

Abstract
10-undecylenic acid (UA) is an OTC antifungal therapy and a nutritional supplement. It is an unsaturated medium chain fatty acid (MCFA) derivative, so our hypothesis was that its 11-mer sodium salt, uC11, would improve intestinal permeation similar to the established enhancer, sodium caprate (C10), but without the toxicity of the parent saturated MCFA, decylenic acid (C11). MTT assay and high-content screening (HCS) confirmed a cytotoxicity ranking in Caco-2 cells: C11 > C10 = uC11. Five to ten millimolars of the three agents reduced TEER and increased the Papp of [14C]-mannitol across Caco-2 monolayers and rat intestinal mucosae, a concentration that matched increases in plasma membrane permeability seen in HCS. Although C11 was the most efficacious enhancer in vitro, it damaged monolayers and tissue mucosae more than the other two agents at similar concentrations and exposure times and was therefore not pursued further. Rat jejunal and colonic in situ intestinal instillations of 100 mM C10 or uC11 with FITC-dextran 4000 (FD4) solutions yielded comparable regional enhancement ratios of ~10 and 30%, respectively, for each agent with acceptable tissue histology. Mini-tablets of uC11 and FD4 however delivered more FD4 compared to C10-FD-4 mini-tablets in both regions, as reflected by a statistically higher AUC, and with no evidence of membrane perturbation. The unsaturated bond in uC11 therefore confers a reduction in lipophilicity and cytotoxicity compared to C11, and the resulting permeation enhancement is on a par with or superior to that of C10, a key component of formulations in current phase II oral peptide clinical trials.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-014-9634-3) contains supplementary material, which is available to authorized users.
KEY WORDS: 10-undecylenic acid, Caco-2 cells, intestinal permeation enhancement, medium chain fatty acids, oral peptides
INTRODUCTION
Improving the intestinal permeation of peptides, proteins and macromolecules remains one of the great challenges in drug delivery. Enabling such oral formulations will improve patient compliance and enable parenteral-to-oral switching for commercial benefit, while the more physiological oral route may reduce potential side effects seen with bolus injection (1). In the past 5 years, major advances have occurred with at least 10 technologies reaching clinical trials for oral peptides (2). Common features of these advanced solid dose formulations are the following: pH-dependent polymer coatings that protect the tablet or capsule until it reaches the duodenum, incorporation of pancreatic peptidase inhibitors and co-formulation with permeation enhancers (PEs) with a history of safe use in man, some of which are listed as excipients. Examples of PEs that have reached the clinic in oral peptide formulations include a broad range of surfactants: carnitines, alkyl maltosides, sodium N-[8-(2-hydroxybenzoyl)amino]caprylate (SNAC), bile salts and medium chain fatty acids (MCFA) (reviewed in 3), while payloads cover molecular weights ranging from 1,000 to 7,000 Da: calcitonin, insulin, PTH (1–32), GLP-1 and octreotide. The most clinically advanced oral peptides are calcitonin (4) and octreotide (5), but these are atypical in that their high potency suggests that the very low oral bioavailabilities achieved may still be commercially viable and they are relatively inexpensive to synthesize compared to other peptides. Many peptides including insulin, however, will require minimum oral bioavailabilities of >15%, so unless oral peptide delivery systems are not to become a narrow commercial niche for one or two products, new formulation strategies are needed. One of the most clinically advanced intestinal PEs is sodium caprate (C10). It is currently a component of Merrion Pharmaceutical’s GIPET™ oral peptide formulation that is in phase II trials (6). C10 has a complex mechanism of action involving increases in epithelial paracellular transport in vitro across tricellular tight junctions (7) as well as transcellular epithelial drug transport due to surfactant detergent-like effects on plasma membranes at concentrations above its critical micellar concentration in vivo (8). It reversibly perturbs epithelial membranes at concentrations of the same order as those required to induce permeation enhancement in rat intestinal instillations (9), suggesting relatively narrow safety margins for repeat oral dosing regimens. This would suggest that new enhancers are needed where, if possible, permeation enhancement and mucosal damage can be more clearly dissociated across a wider dose range.
The 11-mer MCFA sodium salt, undecylenic acid (C11), has physicochemical properties similar to the 10-mer sodium salt, C10, suggesting a capacity for permeation enhancement. A separate mono-unsaturated species of C11 is the sodium salt of 10-undecylenic acid (uC11), which is derived from its parent acid (CH2 = CH(CH2)8COOH (UA)). ClonMedica (Ireland) markets two nonprescription topical antifungal products containing salts of 10-undecylenic acid (UA) as active ingredients: Caldesene® powder for the prevention and treatment of nappy rash and Desenex® powder and ointment for the treatment of athlete’s foot. Salts of UA also inhibit Candida albicans morphogenesis and are used to prevent denture stomatitis by reducing biofilm formation on denture liners (10,11). The salts have a history of oral ingestion in man, spanning over 60 years. They are currently used as a systemic antifungal agents, dosed orally either as the free fatty acid, typically in an oil-based soft gel capsule or as salt powders in a hard gelatin capsules for the alternative treatment of intestinal fungal dysbiosis and chronic rhinosinusitis. The adult daily dosage of UA is 450–750 mg administered in three divided doses (12,13). The oral LD50 of UA in rats was 2.5 g/kg (UA, Material Safety Data Sheet). Thorne Research (USA) supplies soft gel capsules of salts of UA in olive oil (Formula SF722®), as a nutritional supplement “to correct an imbalance in the microbial flora of the digestive tract”. The manufacturer’s recommended regimen is 250 mg UA, administered BID or TID. The extent of Formula SF722 ® use is not publicly available, but the product is described on the company website (14) as “one of our best selling products for over 20 years”, implying extensive human ingestion of UA. Despite debate over the efficacy of UA as a nutritional supplement, continued supply over many years would suggest that ingestion of UA and its salts is generally safe in man. The physicochemical properties of the sodium salt of UA, uC11, therefore lie between those of C10 and C11. The terminal alkene of uC11 reduces its hydrophobicity compared to the saturated C11 salt, with a resultant increase in solubility and critical micelle concentration (CMC) (15,16). UA and its salts have been used as penetration enhancers in transdermal drug delivery (17,18), and there have been attempts to improve oral delivery of testosterone using a UA ester (19,20); however, the use of either UA and/or uC11 specifically as intestinal permeation enhancers has never been investigated. uC11 is a powder, more amenable to commercial-scale solid dose manufacturing than the acid form and was therefore more appropriate for the current oral drug delivery study.
The aim of this study was to assess the efficacy of uC11 as an intestinal permeation enhancer in a range of in vitro and in vivo bioassays and to compare it against C10 and C11. We also used high-content multi-parametric analysis and tissue histology to compare its capacity to damage epithelia in vitro and in vivo, respectively. The data reveal that uC11 is a highly efficacious enhancer with a benign in vitro and in situ toxicity profile and suggest that this molecule can be added to the select range of agents with potential for clinical application as a PE.
MATERIALS AND METHODS
Materials
The acid of C11 was obtained from Sigma Aldrich (Ireland). UA was obtained from Chemos GmbH, (Germany). C11 and uC11 sodium salts were not commercially available and were synthesized by mixing the acids with NaOH at a 1:1 molar ratio in deionized water at 50°C. One percent isopropyl alcohol was included to reduce foaming on mixing. The resulting salts, uC11 and C11, were oven-dried overnight at 65°C to form powders, which were tested by differential scanning calorimetry and thermo-gravimetric analysis to confirm >95% purity (data not shown). Ninety-eight percent pure C10 was obtained from Fluka. Male Wistar rats (Charles River Laboratory, UK) weighing approximately 300 g were used for all animal studies. Caco-2 cells were obtained from the European Collection of Cell Cultures and used between passages 52 and 60.
Critical Micelle Concentration
High-resolution ultrasonic spectroscopy (HR-US) was used to measure the CMC of MCFA salt solutions in Ca2+-free Dulbecco's modified Eagle's medium (DMEM) (21). Ultrasonic wave velocity (υ (m/s)) was measured between 5 and 15 kHz using a HR-US 102 instrument (Ultrasonic Scientific Ltd., Ireland). It was equipped with two sample cells allowing for differential sample measurement to increase velocity measurement resolution (to 0.2 mm/s). MCFA stock solutions were prepared in degassed Ca2+-free DMEM. One millilitre degassed medium was added to the cell and the base υ value determined at 37°C. Stock MCFA solutions were then centrifuged (>10,000g) to ensure that they were completely degassed following preparation. υ was measured, while the sample was titrated by adding MCFA stock solution through a septum in the lid of the measuring cell using a manual Hamilton dispenser and 250 μl Hamilton syringe (Gastight, 1700 series). Each injection from the manual dispenser loaded 4.97 mg of solution into the measuring cell with stirring.
Fluxes Across Caco-2 Monolayers
Caco-2 cells were grown on Corning Costar polyester Transwell® filters (pore size, 0.4 μm; diameter, 12 mm) for 21–28 days in DMEM and incubated at 37°C with 5% CO2. [14C]-mannitol flux across differentiated Caco-2 monolayers and apparent permeability coefficients (Papp) were measured according to previous methods (22). The fluxes of fluorescein isothiocyanate (FITC)-dextrans (FD4, FD10, Sigma Ireland) were measured in the same way as mannitol fluxes with the exception that HBSS (Gibco®, Life Technologies™) supplemented with 12.5 mM d-glucose (Sigma) and buffered with 25 mM HEPES (Gibco®) was used instead of DMEM. To prevent MCFA precipitation, Ca2+-free HBSS was used in the apical compartment. Dextran solutions (5 mg/ml, ×10) were prepared in Ca2+-free HBSS on the day of use (final dextran concentration in apical compartment of 250 μg/ml). For [14C]-mannitol fluxes, a T0 apical sample of 50 μl was taken; all of the basolateral (1,500 μl) media was sampled throughout and completely replaced with fresh HBSS at each sampling point. Samples containing [14C]-mannitol were mixed with scintillation fluid and read in a scintillation counter (Packard Tricarb 2900 TR). FITC-containing samples were transferred onto a Nunc 96 well white Maxisorp® plate (Thermo Scientific). The T0 apical sample was diluted to 1:10 (20 μl in 180 μl HBSS) and then to 1:100 on the plate. The FITC signal intensity was measured (excitation/emission wavelengths of 490/525 nm, respectively) using a Spectra Max Gemini fluorescence intensity microplate reader (Molecular Devices, CA, USA). Papp values for FITC-dextrans were calculated in the same way as mannitol. Each treatment was added in duplicate to each plate and repeated on three separate occasions.
MTT Cytotoxicity Assay
Caco-2 cells were cultured using published methodology (22). The 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) assay was carried out on Caco-2 cells grown on 96 well plates according to previous descriptions (23), but with one modification. At 24 h, DMEM was replaced with Ca2+-free DMEM in order to avoid precipitation of MCFA salts (24). MCFA exposures were carried out for 1, 8 or 24 h. The IC50 was defined as the concentration of test compound required to reduce the absorbance of the MTT-formazan crystals by 50%.
High Content Analysis
In vitro high-content analysis (HCA) was developed to screen discovery candidates for potential hepatic drug toxicity in HepG2 cells (25) and consequently for intestinal cytotoxicity in Caco-2 cells (26). It was used here to study sublethal multiparameteric responses to MCFA exposure in Caco-2 cells. Cells were seeded in 200 μl DMEM per well in 96 well plates (Nunclon®, Thermo Scientific) at a density of 6,000 cells/well. After 24 h, medium was replaced with 300 μl Ca2+-free DMEM containing MCFAs and the plates incubated for 1, 8 and 24 h. One hundred fifty microlitres of medium was removed from each well and a dye mix (150 μl of ×2 stock in Ca2+-free medium) was then added to each well. It contained 0.8-μM Hoechst 33342 (measures cell number (CN), nuclear area (NA) and nuclear intensity (NI); 1 μM Fluo-4 AM (measures intracellular calcium (IC); 20 nM tetramethyl rhodamine methyl ester (TMRM) (measures mitochondrial membrane potential (MMP) and 1 μM TOTO®-3 iodide 642/660 (measures plasma membrane permeability (PMP)). Cells were incubated with the dyes for 50 min before positive controls were added for each parameter—100 μM carbonylcyanide-p-trifluoromethoxyphenylhydrazone (FCCP) (a mitochondrial membrane uncoupler), 20 μM ionomycin (a calcium ionophore) and 0.05% (v/v) Triton® X-100 (membrane perturbant). Positive controls were prepared at ×6 concentrations in ×1 concentration dye mix in Ca2+-free DMEM. Fifty microlitres medium was removed from each positive control well and replaced with 50 μl of the positive control agent. The plate was incubated for a further 10 min before image acquisition using an In-Cell® 1000 High Content Analyzer (GE Healthcare, UK) using multi-target analysis. The excitation/emission wavelengths used were Hoechst 33342: 360/460 nm, Fluo 4-AM: 480/535 nm, TMRM: 535/600 nm and Toto-3®: 620/700 nm. The Hoechst channel was used for cell counts and 10 random fields per well were imaged using a ×10 objective magnification. For each other parameter, eight random fields per well were imaged using a ×20 objective magnification. Exposure times were optimized for Hoechst (150 ms), Fluo-4 AM (500 ms), TMRM (600 ms) and TOTO®-3 iodide (200 ms).
Fluxes Across Isolated Rat Colonic Mucosae
Ussing chamber electrophysiology and flux studies using dissected isolated rat colonic mucosae were carried out according to our most recent description (27) and in accordance with the UCD Animal Research Ethics committee policy on use of animal postmortem tissue. Sheets of intestinal mucosa with underlying lamina propria were mounted in Ussing chambers (WPI, UK) with a circular window surface area of 0.63 cm2. Tissues were bathed bilaterally with 5 ml Krebs-Henseleit buffer (KH) and were continuously gassed with 95% O2/5% CO2 at 37°C. Ca2+ was omitted from KH in the apical chamber in order to avoid MCFA precipitation. The potential difference (PD, (mV)) across the tissue was measured in the open circuit configuration using Ag/AgCl electrodes attached to an automated voltage clamp (EVC 4000, WPI, UK) before being clamped at zero PD to permit recording of short circuit current (Isc, (μA cm−2)). Colonic mucosae with TEER values below 70 Ω cm−2 were excluded to ensure only fully functional tissues were used (28). In addition to monitoring TEER, Papp values for [14C]-mannitol (0.2 μCiml−1) and FD-4 (250 μgml−1) across tissue segments were calculated as for Caco-2 studies. Apical-to-basolateral flux of each paracellular marker was determined by basolateral side sampling (200 μl) at time zero (T0) and every 20 min for 2 h, and the Papp was calculated as above. Volumes were replaced with fresh KH at each sampling point. A T0 apical side sample (100 μl for [14C]-mannitol and 20 μl for FITC-dextrans) was also taken. Capacity to generate an inward electrogenic chloride secretory response to basolateral application of the muscarinic agonist, carbachol (CCh), was used to confirm tissue function following MCFA exposure at 120 min.
Mini-Tablet Production and Characterization
Mini-tablets containing C10, C11 and uC11 salts were formulated with FD4. Microcrystalline cellulose (Avicel® PH102, FMC Biopolymer, UK) was used as a tablet filler and disintegrant. All components were sieved through 150–425 μm screens prior to use. Powder components were weighed directly into a 30 ml skirted tube and mixed by inversion to give a total blend weight of 3 g for ~60 tablets. Each 50-mg tablet contained 20% w/w FD-4; MCFA-containing tablets in addition had 60% w/w C10 or uC11 (i.e. 30 mg) and 20% Avicel®, while control tablets had 80% Avicel® and no MCFA. Tablets were produced using a 3.8-mm round concave multiple-tip tool (Natoli Engineering, MO, USA) on a manual single-station hydraulic press with compression force gauge (MTCM 1, LobePharma, NJ, USA). Tablet powder blend was hand-filled into the die and a compression force of 500 psi applied for each mini-tablet formulation. Tablet hardness was determined using a tablet hardness tester (Pharmatron, Switzerland). An arbitrary method was used to determine relative disintegration times of different mini-tablet formulations. Disintegration time was determined by placing a mini-tablet into an Eppendorf tube containing 1 ml Medium 199 (Sigma, Ireland) at 37°C to ensure that MCFA dissolution was not solubility-limited. The tube was closed, inverted and placed on a plate incubator (37°C) with rotation speed set at 200 rpm. Disintegration time was defined by visual observation as the time taken for complete disintegration of the mini-tablet without any remnants of a core. Tablet content uniformity was assessed by measuring the uniformity of FD4 in tablets from each tablet batch produced. Tablets were dissolved in 10 ml deionized H2O, giving a theoretical content of 1 mg ml−1 FD4. Triplicate samples of the dissolved tablet were then diluted 1:10, yielding a theoretical FD4 concentration of 2 μg ml−1, which was measured as above . Mini-tablet content uniformity, hardness and disintegration time are shown in Suppl. Table 1. The CMC of the C10, C11 and uC11 in calcium-free buffer were obtained as described above (21) and were 26, 6.2 and 17.6 mM, respectively.
In Situ Intestinal Instillation Studies
Procedures were performed under licence B100/4433 from the Irish Department of Health and with the approval of the UCD Animal Research Ethics Committee. Animals were housed under controlled conditions of temperature and humidity with a 12:12 h light/dark cycle. Rats received filtered water and standard laboratory chow ad lib up to 48 h prior to the instillation procedure at which point water was supplemented with 5% (w/v) glucose and animals fasted 16–20 h prior to the procedure with free access to glucose water. Isoflurane-anaesthetized rats were placed on a heat pad, and in situ instillations were performed as previously described (9) with minor modifications. Following midline laparotomy either the upper jejunum or proximal colon was identified and tied off proximally with a size 4 braided silk suture. Following removal of faecal matter, another suture tie was made 5 cm distally from the first tie to create a loop. Treatment solutions were injected into lumens using a 1-ml syringe with 30G needle. Each MCFA was tested at a concentration of 100 mM (equivalent to 27 mg kg−1 C10 and 29 mg kg−1 uC11) and co-administered with FD4 (40 mg kg−1). Solutions were prepared in tissue culture grade PBS without calcium and magnesium. The volume of MCFA/FD4 solution was based on body weight and ranged from 368 to 500 μl per loop. To calculate absolute bioavailability (BA), FD4 (40 mg kg−1) was administered via the tail vain of some rats.
For jejunal mini-tablet instillations in either jejunum or colon, a small incision was made between the vasculature approximately 6.5–7 cm distally from the proximal suture tie to allow the tablet placement using a fine tweezers. A second distal tie proximal to the incision was then made once the mini-tablet was in place. For colonic tablet instillation, it was inserted using a fine tweezers through the same incision initially made to remove faecal matter. Three hundred microlitres PBS was subsequently instilled to aid tablet disintegration and dissolution. The abdomen was closed by suturing the epidermal layer using Mersilk W505 silk sutures with a size 4 cutting needle. Blood samples were taken by retro-orbital bleeds at 0, 5, 15, 30, 45, 60, 90, 120, 150 and 180 min into 0.5-ml Eppendorf tubes and stored at 2–8°C for a minimum of 60 min prior to centrifugation (6,500g, 10 min) and serum collection. Serum was stored at −20°C until analysed. Animals were euthanised at the end of the experiment with intracardiac injection of 0.5 mL pentobarbital sodium (EUTHATALTM, Merial Animal Health Ltd., UK). Based on an average rat weight of 330 g, each mini-tablet represented the administration of 30 mg kg−1 FD4 and 90 mg kg−1 MCFA enhancer.
Rat Serum FD4 Assay
Serum FD4 levels were quantified as previously described (9). Thawed serum was diluted 1:10 in borate buffer on a Nunclon® 96 well white Maxisorp plate. The FD4 standard curve samples included on each plate were prepared in borate buffer and stored at −20°C with the serum samples until required such that the FD4 in the standard curve also underwent a freeze-thaw cycle. The FITC signal intensity was measured as above. Pharmacokinetic (PK) characteristics from serum data were calculated using WinNonLin® software (Pharsight Inc., USA). Non-compartmental analysis was used with the peak concentration (Cmax) and the time taken to reach Cmax (Tmax) calculated from the serum concentration profiles. The area under the serum-concentration curve, AUC0–3 h, was calculated using the linear trapezoidal rule. The BA of FD4 over the 3-h intestinal instillation was calculated using the following:
where AUC0–3 h is the area under the plasma concentration curve over the 3-h instillation period and AUCi.v.∞ is the area under the plasma concentration versus time (0-∞) after i.v. administration of 40 mg kg−1 FD-4 (0.01 mM).
Tissue Histology
Intestinal issue samples for morphological examination following treatments were fixed for 24 h in 10% (w/v) formalin and subsequently embedded in paraffin wax. Tissues from Ussing chambers or instillations were incubated in formalin. Five-micromolar tissue sections were cut on a microtome (Leitz 1512; GMI, USA), mounted on adhesive coated slides, stained with haematoxylin/eosin (H&E) and examined under light microscopy (Nikon Labphoto; Nikon, Japan).
Statistical Analysis
Results from Ussing chamber and instillation studies were analysed by one-way ANOVA with Bonferonni’s ad hoc posttest. HCA was conducted on three independent plates with two treatment replicate wells per plate. All data was normalized to the untreated control, and each treatment time was analysed by one-way ANOVA with Dunnett’s ad hoc posttest. All data is presented as the mean ± SEM with minimum statistical significance set at p < 0.05.
RESULTS
uC11 Permeation Enhancement Across Caco-2 Cell Monolayers
Effects of uC11, C10 and C11 on permeability across monolayers were determined by measuring effects on TEER and the Papp values of flux markers. Minimum concentrations of 3, 8.5 and 10 mM of each of C11, C10 and uC11 induced a statistical reduction in TEER to below 20% of initial value (T0) within 30 min (Fig. 1a, b). This data suggests that TEER is most sensitive to C11, but effective concentrations were of the same order for the three MCFAs. In parallel, there were significant concentration-dependent increases in the Papp of [14C]-mannitol, FD-4 and FD-10 in the presence of C11 and uC11 (Fig. 2a, b). These concentrations mapped to those that caused TEER reductions. Similar increases in Papp were seen for each marker at 5 mM C11 and uC11 and also for 10 mM C10. These data suggest that the potency and efficacy order for inducing permeability across Caco-2 monolayers is similar for the three agents.
Fig. 1.
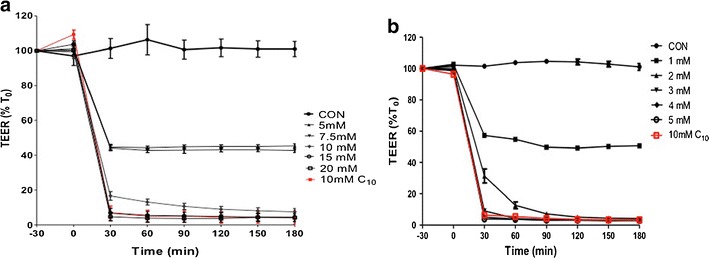
a uC11 and b C11 reduced TEER across Caco-2 monolayers. 10–15 mM uC11 and 3 mM C11 caused a reduction in TEER comparable to 10 mM C10 (red). N = 3
Fig. 2.
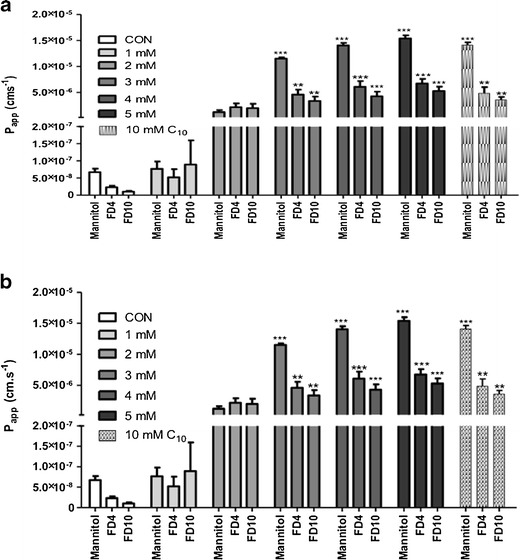
a uC11 and b C11 increased the Papp values obtained over 2 h for [14C]-mannitol, FD-4 and FD-10 across Caco-2 monolayers. C10 data was included for comparison (10 mM). *p < 0.05; **p < 0.01; ***p < 0.001 versus untreated controls. N = 3
Cytotoxicity Assessment of uC11: MTT and HCA
MTT assay results for uC11, C11 and C10 on Caco-2 intestinal epithelial cells over time are shown in Fig. 3a–c. Following 24 h exposure, the IC50 was ~5 mM for both uC11 and C10, whereas it was less than 2.5 mM for C11. Exposure for 1, 8 and 24 h revealed that cells were equally resilient to both 5 mM C10 and uC11 but less resilient to C11. Just 20% loss in viability at 5 mM uC11 and C10 occurred at 8 h, whereas 5 mM C11 killed 20% of cells after only 60 min and 50% at 8 h. C11 was therefore considerably more cytotoxic than both uC11 and C10 in the MTT assay, whereas the latter two agents were equivalent and not lethal even at very high concentrations and exposure times. HCA was then used to examine multiple contemporaneous cellular effects of uC11 and to relate them to efficacy as a permeability enhancer. Initial Caco-2 cell responses to uC11 (1–5 mM, 60 min) were an increase in nuclear intensity (NI) and intracellular Ca2+ concentration (IC) accompanied by mitochondrial hyperpolarization (Fig. 4a–f). Maintenance of high IC preceded a reduction in mitochondrial membrane potential (MMP) and an increase in plasma membrane permeability (PMP) and nuclear fragmentation from 5 to 20 mM, which ultimately lead to death. Importantly, PMP changes seen at 60 min with 8.5 mM uC11 correlated with increased Papp values across monolayers seen at similar concentrations and suggest a mild surfactant effect on the plasma membrane. Similarly, HCA also revealed that 60 min exposure to 8.5 mM C10 and 2.5 mM C11 resulted in significant increases in PMP, confirming the similarity between the concentrations of MCFAs that cause PMP increases in Caco-2 cells and those that increase fluxes across monolayers (data not shown). Of note is that the initial IC increase in response to uC11 occurred at concentrations lower than those that induced PMP increases, and this implicates calcium regulation of tight junction proteins and paracellular transport as an earlier step than perturbation. Most changes in parameters occurred at concentrations of uC11 and exposure times below those that affected viability according to MTT assay. After 8 h exposure to concentrations >5 mM uC11, all parameters were significantly altered as cells began the apoptotic process. The cell number changes are a balance between cell division with cell death and give a more accurate assessment than the MTT snapshot.
Fig. 3.
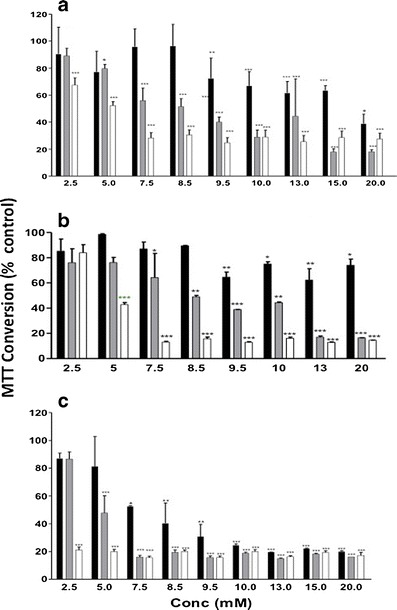
MTT assay on Caco-2 cells over time. a uC11, b C10 and c C11. black bar 1 h, gray bar 8 h and white bar 24 h. *p < 0.05; **p < 0.01; ***p < 0.001 versus medium controls. N = 3 independent plates, each in duplicate
Fig. 4.
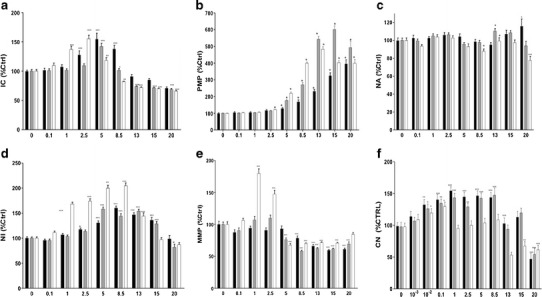
HCA of uC11 on Caco-2 cells exposed for black bar 1 h, gray bar 8 h and white bar 24 h. a intracellular calcium (IC), b plasma membrane potential (PMP), c nuclear area (NA), d nuclear intensity (NI), e mitochondrial membrane potential (MMP) and f cell number (CN)
uC11: Fluxes Across Isolated Rat Colonic Mucosae
uC11 displayed equal capacity as C11 for TEER reduction and Papp increases of [14C]-mannitol and FD-4 (Fig. 5). C11 produced significant increases in mannitol Papp at lower concentrations than uC11. A 5-fold increase in [14C]-mannitol Papp was obtained following exposure to 5 mM C11, whereas 10 mM C10 and uC11 were required to induce similar increases in the [14C]-mannitol Papp, however maximal effects were seen at 20 mM uC11 and C11. For FD4 fluxes, 5 mM C11 was sufficient to induce a significant Papp increase compared to the minimum concentrations of 10 mM C10 and uC11 required, but 20 mM C11 was more efficacious than the same concentration of C10. Histological assessment revealed that 10 mM concentrations of MCFA that increased Papp across colonic tissue also caused some mucosal perturbation compared to controls, (Fig. 6a–d), which is consistent with the relationship between Caco-2 cell PMP and Papp increases following exposures. Notably, C11 caused more damage compared to uC11 and C10. The results obtained on rat colonic mucosae therefore further supported of potential of uC11 as an intestinal permeation PE with similar characteristics to C10 and C11, but the latter is more toxic by several in vitro criteria. Despite the damage caused by all agents, mucosae retained secretory ion transport function since Isc responses to CCh were still substantial for each MCFA (Fig. 6e).
Fig. 5.
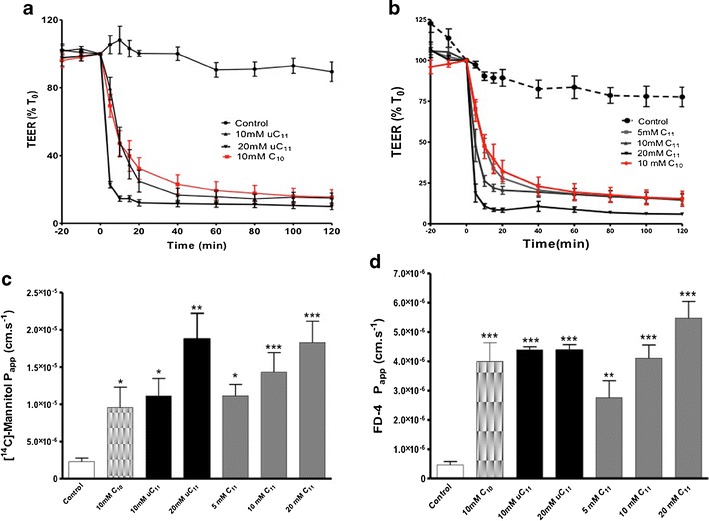
TEER and Papp changes across isolated rat colonic mucosae following MCFA exposure. a TEER: uC11, b TEER: C11, c Papp [14C]-mannitol and d Papp FD-4. Ten mM C10 data is shown for comparison as a positive control. N = 3–6 per group. *p < 0.05; **p < 0.01: ***p < 0.001 versus controls
Fig. 6.
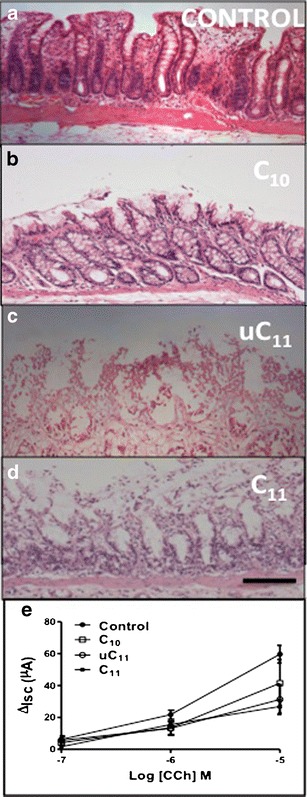
H & E of rat colonic mucosae following 2 h of incubations in chambers. a untreated control, b 10 mM C10, c 10 mM uC11 and d 10 mM C11. Bar = 100 μm. e ΔIsc to CCh in mucosae exposed to MCFAs (10 mM, n = 3)
Rat Intestinal Instillations With MCFA Solutions
C11 was omitted from in vivo study due to its high cytotoxicity. In vivo instillation of FD4 (40 mg kg−1) in solution containing 100 mM uC11 or C10 for 3 h increased FD4 serum concentration in jejunum and colon (Fig. 7a–d). The selection of such a high dose was warranted owing to previous data showing a requirement for 5–10-fold higher doses of C10in vivo compared to in vitro concentrations (9). The possible reasons for such a high dose are many: a more robust epithelium in vivo, dilution, intestinal absorption of the enhancer, and difficulties in estimating free concentrations above the CMC when MCFA vesicles are present. PK data for both agents is summarized (Table I). The similarity in the BA increase achieved by uC11 in both jejunum and colon is further in vivo evidence of its potential application as an intestinal PE. The BA of FD4 following control solution instillation was higher from jejunum (2.1%) compared to the colon (1.7%). BA from colon was more than double that from the jejunum when co-instilled with either MCFA. Enhancement ratios (ER’s) of 10.5 and 10.2 were achieved using C10 and uC11, respectively, in the jejunum, increasing to 28.9 and 29.2 in colon, representing BA values of almost 22 and 50%, respectively, for both agents (Table I). H&E is shown for colonic sections and did not reveal evidence of damage following 3-h instillations with uC11 or C10 (Fig. 7e–g).
Fig. 7.
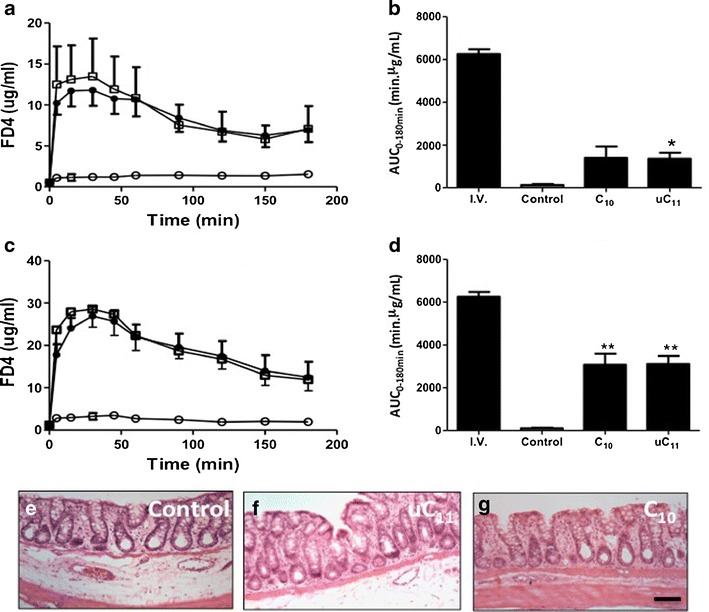
a Serum FD-4 (jejunum), b FD4 AUC (jejunum), c serum FD-4 (colon) and d FD4 AUC (colon) following 3 h of instillations of FD-4 (40 mg kg−1) solutions with or without 100 mM uC11 or C10. white circle control, filled circle uC11,and white square C10. N = 6 per group. *p < 0.02; **p < 0.001. H&E stained sections of rat colon following 3 h of instillations of e FD4 control, f 100 mM uC11 and g 100 mM C10. Bar = 50 μm
Table I.
PK of FD-4 (40 mg kg−1) Solutions in Instillations with 100 mM uC11 or C10
| I.V. | Control—jejunum | C10—jejunum | uC11—jejunum | |
| C max (μg ml−1) | 123.9 ± 22.2 | 3.2 ± 1.2 | 14.1 ± 11.1 | 12.3 ± 4.8 |
| T max (min) | 5.0 ± 0.1 | 30.0 ± 15.0 | 19.2 ± 15.6 | 30.8 ± 17.4 |
| AUC0–180 | 6,250 ± 462 | 134 ± 81 | 1,399 ± 1,311 | 1,360 ± 680 |
| BA (%) | – | 2.1 | 22.4 | 21.8 |
| Enhancement ratio | – | 1 | 10.5 | 10.2 |
| Control—colon | C10—colon | uC11—colon | ||
| C max (μg ml−1) | 2.7 ± 1.2 | 31.3 ± 13.0 | 30.1 ± 5.7 | |
| T max (min) | 60 ± 53 | 24 ± 11 | 38 ± 28 | |
| AUC0–180 | 107 ± 62 | 3,078 ± 1,445 | 3,111 ± 924 | |
| BA (%) | 1.7 | 49.2 | 49.8 | |
| Enhancement ratio | 1 | 28.9 | 29.2 |
Rat Intestinal Instillations With MCFA Mini-Tablets
FD-4/uC11 and FD-4/C10 mini-tablets were instilled to rat jejunum and colon at a dose level of 120 mg kg−1 (100 mM) and the data is shown in Fig. 8 and Table II. In jejunum, C10 tablets surprisingly did not alter FD4 BA, whereas a significant increase was obtained from uC11 tablets and there was less variability from the uC11 tablet compared to C10. Similar to solution studies, basal FD4 absorption from control tablets was higher in jejunum compared to colon. uC11 and C10 mini-tablet formulations were effective in enhancing FD4 absorption across colon, but uC11 tablets increased FD4 absorption across both jejunum and colon to a greater extent than C10 tablets (Fig. 8c, d). H&E-stained colonic sections following exposure to equal concentrations of C10 and uC11 in tablet instillations did not reveal mucosal perturbation following exposure to uC11 or C10 compared to control (Fig. 8d–f). Importantly, the aqueous solubility was somewhat lower for uC11 (155 mg ml−1 or 0.75 M) compared to C10 (385 mg ml−1 or 1.98 M), resulting in disintegration times of 34.3 and 26.4 min, respectively (Suppl. Table 1). The lower aqueous solubility may have resulted in a slower colonic release of uC11, prolonging mucosal perturbation and increasing FD4 absorption. This could explain the longer Tmax (84 min) in the colon for the uC11/FD-4 tablet compared to the C10/FD-4 tablet (45 min) (Table II).
Fig. 8.
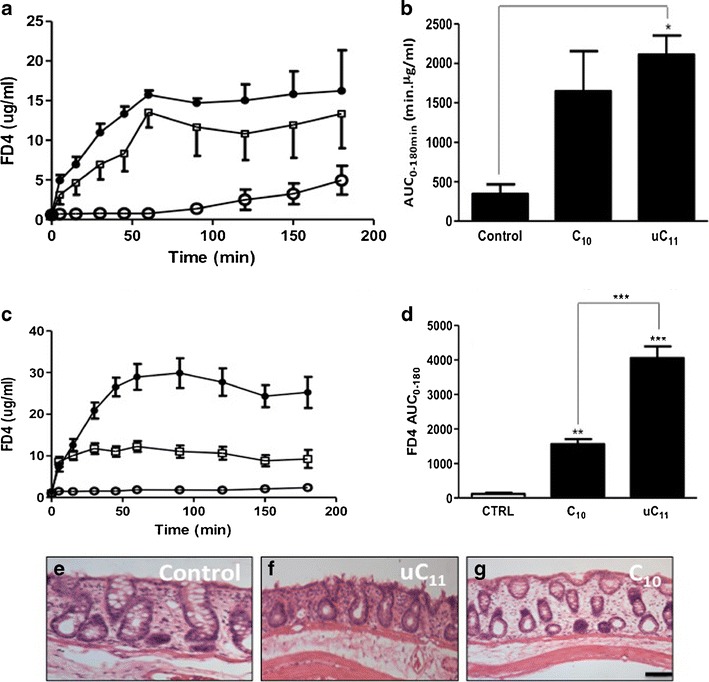
a Serum FD-4 (jejunum), b FD4 AUC (jejunum), c serum FD-4 (colon) and d FD4 AUC (colon) following 3 h of instillations of FD-4 (40 mg kg−1) mini-tablets with or without 100 mM uC11 or C10, each at 60% w/w white circle control, filled circle uC11 and white square C10. N = 6 per group. *p < 0.05; **p < 0.01; ***p < 0.001. H&E stained sections of rat colon following 3 h of instillations of e FD4 control, f 100 mM uC11 and g 100 mM C10. Bar = 50 μm
Table II.
PK of FD-4/uC11 or FD-4/C10 Mini-Tablets in Instillations
| Control tablets | FD-4/C10 | FD-4/uC11 | |
|---|---|---|---|
| Jejunum | Jejunum | Jejunum | |
| C max (μg ml−1) | 5.0 ± 3.2 | 16.6 ± 6.6 | 20.4 ± 10.2 |
| T max (min) | 180 ± 0 | 132 ± 66 | 78 ± 51 |
| AUC0–180 | 343 ± 213 | 1,645 ± 1,135 | 2,072 ± 651 |
| BA (%) | 6.7 | 26.3 | 40.2 |
| Enhancement ratio | 1 | 4.8 | 6.0 |
| Colon | Colon | Colon | |
| C max (μg ml−1) | 2.5 ± 1.0 | 12.4 ± 2.5 | 32.2 ± 7.0 |
| T max (min) | 105 ± 57 | 45 ± 21 | 84 ± 25 |
| AUC0–180 | 115 ± 64 | 1,562 ± 281 | 4,213 ± 767 |
| BA (%) | 2.2 | 30.3 | 67.4 |
| Enhancement ratio | 1 | 13.6 | 36.6 |
DISCUSSION
uC11 was identified as a new intestinal PE with the advantage of having an established history of safe oral ingestion in man due to its use for over 20 years as a nutritional supplement and alternative medicine at doses in excess of 500 mg per day. The unsaturated carbon double bond in uC11 reduces lipophilicity, increases the CMC and solubility relative to the saturated form, C11. Thus, the physicochemical properties of uC11 lie between those of the C10 and C11 as indicated by a CMC value of 17.6 mM compared to values of 26 and 6.2 mM, respectively, and this is likely to be the key factor contributing to the balance between efficacy as a PE and cytotoxicity.
C10 is advanced in the clinic as a major component of the GIPET™ solid dose formulation for oral peptides being developed by Merrion Pharmaceuticals (Ireland) (6). Still, while C10 is an approved food additive without a recommended maximum limit of ingestion, it is still not yet incorporated into an approved oral therapeutic dosage form nor are humans normally exposed to the high dose levels required in oral peptide clinical trials compared to those in food and drink. Of relevance is that similar to C10, uC11 is a solid at room temperature and is suitable for commercial-scale solid dose pharmaceutical manufacturing. Initial assessment of C10 and uC11 by HCA in Caco-2 cells indicated that both MCFAs have very similar sublethal cytotoxicity profiles, and this tallied with the MTT assay revealing a high IC50 for C10 and uC11 even after 24 h. In contrast, C11 was more cytotoxic than the other two molecules in HCA and MTT assays, where 50% of cells were killed with 5 mM concentrations for 8 h compared to 20% for uC11. Taken together with the more damaging histology seen in isolated intestinal mucosae following exposure to C11 compared to uC11, such toxicity potential ruled out further study of C11 as a PE due to its likely overly robust surfactant effects.
The intestinal PE capacity of uC11 was similar to C10 across several in vitro and in vivo intestinal models. Initial studies demonstrated uC11’s capacity to increase mannitol and FD fluxes in the MW range of 0.182–40 kDa across Caco-2 monolayers. However, despite demonstration by HCA of a similar capacity of the two agents to increase Caco-2 plasma membrane permeability in HCA assays, flux data suggested that uC11 has a lower capacity to increase mannitol flux than C10, with an EC50 (concentration to increase Papp by 50%) ~1.7-fold higher. Surprisingly, FD flux studies across monolayers demonstrated that the capacity of uC11 to increase relative flux was proportional to the MW of the ad-mixed FD, i.e. the EC50 values decreased with increasing MW (R2 = 0.99). This is understood, however, in the context of a reduced basal Papp across monolayers with increased flux marker MW (29). Solution studies on rat colonic tissue mounted in Ussing chambers and in vivo instillations to rat jejunum and colon produced additional evidence of uC11’s intestinal PE efficacy, but importantly, compared to Caco-2 monolayers, its efficacy in these models was equal to C10.
Instillations of solutions of uC11 and C10 to colonic loops yielded >2-fold increase in FD4 relative BA compared to jejunal, but again, this is in the context of a higher basal FD4 BA across jejunal loops compared to colonic, probably due to the larger microvilli surface area. The regional difference in MCFA enhancement action may also be due to differences in epithelial membrane structure between the upper and lower GI tract, with the apical membrane of the small intestinal epithelia designed to better withstand surfactant perturbation than the colon due to periodic jejunal exposure to 30 mM bile salts in the fed state (30). Increased PE effects of C10 and other surfactants in the colon compared to small intestine has been widely reported (31–33). If a formulation containing surfactant-type PEs can be designed to reach the colon, it may have potential for significant systemic delivery of normally impermeable payloads. Concerns over colonic co-absorption of bystander pathogens, including bacteria, bacterial toxins and viruses in the presence of PEs have thus far not been vindicated, but studies have been largely restricted to in vitro models (34). Of interest is that similar C10 concentrations required for PE in vivo inhibited adhesion of Salmonella typhimurium in isolated rat intestinal mucosae and indeed were bactericidal against S. typhimurium and several gram-positive and -negative bacteria (35). Overall, it would be preferable for MCFAs to have no effect on the intestinal microbiota, either toxic, innocuous or probiotic species.
uC11 and C10 were incorporated into mini-tablets to assess if a solid dosage form could enable permeation enhancement. MCFA lipophilicity and capacity to perturb biological membranes increases with alkyl chain length, and this is associated with reduced aqueous solubility, which prolongs the time required for the MCFA dissolve out from tablets. This means that there is greater intestinal absorption MCFA from solutions than tablets in vivo and therefore that assumptions cannot be made on designating one PE as superior to another on the basis of ad-mixed solutions. Since increasing the alkyl chain length results in a lower CMC, as was experimentally detected, it will also reduce the maximum free monomer concentration in solution before forming micelles. Importantly, it is the monomer that is thought to be the species that perturbs membranes (15). Adsorption to epithelial membranes also reduces monomer concentration. Thus, MCFA concentration in the GI lumen from a tablet formulation is dissolution-rate limited. A high level of MCFA (60% w/w) was used in the mini-tablet formulation to ensure sufficient local concentrations were achieved in vivo in the intestinal lumen to perturb mucosal surfaces. This high MCFA concentration in the tablet imparts a low surface porosity. The use of Avicel®, an insoluble microcrystalline cellulose binder, ensured that mini-tablet disintegration was predominantly controlled by the intrinsic dissolution rate of the MCFA, allowing for subsequent study of its effect on permeation enhancement. Tablets therefore disintegrated by dissolution and erosion of MCFA at their surface, forming a matrix by which MCFA and FD4 release were subsequently controlled by MCFA dissolution.
Comparing C10 solution and tablet colonic instillations suggested approximately 60% relative BA from the tablet formulation compared to solution. PE from the tablets may be due to altered fluid osmolality and mucosal dehydration in addition to surfactant action. It should be noted that the dehydration of mucosal surfaces may be more pronounced in rat loop studies than in Ussing chambers. The average diameters of the rat jejunal and colonic lumen are 4–5 mm and 3–10 mm, respectively, approximately one tenth that of human (36). Instillation of a 3.8-mm mini-tablet to rat intestinal region segments permits intimate contact of hygroscopic tablet components with the entire circumference of the mucosal surface. Dissolution of the 50 mg powder components also alters fluid osmolarity and the effect of hyper-osmolarity on C10 interaction with the rectal GI epithelium has also been demonstrated in a suppository of ampicillin (37). Overall, data generated suggests that uC11 may be able to strike the dissolution-perturbation balance in a tablet formulation more effectively than C10 since the aqueous solubility and CMC of uC11 lies between that of C10 and C11. Intestinal instillations revealed major differences between C10 and uC11 mini-tablet capacities to increase FD4 BA: the C10 formulation did not increase FD4 serum levels following jejunal instillation, whereas uC11 mini-tablets did. The greatest difference between the two MCFAs was found in the colon, and may be due to the prolonged perturbation resulting from slower dissolution of uC11, leading to its superior efficacy. The uC11 mini-tablets also had a much longer Tmax (84 min) in the colon compared to C10 (45 min). Still, a higher level of initial mucosal perturbation induced by uC11 compared to C10 cannot be ruled out as the cause of increased FD4 bioavailability. The novel application of uC11 as a jejunal and colonic permeation enhancer suitable for a solid dosage form warrants further investigation.
The solution and mini-tablet studies using MCFAs on colonic loops demonstrated a concomitant increase in drug flux associated with mucosal perturbation. This was also observed by other investigators studying amphiphilic PEs (38) and was a likely finding consistent with HCA assessment. While isolated mucosae from Ussing chamber studies showed evidence of irreversible epithelial perturbation following 2-h exposure to 10 mM uC11 and C10, this was not observed in colonic instillations following 3-h exposure to 100 mM of both agents, in line with other instillation studies with surfactant challenge (39,40). This would suggest that a degree of membrane damage is inevitable with all effective PEs: screening should be on assessing whether adequate efficacy is achieved with an acceptable degree of reversible perturbation in concentration ranges that can be formulated in solid dosage forms for co-release with payload. This study confirms that this is the case for uC11 and C10, but not C11. In conclusion, an alternative efficacious PE to C10 has been discovered which may have some advantages over it, including its history of oral use in man with alternative medicines. Further studies with peptide cargoes (as against marker molecules) will elucidate the potential of uC11 in solid dose formulations and in nanoparticle and conjugated formats. As to the issue of separating efficacy and toxicity of MCFAs, human studies to date with C10 suggest that efficacy for this class can be achieved without significant reports of toxicity, likely due to rapid epithelial restoration (6,8). This does not address repeat dose administration risks nor does it reveal in depth analysis of the potential for lipopolysaccharide fragments to permeate compromised intestine to stimulate production of Type 1 cytokines (41). In vitro toxicity studies encompassing tissue histology, cytotoxicity assays and HCA in parallel with permeability assays will allow a mechanistic basis for rank ordering within and between classes of PE in respect of selection for in vivo study.
Electronic Supplementary Material
(DOC 29 kb)
ACKNOWLEDGMENTS
This work was co-funded by Science Foundation Ireland Strategic Research Cluster grant 07/SRC/B1154 and an industry partnership award to EW from the Irish Research Council with sponsorship from Merrion Pharmaceuticals, Ireland. We thank Margaret Coady for assistance with histopathology.
REFERENCES
- 1.Pinto-Reis C, Silva C, Martinho N, Rosado C. Drug carriers for oral delivery of peptides and proteins: accomplishments and future perspectives. Ther Deliv. 2013;4:251–265. doi: 10.4155/tde.12.143. [DOI] [PubMed] [Google Scholar]
- 2.Maher S, Brayden DJ. Overcoming poor permeability: translating intestinal permeation enhancers for oral peptide delivery. Drug Discov Today Technol. 2012;9:e113–e119. doi: 10.1016/j.ddtec.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 3.Aungst BJ. Intestinal permeation enhancers. AAPS J. 2012;14:10–18. doi: 10.1208/s12248-011-9307-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Binkley N, Bolognese M, Sidorowicz-Bialynicka A, Vally T, Trout R, Miller C, et al. A phase 3 trial of the efficacy and safety of oral recombinant calcitonin: the oral calcitonin in postmenopausal osteoporosis (ORACAL) trial. J Bone Miner Res. 2012;27:1821–1829. doi: 10.1002/jbmr.1602. [DOI] [PubMed] [Google Scholar]
- 5.Tuvia S, Atsmon J, Teichman SL, Katz S, Salama P, Pelled D, et al. Oral octreotide absorption in human subjects: comparable pharmacokinetics to parenteral octreotide and effective growth hormone suppression. J Clin Endocrinol Metab. 2012;97:2362–2369. doi: 10.1210/jc.2012-1179. [DOI] [PubMed] [Google Scholar]
- 6.Walsh E, Adamczyk B, Chalasani KB, Maher M, O’Toole EB, Fox J, et al. Oral delivery of macromolecules: rationale underpinning Gastrointestinal Permeation Enhancement Technology (GIPET®) Ther Deliv. 2011;2:1595–1610. doi: 10.4155/tde.11.132. [DOI] [PubMed] [Google Scholar]
- 7.Krug SM, Amasheh M, Dittman I, Christoffel I, Fromm M, Amasheh S. Sodium caprate as enhancer of macromolecule permeation across tricellular tight junctions of intestinal cells. Biomaterials. 2013;34:275–282. doi: 10.1016/j.biomaterials.2012.09.051. [DOI] [PubMed] [Google Scholar]
- 8.Maher S, Leonard TW, Jacobsen J, Brayden DJ. Safety and efficacy of sodium caprate in promoting oral drug absorption: from in vitro to the clinic. Adv Drug Deliv Rev. 2009;61:1427–1449. doi: 10.1016/j.addr.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 9.Wang X, Maher S, Brayden DJ. Restoration of rat colonic epithelium after in situ intestinal instillation of the absorption promoter, sodium caprate. Ther Deliv. 2010;1:75–82. doi: 10.4155/tde.10.5. [DOI] [PubMed] [Google Scholar]
- 10.Mclain N, Ascanio R, Baker C, Strohaver RA, Dolan JW. Undecylenic acid inhibits morphogenesis of Candida albicans. Antimicrob Agents Chemother. 2000;44:2873–2875. doi: 10.1128/AAC.44.10.2873-2875.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonçalves LM, Del Bel Cury AA, Sartoratto A, Garcia Rehder VL, Silva WJ. Effects of undecylenic acid released from denture liner on Candida biofilms. J Dent Res. 2012;91:985–989. doi: 10.1177/0022034512458689. [DOI] [PubMed] [Google Scholar]
- 12.Helms S, Miller A. Natural treatment of chronic rhinosinusitis. Altern Med Rev. 2006;11:196–207. [PubMed] [Google Scholar]
- 13.Undecylenic acid. Monograph Altern Med Rev. 2002; 7:68–70. http://www.altmedrev.com/publications/7/1/68.pdf. Accessed 16 June 2014. [PubMed]
- 14.Thorne Research. http://www.drvita.com/product/thorne-research-formula-sf722-250-fish-gelatin-gelcaps/8835. Accessed 7 October 2013.
- 15.Helenius A, Simons K. Solubilization of membranes by detergents. Biochim Biophys. Acta Rev Biomembr. 1975;415:29–79. doi: 10.1016/0304-4157(75)90016-7. [DOI] [PubMed] [Google Scholar]
- 16.Lichtenberg D, Robson RJ, Dennis EA. Solubilization of phospholipids by detergents structural and kinetic aspects. Biochim Biophys Acta. 1983;737:285–304. doi: 10.1016/0304-4157(83)90004-7. [DOI] [PubMed] [Google Scholar]
- 17.Aungst BJ, Rogers NJ, Shefter E. Enhancement of naloxone penetration through human skin in vitro using fatty acids, fatty alcohols, surfactants, sulphoxides and amides. Int J Pharm. 1986;33:225–234. doi: 10.1016/0378-5173(86)90057-8. [DOI] [Google Scholar]
- 18.US2012/0269881(A9) (Noven Pharmaceuticals Inc. USA). Trandermal drug delivery device including an occlusive backing.
- 19.Muchow M, Maincent P, Müller RH, Keck CM. Production and characterization of testosterone undecanoate-loaded NLC for oral bioavailability enhancement. Drug Dev Ind Pharm. 2011;37:8–14. doi: 10.3109/03639045.2010.489559. [DOI] [PubMed] [Google Scholar]
- 20.Yin AY, Htun M, Swerdloff RS, Diaz-Arjonilla M, Dudley RE, Faulkner S, et al. Reexamination of pharmacokinetics of oral testosterone undecanoate in hypogonadal men with a new self-emulsifying formulation. J Androl. 2012;33:190–201. doi: 10.2164/jandrol.111.013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hickey S, Hagan SA, Kudryashov E, Buckin V. Analysis of phase diagram and microstructural transitions in an ethyl oleate/water/Tween 80/Span 20 microemulsion system using high-resolution ultrasonic spectroscopy. Int J Pharm. 2010;388:213–222. doi: 10.1016/j.ijpharm.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 22.Hubatsch I, Ragnarsson EG, Artursson P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat Protoc. 2007;2:2111–2119. doi: 10.1038/nprot.2007.303. [DOI] [PubMed] [Google Scholar]
- 23.Whitehead K, Mitragotri S. Mechanistic analysis of chemical permeation enhancers for oral drug delivery. Pharm Res. 2008;25:1412–1419. doi: 10.1007/s11095-008-9542-2. [DOI] [PubMed] [Google Scholar]
- 24.Anderberg EK, Nyström C, Artursson P. Epithelial transport of drugs in cell culture. VII: effects of pharmaceutical surfactant excipients and bile acids on transepithelial permeability in monolayers of human intestinal epithelial (Caco-2) cells. J Pharm Sci. 1992;81:879–887. doi: 10.1002/jps.2600810908. [DOI] [PubMed] [Google Scholar]
- 25.O’Brien PJ, Irwin W, Diaz D, Howard-Cofield E, Krejsa CM, Slaughter MR, et al. High concordance of drug-induced human hepatotoxicity with in vitro cytotoxicity measured in a novel cell-based model using high content screening. Arch Toxicol. 2006;80:580–604. doi: 10.1007/s00204-006-0091-3. [DOI] [PubMed] [Google Scholar]
- 26.Rawlinson LA, O’Brien PJ, Brayden DJ. High content analysis of cytotoxic effects of pDMAEMA on human intestinal epithelial and monocyte cultures. J Control Release. 2010;146:84–92. doi: 10.1016/j.jconrel.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 27.Bzik VA, Medani M, Baird AW, Winter DC, Brayden DJ. Mechanisms of action of zinc on rat intestinal epithelial electrogenic ion secretion: insights into its antidiarrhoeal actions. J Pharm Pharmacol. 2012;64:644–653. doi: 10.1111/j.2042-7158.2011.01441.x. [DOI] [PubMed] [Google Scholar]
- 28.Ungell AL, Nylander S, Bergstrand S, Sjöberg Å, Lennernäs H. Membrane transport of drugs in different regions of the intestinal tract of the rat. J Pharm Sci. 1998;87:360–366. doi: 10.1021/js970218s. [DOI] [PubMed] [Google Scholar]
- 29.Lindmark T, Schipper N, Lazorová L, De Boer AG, Artursson P. Absorption enhancement in intestinal epithelial Caco-2 monolayers by sodium caprate: assessment of molecular weight dependence and demonstration of transport toutes. J Drug Target. 1998;5:215–223. doi: 10.3109/10611869808995876. [DOI] [PubMed] [Google Scholar]
- 30.Maroni A, Zema L, Del Curto MD, Foppoli A, Gazzaniga A. Oral colon delivery of insulin with the aid of functional adjuvants. Adv Drug Deliv Rev. 2012;64:540–556. doi: 10.1016/j.addr.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 31.Uchiyama T, Sugiyama T, Quan YS, Kotani A, Okada N, Fujita T, et al. Enhanced permeability of insulin across the rat intestinal membrane by various absorption enhancers: the intestinal mucosal toxicity and absorption-enhancing mechanism of n-lauryl-β-d-maltopyranoside. J Pharm Pharmacol. 1999;51:1241–1250. doi: 10.1211/0022357991776976. [DOI] [PubMed] [Google Scholar]
- 32.Tomita M, Sawada T, Ogawa T, Ouchi H, Hayashi M, Awazu S. Differences in the enhancing effects of sodium caprate on colonic and jejunal drug absorption. Pharm Res. 1992;9:648–653. doi: 10.1023/A:1015854127486. [DOI] [PubMed] [Google Scholar]
- 33.Petersen SB, Nolan G, Maher S, Rahbek UL, Guldbrandt M, Brayden DJ. Evaluation of alkylmaltosides as intestinal permeation enhancers: comparison between rat intestinal mucosal sheets and Caco-2 monolayers. Eur J Pharm Sci. 2012;47:701–712. doi: 10.1016/j.ejps.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 34.Brayden DJ, Mrsny R. Oral peptide delivery: prioritizing the leading technologies. Ther Deliv. 2011;2:1567–1573. doi: 10.4155/tde.11.114. [DOI] [PubMed] [Google Scholar]
- 35.Cox A, Rawlinson LA, Baird A, Bzik V, Brayden D. In vitro interactions between the oral absorption promoter, sodium caprate (C10) and S. typhimurium in rat intestinal ileal mucosae. Pharm Res. 2008;25:114–122. doi: 10.1007/s11095-007-9354-9. [DOI] [PubMed] [Google Scholar]
- 36.Kararli TT. Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm Drug Dispos. 1995;16:351–380. doi: 10.1002/bdd.2510160502. [DOI] [PubMed] [Google Scholar]
- 37.Lindmark T, Söderholm J, Olaison G, Alván G, Ocklind G, Artursson P. Mechanism of absorption enhancement in humans after rectal administration of ampicillin in suppositories containing sodium caprate. Pharm Res. 1997;14:930–935. doi: 10.1023/A:1012112219578. [DOI] [PubMed] [Google Scholar]
- 38.Cano-Cebrián MJ, Zornoza T, Granero L, Polache P. Intestinal absorption enhancement via the paracellular route by fatty acids, chitosans and others: a target for drug delivery. Curr Drug Deliv. 2005;2:9–22. doi: 10.2174/1567201052772834. [DOI] [PubMed] [Google Scholar]
- 39.Swenson ES, Milisen W, Curatolo W. Intestinal permeability enhancement: efficacy, acute local toxicity, and reversibility. Pharm Res. 1994;11:1132–1142. doi: 10.1023/A:1018984731584. [DOI] [PubMed] [Google Scholar]
- 40.Fan D, Wu X, Dong W, Sun W, Li J, Tang X. Enhancement by sodium caprate and sodium deoxycholate of the gastrointestinal absorption of berberine chloride in rats. Drug Dev Industrial Pharmacy. 2013;39:1447–1456. doi: 10.3109/03639045.2012.723219. [DOI] [PubMed] [Google Scholar]
- 41.Leclercq S, Cani PD, Neyrinck AM, Stärkel P, Jamar F, Mikolajczak M, et al. Role of intestinal permeability and inflammation in the biological and behavioral control of alcohol-dependent subjects. Brain Behav Immun. 2012;26:911–918. doi: 10.1016/j.bbi.2012.04.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 29 kb)