

Abstract
Background
One of the most important producers of high quality industrial enzymes is the Gram-positive bacterium, Bacillus subtilis (B. Subtilis). One major limitation that hinders the wide application of B. subtilis is the secretion of high levels of extracellular proteases which degrade the secreted foreign proteins. In this study, homologus recombination technique was used to knock out its protease gene, aprE.
Methods
The internal segment of the pro-sequence of aprE gene of B. subtilis 168 with a length of 80 bps and its complementary sequence were synthesized and ligated into pUB110 at EcoR1 and XbaI restriction sites. Competent cells of B. subtilis 168 were prepared and transformed by electroporation using Bio Rad gene pulser as explained in the methods section. Transformants carrying the recombinant plasmid were selected for resistance to neomycin. The success of homologous recombination was checked by PCR amplification of the neomycin gene which was part of the vector and did not exist in the genome of B. subtilis 168. The protease activity was measured using the Protease Fluorescent Detection Kit based on the proteolytic hydrolysis of fluorescein isothiocyanate (FITC)–labeled casein-substrate.
Results
The results demonstrated that aprE gene would not be able to produce further active subtilisin E. The reduction of protease activity also confirmed the efficacy of the induced mutation in this gene.
Conclusion
It will therefore be a major challenge for future research to identify and modulate quality control systems of B. subtilis which limit the production of high quality protease- sensitive products such as lipase.
Keywords: aprE gene, Bacillus subtillis, Subtilisins
Introduction
Bacillus subtilis is a well-known Gram-positive soil bacterium that naturally produces and secrets high concentrations of proteins into the medium (1). The absence of an outer membrane in B. subtilis can simplify the protein secretion pathways and allow the organism to secrete high levels of extracellular proteins. In contrast to the Gram-negative bacterium Escherichia coli, B. subtilis is considered as a GRAS organism (Generally Recognized As Safe). Thus, scientists have widely used B. subtilis for commercial exploitation as a major “cell factory” for the secretion of heterologous proteins (2, 3). As far as the capability of secreting extracellular enzymes directly into the culture medium is concerned, B. subtilis can potentially serve as an efficient expression host (4, 5). The secreted foreign proteins usually remain in biologically active forms, and the downstream purification is greatly simplified (6, 7).
One of the major limitations that hinder the wide application of B. subtilis is the secretion of high levels of extracellular proteases which degrade the secreted foreign proteins. It is well established that B. subtilis has six extracellular proteases including neutral protease A, subtilisin (also known as alkaline protease), extracellular protease, metalloprotease, bacillopeptidase F, and neutral protease B (8). In view of the fact that the protease production is limited, protease deficient B. subtilis strains have been developed by genome engineering techniques (9). For example, B. subtilis WB800, deficient in eight extracellular proteases can serve as an excellent host for the expression of heterologous proteins (10).
In order to improve the production of heterologous proteins in B. subtilis 168, it was decided to inactivate the aprE gene of this bacterium encoding one of the major extracellular alkaline serine proteases, Subtilisin E, by site directed mutation of this gene using homologous recombination techniques. For this purpose, the pro-sequence in the aprE gene was targeted. Subtilisin E is first made as pre-pro-subtilisin (9, 11, 12). This enzyme consists of a signal peptide for protein secretion (pre-sequence) and a peptide extension of 77 amino acid residues (pro-sequence) located between the signal peptide and the mature protease (13). Studies have indicated that the pro-sequence is essential for guiding appropriate folding of the enzymatically active conformation of Subtilisin E (12, 13). Inactivating the pro-sequence of the aprE gene, therefore, should impair the production of an active subtilisin resulting in a genetically engineered strain deficient in one of the major extracellular proteases making an appropriate host for the expression of protease sensitive products.
Materials and Methods
Bacterial strains, plasmids, and growth conditions
The bacterial strain used in this study was B. subtilis strain 168 (DSMZ, Germany) containing the plasmid pUB110. The medium consisted of 7% maltose (w/v), 0.05% yeast extract (Difco), 4.6% dried corn steep liquor, 0.5% fish meal extract, 0.1% KH2PO4, 0.02% MgSO4· 7H2O, and 15 μg/ml tetracycline (pH=6.8). The supernatant was collected from the culture broth by centrifugation at 10,000 g for 20 min and after that the pellet was resuspended in 1 mM EDTA, 1 mM PMSF, 1 mM DTT, and 5% glycerol in 50 mM potassium phosphate buffer (pH=7.4). Next, it was sonicated (model 7500; BIOMIC). The supernatants were then collected following centrifugation. Plasmid DNA from B. subtilis 168 was prepared as described by Sambrook et al (14).
Insert preparation
The internal segment of the pro-sequence of the aprE gene of B. subtilis 168 with a length of 80 bps (nucleotides 96 to 176 upstream from the initiation codon) and its complementary sequence were synthesized by Kawsar Biotech Company (Iran). Restriction sites for the enzymes EcoRI and XbaI were designed on both ends of the single strands as outlined below:
5´AATTCagcagtacagaaaagaaatacattgtcgtcggatttaaacagacaatgagtgccatgagttccgccaagaaaaaggatgtT3´ and 5´CTAGAacatcctttttcttggcggaactcatggcactcattgtctgtttaaatccgacaatgtatttcttttctgtactgctG3´
Vector preparation
Twenty five μl of PUB110 was cross digested with the enzymes EcoRI and XbaI in a total volume of 100 μl according to the manufacturer’s recommended conditions (Germany). Gel electrophoresis was also run to verify complete digestion of the plasmid PUB110.
Ligation reaction and transformation
The ligation reaction was carried out using an appropriate amount of vector and insert (a ratio of 3:1 insert to vector). One μl of 10×ligation buffer and 1 μl of Ligase were added to the mixture adjusting the final volume to 10 μl with ddH2O. The ligation mixture was incubated overnight at 16°C.
Competent cells of B. subtilis 168 were prepared and transformed by electroporation using Bio Rad gene pulser. Electrocompetent cells were prepared by diluting 2 ml of the overnight culture of B. subtilis 168 in 50 ml of fresh LB culture medium and grown at 37°C to reach OD value of 0.3 at 600 nm. Then the cell suspension was cooled on ice for 10 min and harvested by centrifugation at 4°C and 10000 g for 10 min. The cells were suspended in 5 ml ice-cold electroporation buffer and spun at 4°C at 10000 g for 10 min. Finally, the cells were suspended in 1.5 ml of ice-cold electroporation solution and the competent cells were kept on ice until use. Ten μl (5 ng/μl) of the ligation mixture containing the recombinant plasmid pUB110 was added to 100 μl of electrocompetent cells and homogenized by gently mixing with pipette several times. The mixture was then transferred into a pre-chilled cuvette and an electric shock with a voltage of 1000 V and a time constant of 8.6 ms was applied. Following the electric shock, 2 ml of LB culture medium was immediately added to the electroporated cells followed by incubation for 1.5 hr at 37°C.
Selection for neomycin resistance
Transformants carrying the recombinant plasmid were selected for resistance to neomycin.
Primer design for the neomycin gene and PCR amplification
To confirm the incidence of homologous recombination, PCR amplification of the neomycin gene was carried out. Neomycin gene was part of the plasmid pUB110 used as the vector and it did not exist in the genome of B. subtilis 168. The neomycin gene was amplified using the genomic DNA from transformed B. subtilis 168 as a template with a pair of primers, F (5΄-TCGGAAAGTTGACCAGA-3΄) and R (5΄-TTTGTGCCCTTATCGTAG-3΄). PCR was conducted by subjecting the reaction mixture to an initial denaturation at 94°C for 5 min, followed by 30 cycles of 94°C for 1 min, 55°C for 2 min, 72°C for 3 min and a final extension step at 72°C for 5 min.
Extracellular protease activity assay
To quantitatively compare the protease activity of wild and mutant strains deficient in active subtilisin E, the Protease Fluorescent Detection Kit (Sigma-Aldrich, Germany) based on the proteolytic hydrolysis of a fluorescein isothiocyanate (FITC)-labeled casein-substrate was used according to the manufacturer’s protocol. FITC-casein is native casein that has been labeled using fluorescein isothiocyanate. The UV absorbance of this heavily-labeled, intact protein substrate at 280 nm changed dramatically upon digestion by proteases, resulting in a measurable indication of proteolysis. Different concentrations of FITC-casein were also prepared by serial dilutions and their absorbance at 280 nm was measured to determine the concentration ranges across which the Beer-Lambert law was obeyed.
Results
Construction of recombinant plasmid, pUB110
To prepare the pUB110for ligation, the plasmid was first subjected to restriction digestion with the enzymes EcoRI and XbaI. Digested plasmid was then dephosphorylated by alkaline phosphatase and recovered from the gel. The internal segment of pro-sequence in the aprE gene of B. subtilis was constructed with the same restriction enzyme sites (EcoRI and XbaI) as the vector. Agarose gel analysis of the pUB110digested plasmid after dephosphorylation and EcoRI and XbaI digested insert is depicted in Figure 1. Constructed insert and vector had the lengths of 80 and 4000 bps, respectively. An appropriate amount of prepared plasmid (pUB110) and insert were mixed for ligation reaction with T4 DNA ligase. Then recombinant plasmid was transformed into B. subtilis 168. Figure 1 shows the pUB110plasmid digested with EcoRI and XbaI and also the 80 bp insert. The integrity of the plasmid (4 kb) of cut plasmid and the purchased insert both were verified.
Figure 1.
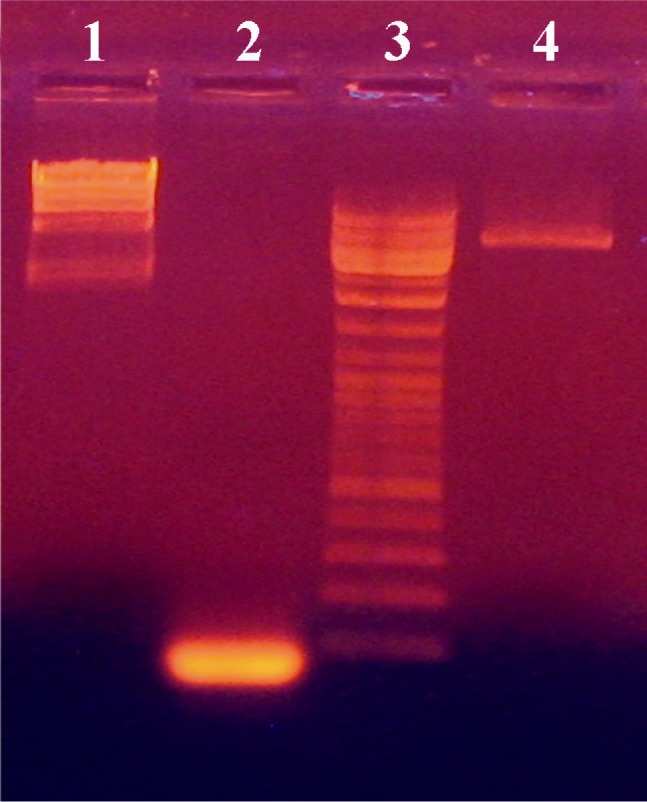
PCR amplification of 80 bp insert from digested plasmid pUB110 with EcoRI and XbaI. HindIII digested lambda DNA, the 80 bp insert, DNA ladder and pUB110plasmid digested with EcoRI and XbaI are shown in lanes 1, 2, 3 and 4, respectively
Transformation of recombinant pUB110plasmid into Bacillus subtilis 168
Screening the electroporated cells was done for antibiotic resistance and performed on nutrient agar plates supplemented with 5 and 10 mg/ml of neomycin (14). The colonies of transformed cells from the plasmid containing neomycin resistance gene successfully grew and formed a bacterial lawn on LB media as shown in Figure 2. DNA extracted from the transformed cells was compared with non-transformed bacteria. Figure 3 shows the pattern of DNA extracted from the transformed and non-transformed bacteria.
Figure 2.
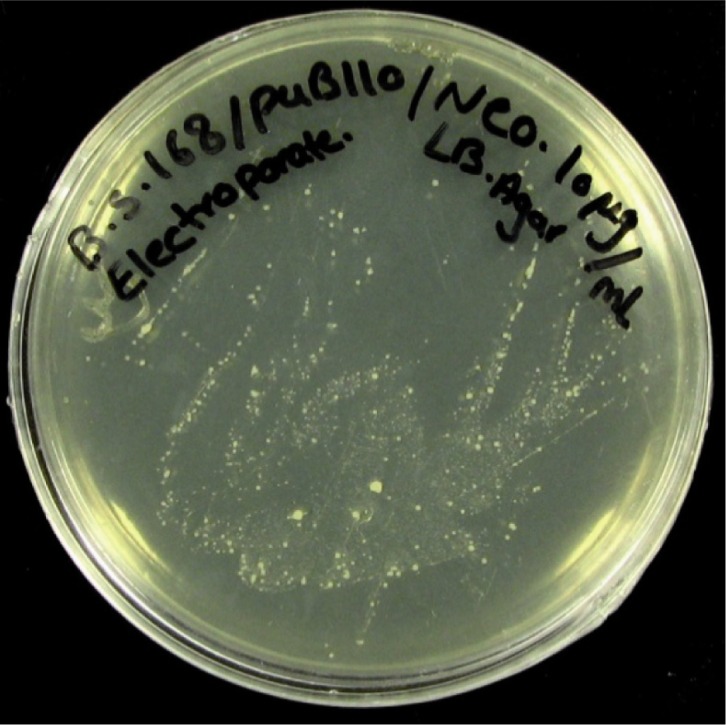
The colonies of transformed cells from the plasmid containing neomycin resistance gene streaked out on LB plates
Figure 3.
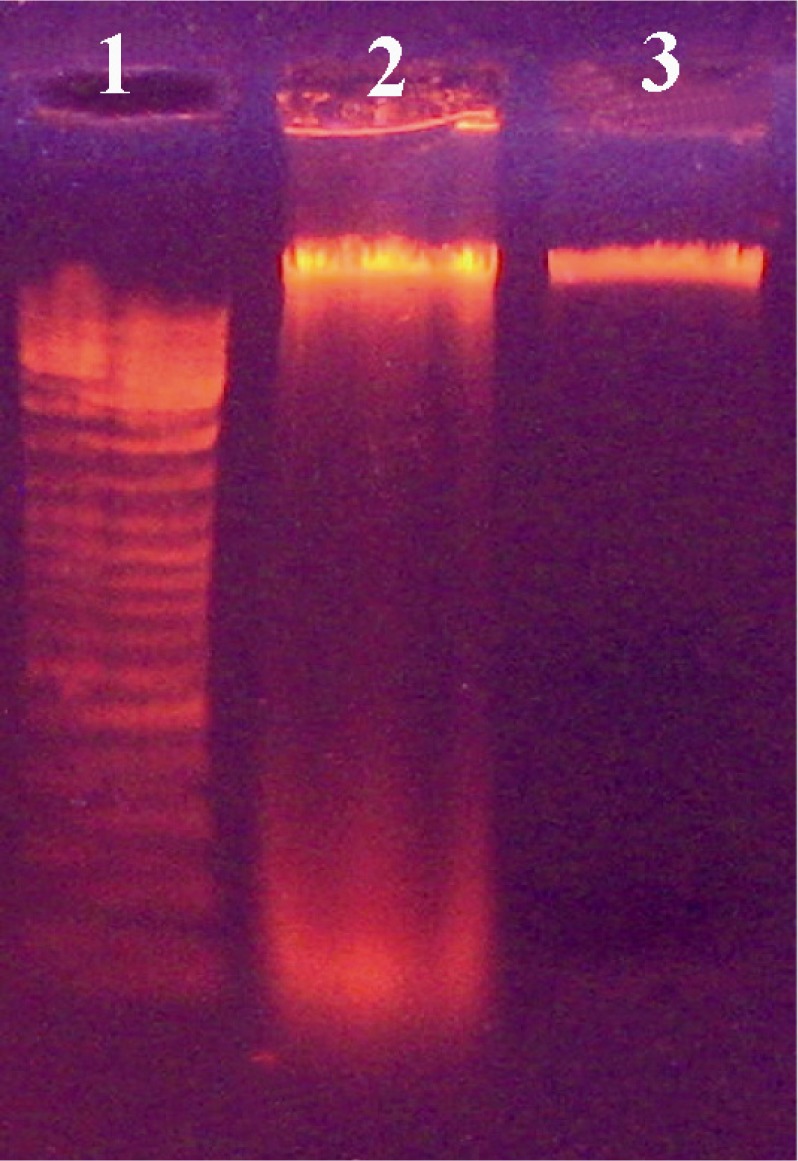
Electrophoretic analysis of DNA from transformed and non-transformed B. subtilis 168. Lanes; 1) DNA size marker; 2) Non-transformed cells and 3) Transformed cells
PCR amplification of neomycin gene
To confirm the incidence of homologous recombination, PCR amplification of the neomycin gene was carried out. Neomycin gene was part of pUB110and it did not exist in the genome of B. subtilis 168. Integration of this plasmid in the chromosome by double cross-over replaced the neomycin resistant gene in chromosomal DNA of transformed B. subtilis 168. The neomycin gene was amplified using genomic DNA from transformed B. subtilis 168 as a template with a pair of designed primers. A sharp band in the 300 bps region (Figure 4) in the transformed cells proved the integration of the plasmid DNA into chromosomal DNA of transformed bacteria.
Figure 4.
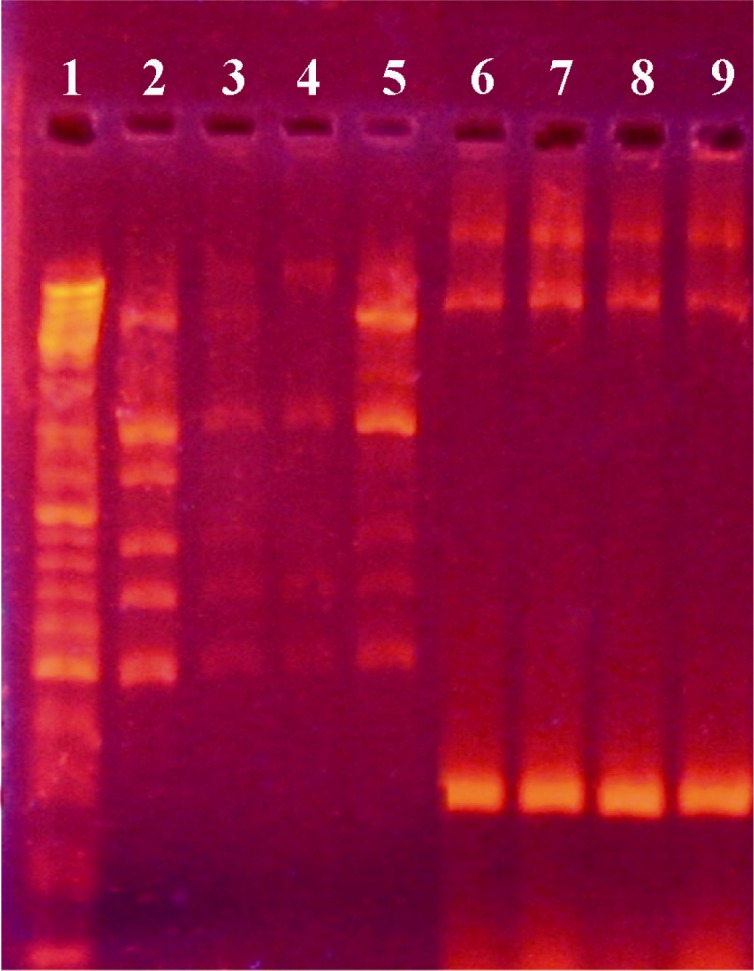
Agarose gel electrophoresis showing PCR amplification products of neomycin gene (300 bp). First lane is a DNA size marker (100 bp DNA ladder). Lanes 2-5 and lanes 6-9 are PCR products from the neomycin gene in non-transformed and transformed cells, respectively
Extracellular protease activity assay in wild and mutant strains
Protease activity of samples was determined by measuring the residue of FITC-Casein in media. Activity measurement at the examined condition indicated that in mutant samples, the UV absorbance was increased in comparison with wild samples at 280 nm. These changes demonstrated that the aprE gene in B. subtilis 168 would not further be able to produce active subtilisin E and finally would decrease the protease activity.
Discussion
B. subtilis bacteria utilize especial systems that secrete proteins out of cells. Subtilisin E is one of typical examples which functions similarly (15). This extracellular alkaline serine protease encoded with aprE gene was produced at the end of exponential growth phase (16). Subtilisin E leads to degradation of other important proteins like lipase enzymes. It is known that if aprE gene is knocked out, B. subtilis will no longer be able to produce active subtilisin E and therefore, it can help to produce active proteins which are sensitive to proteases (13).
The aim of the present study was the inactivation of aprE gene using site directed mutation at pro-sequence region. For this purpose, the recombinant plasmid was constructed with mixing appropriate amount of insert and pUB110 plasmid as a vector. After transformation of recombinant plasmid into B. subtilis 168, screening results showed that electroporated cells can grow in supplemented media containing neomycin antibiotic. Comparison between transformed and non-transformed cells demonstrated that digestion, ligation and transformation were done successfully. The results of PCR amplification of neomycin gene further confirmed the homologous recombination. Finally, in accordance with our finding, the reduction of protease activity demonstrated that the mutation in aprE gene has indeed taken place at the right site.
Conclusion
As previously mentioned, B. subtilis is one of the most widely used bacterium for the production of industrial enzymes but its applications is limited due to its proteases. According to the results obtained in this study, homologous recombination techniques can help to inactivate protease encoding genes and improve production of other protease-sensitive- proteins. It will therefore be a major challenge for future research to identify and modulate quality control systems of B. subtilis which limit the production of high quality protease- sensitive products such as lipase.
References
- 1.Harwood CR. Bacillus subtilis and its relatives: molecular biological and industrial workhorses. Trends Biotechnol. 1992;10(7):247–256. doi: 10.1016/0167-7799(92)90233-l. [DOI] [PubMed] [Google Scholar]
- 2.Schallmey M, Singh A, Ward OP. Developments in the use of Bacillus species for industrial production. Can J Microbiol. 2004;50(1):1–17. doi: 10.1139/w03-076. [DOI] [PubMed] [Google Scholar]
- 3.Westers L, Westers H, Quax WJ. Bacillus subtilis as cell factory for pharmaceutical proteins: a biotechnological approach to optimize the host organism. Biochim Biophys Acta. 2004;1694(1-3):299–310. doi: 10.1016/j.bbamcr.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 4.Fu LL, Xu ZR, Li WF, Shuai JB, Lu P, Hu CX. Protein secretion pathways in Bacillus subtilis: Implication for optimization of heterologous protein secretion. Biotechnol Adv. 2007;25(1):1–12. doi: 10.1016/j.biotechadv.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 5.Zhang XZ, Cui ZL, Hong Q, Li SP. High-level expression and secretion of methyl parathion hydrolase in Bacillus subtilis WB800. Appl Environ Microbiol. 2005;71(7):4101–4103. doi: 10.1128/AEM.71.7.4101-4103.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Himanen JP, Taira S, Sarvas M, Saris P, Runeberg-Nyman K. Expression of pertussis toxin subunit S4 as an intracytoplasmic protein in Bacillus subtilis. Vaccine. 1990;8(6):600–604. doi: 10.1016/0264-410x(90)90017-g. [DOI] [PubMed] [Google Scholar]
- 7.Bornscheuer UT, Bessler C, Srinivas R, Krishna SH. Optimizing lipases and related enzymes for efficient application. Trends Biotechnol. 2002;20(10):433–437. doi: 10.1016/s0167-7799(02)02046-2. [DOI] [PubMed] [Google Scholar]
- 8.Veening JW, Igoshin OA, Eijlander RT, Nijland R, Hamoen LW, Kuipers OP. Transient heterogeneity in extracellular protease production by Bacillus subtilis. Mol Syst Biol. 2008;4:184. doi: 10.1038/msb.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murashima K, Chen CL, Kosugi A, Tamaru Y, Doi RH, Wong SL. Heterologous production of Clostridium cellulovorans engB, using protease-deficient Bacillus subtilis, and preparation of active recombinant cellulosomes. J Bacteriol. 2002;184(1):76–81. doi: 10.1128/JB.184.1.76-81.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho HY, Yukawa H, Inui M, Doi RH, Wong SL. Production of minicellulosomes from Clostridium cellulovorans in Bacillus subtilis WB800. Appl Environ Microbiol. 2004;70(9):5704–5707. doi: 10.1128/AEM.70.9.5704-5707.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu SC, Yeung JC, Duan Y, Ye R, Szarka SJ, Habibi HR, et al. Functional production and characterization of a fibrin-specific single-chain antibody fragment from Bacillus subtilis: effects of molecular chaperones and a wall-bound protease on antibody fragment production. Appl Environ Microbiol. 2002;68(7):3261–3269. doi: 10.1128/AEM.68.7.3261-3269.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rattray AJ, Symington LS. Multiple pathways for homologous recombination in Saccharomyces cerevisiae. Genetics. 1995;139(1):45–56. doi: 10.1093/genetics/139.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ikemura H, Takagi H, Inouye M. Requirement of pro-sequence for the production of active subtilisin E in Escherichia coli. J Biol Chem. 1987;262(16):7859–7864. [PubMed] [Google Scholar]
- 14.Sambrook J, Russell DW. 2nd ed. New York: Cold Spring Harbor Laboratory Press; 2001. Molecular cloning: A laboratory manual. [Google Scholar]
- 15.Hata M, Ogura M, Tanaka T. Involvement of stringent factor RelA in expression of the alkaline protease gene aprE in Bacillus subtilis. J Bacteriol. 2001;183(15):4648–4651. doi: 10.1128/JB.183.15.4648-4651.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berger E, du Plessis E, Gerber I, Crampton M, Nxumalo N, Louw M. Impact of inactivated extracellular proteases on the modified flagellin type III secretion pathway of Bacillus halodurans. Appl Environ Mircrobiol. 2009;75(1):271–274. doi: 10.1128/AEM.02430-08. [DOI] [PMC free article] [PubMed] [Google Scholar]