated that methylation of MGMT has been linked to low levels of MSI (MSI-L) and Samowitz et al[33] demonstrated that the appearance of K-ras mutations is rare in high level MSI-H CRC. It is also known that MSI-H CRC associated with a CIMP+ phenotype is mostly located within the proximal colon[27,29]. In our study, all but three tumors have been located within the distal colon, so we can only speculate that they may not belong to the MSI-H group. We propose that in our group of analyzed patients who mostly have distal CRC, comethylation of p16 and MGMT genes together with other molecular changes suggest a pathway in signal transduction that could increase survival. Importantly this is in spite of the opposing effect of K-ras mutations. It would, therefore, be interesting to determine the MSI phenotype in this patient population in order to establish a possible association with observed genetic and epigenetic alteration.
Though all these questions cannot be fully resolved in the present study, our results confirm the multifactorial nature of cancer development, highlighting the molecular differences across the various classes of lesions, and exclude a single linear model of accumulating genetic alterations. In this report, we describe the association of concurrent methylation of p16 and MGMT genes with the course of disease in unselected group of CRCs. At this point, it must be noted that we did not analyze a large group of patients. However, the size of our study is still large enough to interpret the results correctly.
While some studies reported a correlation between promoter hypermethylation of the p16 gene and prognosis in patients with colorectal cancers[34-38], others observed no significant role of p16 methylation as a prognostic factor[30]. In addition, Nagasaka et al[39] found that methylated MGMT was significantly related to lower risk of recurrence in CRC. They proposed that colorectal tumors with methylated MGMT are less aggressive than tumors without such epigenetic change. Liang et al[34] proposed that geographical differences or other unknown factors supplementary to p16 methylation might increase tumor aggressiveness. Considering these and our previous results[14], we speculate that different geographic, environmental, and lifestyle factors could affect the genesis and clinical behavior of CRCs with different molecular profiles. Nevertheless, prospective studies that incorporate a larger number of genes and environmental factors are required to determine the relative contribution of each factor to the risk of developing CRCs with specific epigenetic and genetic profiles. We further support the notion that precise determination of molecular changes in CRCs is needed in order to overcome the risk of oversimplifying the role of a single or few genetic or epigenetic changes in tumorigenesis. Such analysis, supplemented by the conventional study of prognostic factors, introduces quality to prognostication of patients with CRC and aids the design of more efficient treatment modalities.
ACKNOWLEDGMENTS
We thank Dr. Nadezda Urosevic for reviewing the manuscript.
COMMENTS
Background
Colorectal carcinogenesis is a multistep process in which the progressive accumulation of genetic and epigenetic changes leads to a malignant transformation of normal epithelial cells of colon and rectum. Point mutations in K-ras gene are among the most common genetic features of colorectal cancers. In the past decade it has been shown that methylation of the promoter region of many tumor suppressor and DNA repair genes, such as p16 and MGMT has the important role in pathogenesis of this tumor type too, but its influence on disease progression remain inconclusive.
Research frontiers
Epigenetic changes usually begin very early in carcinogenesis, they are potentially reversible, and they can advance to gene alterations. For this reason, detection of aberrant methylation can be important for early diagnosis, prognosis and treatment of patients affected by this disease.
Innovations and breakthroughs
Comethylation of p16 and MGMT genes was significantly associated with lower aggressiveness of disease within two-year period of observation (P < 0.05), and showed the trend toward the association with longer survival in patients with CRCs (P = 0.072). On the other hand, the presence of K-ras mutations was associated with higher aggressiveness and shortened overall survival (P < 0.001). The comethylation of p16 and MGMT genes was significantly associated with lower aggressiveness of disease even when K-ras mutations were included in analysis as an independent variable (P < 0.01). This is the first attempt to assess the cumulative and individual effect of these genes on the disease progression.
Applications
The results presented in this article have demonstrated that the precise determination of molecular changes in CRCs is needed in order to overcome the risk of oversimplifying the role of single or a few genetic or epigenetic changes in tumorigenesis. In addition, this study underlines the need for considering the different geographic, environmental, and lifestyle factors that could affect the genesis and clinical behavior of CRCs with different molecular profiles. Prospective studies supplemented by the conventional study of prognostic factors, could improve the quality and accuracy of patients prognosis and aid design of the more efficient treatment modalities.
Terminology
Epigenetic changes: heritable changes in gene function that do not include the changes in DNA sequence. DNA methylation: the addition of a methyl group to a cytosine residue that lies next to guanine within CpG dinucleotides. Aberrant de novo methylation of CpG islands within the promoter region may lead to silencing of gene transcription through a complex process involving chromatin condensation and histone deacetylation. CpG islands: CpG rich areas located in the promoter regions of many genes. Methylation-specific PCR (MSP): amplification of modified DNA by sodium bisulfite that converts unmethylated cytosines to uracils, while methylated cytosines remains unmodified.
Peer review
The authors found some interesting findings where they showed that patients with tumors where both promoters were methylated showed less death and had longer survival rates than patients with unmethylated promoters. The data is straightforward and convincing. The authors address a clinically relevant issue on CRC prognosis. They have also demonstrated initial and unique findings examining K-ras, p16 and MGMT modifications and patient outcome.
Footnotes
Supported by the grant 143010 from the Ministry of Science and Environment Protection of the Republic of Serbia
S- Editor Liu Y L- Editor Lutze M E- Editor Zhou T
References
- 1.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 2.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 3.Wade PA, Gegonne A, Jones PL, Ballestar E, Aubry F, Wolffe AP. Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat Genet. 1999;23:62–66. doi: 10.1038/12664. [DOI] [PubMed] [Google Scholar]
- 4.Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res. 1998;72:141–196. [PubMed] [Google Scholar]
- 5.Baylin SB, Esteller M, Rountree MR, Bachman KE, Schuebel K, Herman JG. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet. 2001;10:687–692. doi: 10.1093/hmg/10.7.687. [DOI] [PubMed] [Google Scholar]
- 6.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 7.Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE, Sidransky D, Baylin SB. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995;55:4525–4530. [PubMed] [Google Scholar]
- 8.Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999;59:793–797. [PubMed] [Google Scholar]
- 9.Pegg AE. Mammalian O6-alkylguanine-DNA alkyltransferase: regulation and importance in response to alkylating carcinogenic and therapeutic agents. Cancer Res. 1990;50:6119–6129. [PubMed] [Google Scholar]
- 10.Coulondre C, Miller JH. Genetic studies of the lac repressor. IV. Mutagenic specificity in the lacI gene of Escherichia coli. J Mol Biol. 1977;117:577–606. doi: 10.1016/0022-2836(77)90059-6. [DOI] [PubMed] [Google Scholar]
- 11.Wiencke JK, Zheng S, Lafuente A, Lafuente MJ, Grudzen C, Wrensch MR, Miike R, Ballesta A, Trias M. Aberrant methylation of p16INK4a in anatomic and gender-specific subtypes of sporadic colorectal cancer. Cancer Epidemiol Biomarkers Prev. 1999;8:501–506. [PubMed] [Google Scholar]
- 12.Esteller M. Epigenetic lesions causing genetic lesions in human cancer: promoter hypermethylation of DNA repair genes. Eur J Cancer. 2000;36:2294–2300. doi: 10.1016/s0959-8049(00)00303-8. [DOI] [PubMed] [Google Scholar]
- 13.Verma M, Srivastava S. Epigenetics in cancer: implications for early detection and prevention. Lancet Oncol. 2002;3:755–763. doi: 10.1016/s1470-2045(02)00932-4. [DOI] [PubMed] [Google Scholar]
- 14.Urosević N, Krtolica K, Skaro-Milić A, Knezević-Usaj S, Dujić A. Prevalence of G-to-T transversions among K-ras oncogene mutations in human colorectal tumors in Yugoslavia. Int J Cancer. 1993;54:249–254. doi: 10.1002/ijc.2910540215. [DOI] [PubMed] [Google Scholar]
- 15.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 16.Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grunau C, Clark SJ, Rosenthal A. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res. 2001;29:E65–E65. doi: 10.1093/nar/29.13.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bos JL, Fearon ER, Hamilton SR, Verlaan-de Vries M, van Boom JH, van der Eb AJ, Vogelstein B. Prevalence of ras gene mutations in human colorectal cancers. Nature. 1987;327:293–297. doi: 10.1038/327293a0. [DOI] [PubMed] [Google Scholar]
- 19.Burmer GC, Loeb LA. Mutations in the KRAS2 oncogene during progressive stages of human colon carcinoma. Proc Natl Acad Sci USA. 1989;86:2403–2407. doi: 10.1073/pnas.86.7.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andreyev HJ, Norman AR, Cunningham D, Oates JR, Clarke PA. Kirsten ras mutations in patients with colorectal cancer: the multicenter "RASCAL" study. J Natl Cancer Inst. 1998;90:675–684. doi: 10.1093/jnci/90.9.675. [DOI] [PubMed] [Google Scholar]
- 21.Maruyama K, Ochiai A, Nakamura S, Baba S, Hirohashi S. Dysfunction of E-cadherin-catenin system in invasion and metastasis of colorectal cancer. Nihon Geka Gakkai Zasshi. 1998;99:402–408. [PubMed] [Google Scholar]
- 22.Bhatavdekar JM, Patel DD, Chikhlikar PR, Trivedi TI, Gosalia NM, Ghosh N, Shah NG, Vora HH, Suthar TP. Overexpression of CD44: a useful independent predictor of prognosis in patients with colorectal carcinomas. Ann Surg Oncol. 1998;5:495–501. doi: 10.1007/BF02303641. [DOI] [PubMed] [Google Scholar]
- 23.Offerhaus GJ, Giardiello FM, Bruijn JA, Stijnen T, Molyvas EN, Fleuren GJ. The value of immunohistochemistry for collagen IV expression in colorectal carcinomas. Cancer. 1991;67:99–105. doi: 10.1002/1097-0142(19910101)67:1<99::aid-cncr2820670119>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 24.Choi HJ, Jung IK, Kim SS, Hong SH. Proliferating cell nuclear antigen expression and its relationship to malignancy potential in invasive colorectal carcinomas. Dis Colon Rectum. 1997;40:51–59. doi: 10.1007/BF02055682. [DOI] [PubMed] [Google Scholar]
- 25.Olschwang S, Hamelin R, Laurent-Puig P, Thuille B, De Rycke Y, Li YJ, Muzeau F, Girodet J, Salmon RJ, Thomas G. Alternative genetic pathways in colorectal carcinogenesis. Proc Natl Acad Sci USA. 1997;94:12122–12127. doi: 10.1073/pnas.94.22.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 27.Toyota M, Ohe-Toyota M, Ahuja N, Issa JP. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proc Natl Acad Sci USA. 2000;97:710–715. doi: 10.1073/pnas.97.2.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:558–561. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- 29.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Rijnsoever M, Grieu F, Elsaleh H, Joseph D, Iacopetta B. Characterisation of colorectal cancers showing hypermethylation at multiple CpG islands. Gut. 2002;51:797–802. doi: 10.1136/gut.51.6.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wynter CV, Walsh MD, Higuchi T, Leggett BA, Young J, Jass JR. Methylation patterns define two types of hyperplastic polyp associated with colorectal cancer. Gut. 2004;53:573–580. doi: 10.1136/gut.2003.030841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whitehall VL, Walsh MD, Young J, Leggett BA, Jass JR. Methylation of O-6-methylguanine DNA methyltransferase characterizes a subset of colorectal cancer with low-level DNA microsatellite instability. Cancer Res. 2001;61:827–830. [PubMed] [Google Scholar]
- 33.Samowitz WS, Holden JA, Curtin K, Edwards SL, Walker AR, Lin HA, Robertson MA, Nichols MF, Gruenthal KM, Lynch BJ, et al. Inverse relationship between microsatellite instability and K-ras and p53 gene alterations in colon cancer. Am J Pathol. 2001;158:1517–1524. doi: 10.1016/S0002-9440(10)64102-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang JT, Chang KJ, Chen JC, Lee CC, Cheng YM, Hsu HC, Wu MS, Wang SM, Lin JT, Cheng AL. Hypermethylation of the p16 gene in sporadic T3N0M0 stage colorectal cancers: association with DNA replication error and shorter survival. Oncology. 1999;57:149–156. doi: 10.1159/000012023. [DOI] [PubMed] [Google Scholar]
- 35.Esteller M, González S, Risques RA, Marcuello E, Mangues R, Germà JR, Herman JG, Capellà G, Peinado MA. K-ras and p16 aberrations confer poor prognosis in human colorectal cancer. J Clin Oncol. 2001;19:299–304. doi: 10.1200/JCO.2001.19.2.299. [DOI] [PubMed] [Google Scholar]
- 36.Yi J, Wang ZW, Cang H, Chen YY, Zhao R, Yu BM, Tang XM. p16 gene methylation in colorectal cancers associated with Duke's staging. World J Gastroenterol. 2001;7:722–725. doi: 10.3748/wjg.v7.i5.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maeda K, Kawakami K, Ishida Y, Ishiguro K, Omura K, Watanabe G. Hypermethylation of the CDKN2A gene in colorectal cancer is associated with shorter survival. Oncol Rep. 2003;10:935–938. [PubMed] [Google Scholar]
- 38.Sanz-Casla MT, Maestro ML, Vidaurreta M, Maestro C, Arroyo M, Cerdán J. p16 Gene methylation in colorectal tumors: correlation with clinicopathological features and prognostic value. Dig Dis. 2005;23:151–155. doi: 10.1159/000088597. [DOI] [PubMed] [Google Scholar]
- 39.Nagasaka T, Sharp GB, Notohara K, Kambara T, Sasamoto H, Isozaki H, MacPhee DG, Jass JR, Tanaka N, Matsubara N. Hypermethylation of O6-methylguanine-DNA methyltransferase promoter may predict nonrecurrence after chemotherapy in colorectal cancer cases. Clin Cancer Res. 2003;9:5306–5312. [PubMed] [Google Scholar]