

Abstract
AIM: To observe changes of mitochondria and investigate the effect of ethanol on mitochondrial perme-ability transition pore (PTP), mitochondrial membrane potential (MMP, ΔΨm) and intracellular calcium concentration in hepatocytes by establishing an animal model of alcoholic liver disease (ALD).
METHODS: Fourty adult male Wistar rats were randomly divided into two groups, the model group (20) was administered alcohol intragastrically plus an Oliver oil diet to establish an ALD model, and the control group (20) was given an equal amount of normal saline. The ultramicrostructural changes of mitochondria were observed under electron microscopy. Mitochondria of liver was extracted, and patency of PTP, mitochondrial membrane potential (ΔΨm), mitochondrial mass and intracellular calcium concentration of isolated hepacytes were detected by flow cytometry using rhodamine123 (Rh123), Nonyl-Acridine Orange and calcium fluorescent probe Fluo-3/AM, respectively.
RESULTS: Membrane and cristae were broken or disappeared in mitochondria in different shapes under electron microscopy. Some mitochondria showed U shape or megamitochondrion. In the model group, liver mitochondria PTP was broken, and mitochondria swelled, the absorbance at 450 nm, A540 decreased (0.0136 ± 0.0025 vs 0.0321 ± 0.0013, model vs control, P < 0.01); mitochondria transmembrane potential (239.4638 ±12.7263 vs 377.5850 ± 16.8119, P < 0.01) was lowered; mitochondrial mass (17.4350 ± 1.9880 vs 31.6738 ± 3.4930, P < 0.01); and [Ca2+]i was increased in liver cells (7.0020 ± 0.5008 vs 10.2050 ± 0.4701, P < 0.01).
CONCLUSION: Chronic alcohol intake might lead to broken mitochondria PTP, decreased mitochondria membrane potential and injury, and elevated intracellular Ca2+ production. Ethanol-induced chondriosome injury may be an important mechanism of alcoholic diseases.
Keywords: Alcoholic liver disease, Chondriosome, Apoptosis, Ultra microstructure, Membrane potentials, Permeability transition pore, Transmembrane potential, chondriosome mass, Ca2+
INTRODUCTION
Alcohol abuse used to be a principal factor for the cause of serious liver disease in western countries. In China the incidence of alcoholic liver disease (ALD) has increased. Alcohol abuse has been considered the second leading cause of hepatic lesion after virus hepatitis. The accurate mechanism of ethanol-induced hepatic cells injury is still not completely clear. Some researchers found that ethanol could activate lipid peroxidation, leading to liver injury. Since the mitochondrion is one of the fundamental parts of reactive oxygen species (ROS) formation, we investigated mitochondrial changes in ALD in order to prove the correlation between mitochondrion injury and hepatocytes apoptosis induced by ethanol and reveal the role of ethanol-induced mitochondrion injury in the pathogenesis of ALD, hoping that by this study we could improve the prevention and treatment of ALD.
MATERIALS AND METHODS
Materials
Healthy male Wistar rats, weighing 150 ± 5 g,were purchased from the Experimental Animal Center of Shandong University. Olive oil (unsaturated fatty acid content > 90%) was a product of Algemesi Co., Spain. Erguotou (a liquor of 60°) was a product of Beijing Hongxing Erguotou Alcohol Factory. HEPES, Rhodamin123 and Fluo-3/AM were purchased from Sigma Co., USA. DMEM was obtained from GIBCO. FBS was purchased from Hangzhou Sijiqing. NAO was obtained from GENMED.
Animals and protocol
Forty adult Wistar rats (4 wk old), weighing (150 ± 5) g, were fed in separate cages with 5 in each. After feeding normally for a week, these rats were randomly divided into two groups. Twenty were given intragastric alcohol as the ALD model group whose food was composed of ethanol 46% (intragastric administration twice a day at an eight-hour interval), protein 18%, carbohydrate 12%, fat 14% (mainly olive oil, unsaturated fatty acid 9%) by energy as well as vitamin complex and essential minerals at the same time. In the first four weeks, the rats were lavaged with 30% ethanol (4 g/kg per day), 40% ethanol (5 g/kg per day) in the following four weeks, 50% ethanol (6 g/kg per day ) from wk 9 to 12. Another twenty rats in the control group were given an equal volume of normal saline and the composition of food was the same as the model group until the end of the 12th wk.
Preparation and observation of samples under electron microscopy
Liver tissue (1 mm3) from the right lobe of the liver near the porta hepatis was removed and pre-fixed for 2 h at 4°C in 4% paraformaldehyde solution with 0.1 mol/L phosphate buffer (pH 7.3) and 2.5% glutaraldehyde. The tissue was then rinsed in the same buffer and post-fixed in 1% osmium tetroxide in 0.1 mol/L phosphate buffer (pH 7.3). Post-fixation was followed by dehydration in ethanol, embedding in Epon 812 and polymerization. Tissues were cut using an LKB 2088 ultramicrotome, stained with 1% uranyl acetate and lead citrate, and examined under a transmission electron microscope (JEM-2000 EXII, 80 KV, Japan).
Patency of mitochondrial PTP
Isolation and purification of mitochondria: Approxi-mately 5 g of the liver harvested from sacrificed rats was homogenized in a ten-fold volume of isolation buffer (0.33 mmol/L sucrose, 0.025 mmol/L EDTA). After filtering with double-ply gauze to remove tissue dregs, the liver homogenate was centrifuged for 10 min at 800 × g. The supernatant was further centrifuged for 10 min at 8200 × g.The second supernatant was discarded and the sediment was resuspended with washing buffer (0.33 mmol/L sucrose). The suspension obtained was centrifuged for another 10 min at 8200 × g to get the purified mitochondria.
Detection of mitochondrial PTP opening: The purified mitochondria were diluted to 0.3 mg/mL with test solution (230 mmol manicol, 70 mmol sucrose, 3 mmol Hepes, pH 7.4). The absorbance of mitochondria was detected at 570 nm using an ultraviolet spectrophotometer at 20°C to find the state of mitochondrial PTP opening.
Isolation and purification of hepatocytes
Hepatocytes were isolated with the procedure of in situ two-step perfusion of Seglen[1]. After anesthetizing with pentobarbital sodium, the abdominal cavity of the rats was opened from the middle abdominal wall. Plastic duct was inserted into the portal vein after exposure. The livers were perfused first with Ca2+- and Mg2+- free D-Hanks solution for 20 min (20-40 mL/min) at 37°C to remove blood and Ca2+ in the liver, and next with 0.01% (w/v) collagenase solution at 37°C when the fluid flowed from the inferior vena cava became clear. The membrane of gently fully digested liver was removed with curved forceps, then gently the hepatocytes and sinusoid endothelial cells were dispersed in Dulbecco's modified Eagle's medium. After filtering through 200-screen mesh to remove cell aggregates and tissue fibers, the cell suspension was washed thrice in DMEM/H with low-speed centrifugation (50 × g, 5 min × 3) to obtain purified hepatocytes.
Detection of mitochondrial membrane potential of hepatocytes
Rh123 powder was prepared with methanol stored at -20°C as a 1 g/L solution. The storing solution was diluted to 5 mg/L with PBS when used. After incubating in Rhodamin123 (final concentration 2.5 mg/L) for 1 h at 37°C, hepatocytes (1 × 106) were washed thrice in preheating PBS. After resuspending in 300 μL PBS, filtered through 200-screen mesh, 104 cells were measured with flow cytometry using the CellQuest software (maximum absorbing wave length 590 nm, excitation wave length 488 nm).
Detection of mitochondrial mass
After incubated in 1 mL NAO staining solution at 37°C for 20 min, the hepatocytes (1 × 106) were centrifuged at 300 × g for 5 min. The precooling GENMED preserving fluid was added into the supernatant, mixed with cell granulations and analyzed with flow cytometry using the CellQuest software (maximum absorbing wave length 519 nm, and excitation wave length 495 nm).
Detection of intracellular calcium concentration
After incubating in 1 mL Fluo-3/AM (5 μmol/L) at 37°C for 40 min, the hepatocytes (1 × 106) were washed 3 times with PBS, resuspended in 300 μL PBS and filtered through a 200-screen mesh, and 104 cells were collected and detected with flow cytometry using the CellQuest software (maximum absorbing wave length 506 nm, and excitation wave length 490 nm).
Statistical analysis
All values were expressed as mean ± SD and analyzed with the SPSS Version. All experiments were repeated at least three times. P < 0.05 was considered statistically significant.
RESULTS
Ultrastructural changes of hepatocyte mitochondria under electron microscopy
There were rich organelles with lots of ribosomes, endoplasmic reticulum, mitochondria and few lipid droplets in hepatocytes in the control group under electron microscopy. Nuclei of hepatocytes were round with clear membranes and well-distributed nuclear chromatin. The perinuclear cisterna and nucleopore were clear (Figure 1A).
Figure 1.
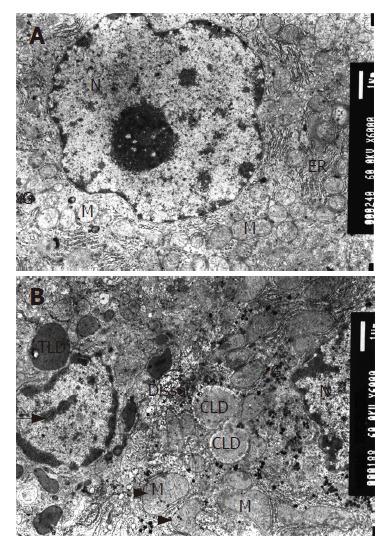
Mitochondria appearance under electron microscope (EM × 6000); A: Mitochondria in normal group; B: Mitochondria in model group. M: mitochondria, G: glycogen, N nucleus, ER: endoplasmic reticulum, LD: lipid droplet. The long arrow shows abnormally distributed chromatin in nuclei, the short one is megamitochondrion and the arrow head is U-type mitochondria.
Many lipid droplets with equal size and electron density and chromaffin lipid droplets could be seen in the model group. The endoplasmic reticulum became swollen. The membrane was ruptured and cristae were broken or disappeared in the mitochondria resulting in different shapes, and the electron density of their ground substance decreased. Some mitochondria were U-type or megamitochondrion. The glycometabolism was abnormal, and glycogen increased with various densities. Many myelin figures of metabolites and few lysosomes could be observed. Nuclear material presented irregular profiles with a pseudo-inclusion body found within. Nuclear chromatin was abnormal in distribution and occasionally gathered at the perimeter. The number of plasmocytes increased (Figure 1B).
We had established the ALD model successfully, and confirmed our viewpoint that alcohol could cause structural and morphological damage in mitochondria.
Patency of PTP in hepatocytes mitochondria
The results of A540 detected in model group (0.0136 ±0.0025) are obviously lower than in the control group (0.0321 ± 0.0013), P < 0.01, which demonstrated that the PTP in liver mitochondria of rats were opened in the model group (Table 1).
Table 1.
Absorbance comparison of liver mitochondria in two groups of rats
| Groups | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | χ bar ± s |
| Control | 0.0305 | 0.0304 | 0.0331 | 0.0334 | 0.032 | 0.0314 | 0.0325 | 0.0336 | 0.0136 ± 0.0025 |
| Model | 0.0167 | 0.0169 | 0.0161 | 0.0123 | 0.0119 | 0.012 | 0.0113 | 0.0116 | 0.0321 ± 0.0013b |
P < 0.01 vs control group.
The data indicated that ethanol could increase mitochondria membrane permeability, resulting in mitochondria swelling and damage.
Mitochondrial membrane potential of hepatocytes dete-cted by Rh123
The mean fluorescence intensity (MFI) of Rh123 in the model group was 239.464% ± 12.726% while in the control group was 377.585% ± 16.812%, which demonstrated that the mitochondrial membrane potential (Δψm) in the model group decreased evidently with a statistically significant difference (P < 0.01) (Figure 2).
Figure 2.
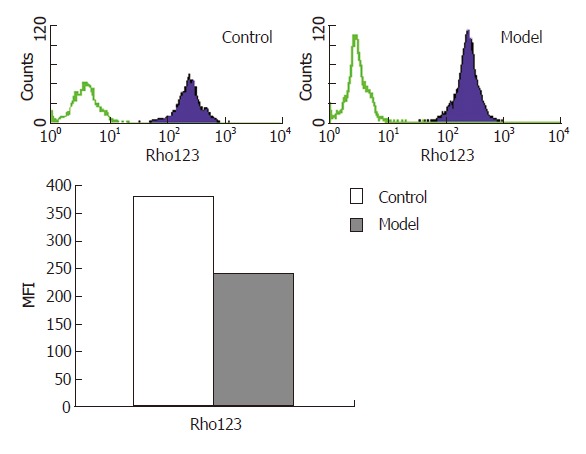
Mitochondrial membrane potential detected by flow cytometry.
Changes of liver mitochondria mass
The MFI of diphosphatidyl glycerol in the model group was 31.6738% ± 3.4930% while the control group was 17.4350% ± 1.9882%, P < 0.01 (Figure 3).
Figure 3.
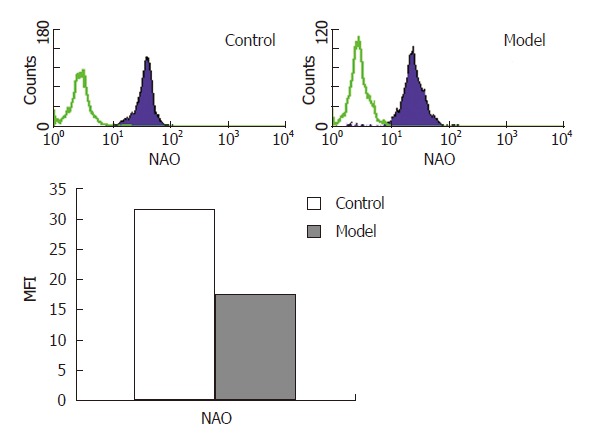
Mitochondria mass detected by flow cytometry.
Intracellular calcium concentration in hepatocytes
The MFI of Fluo-3/AM in the model group was 7.0020% ± 0.5008% while the control group was 10.2050% ± 0.4701%, which demonstrated that the intracellular calcium concentration in hepatocytes in the model group increased evidently with a statistically significant difference (P < 0.01) (Figure 4).
Figure 4.
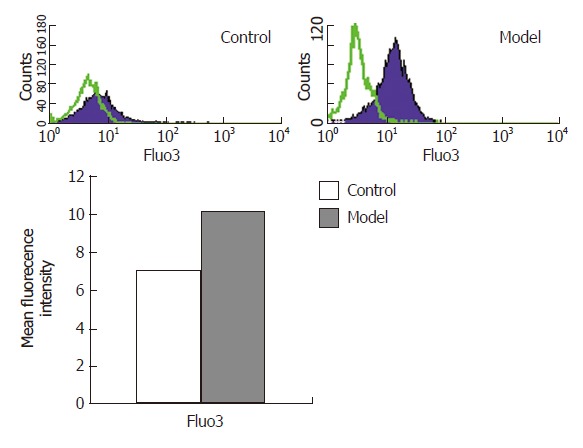
Intracellular calcium concentrations in hepatocytes detected by flow cytometry.
DISCUSSION
Excessive alcohol consumption can result in multiple organ injury, of which alcoholic liver disease (ALD) is the most common. With economic development and improvement of living conditions of the people, the incidence of liver diseases caused by alcohol abuse has been increasing in China, although its pathogenesis remains obscure. In the past, most researches about ALD focused on oxidative stress, cytokines unbalance, level alteration of proteins and genes and so on. Recently, more attention has been on the function of mitochondria in the pathogenesis of ALD, but the mode of mitochondrial damage has not been clarified. In our study, we presented data showing structure and function damage, membrane permeability transition as well as membrane potential alteration of mitochondria, hoping to reveal the correlation between mitochondrial damage and the early stage of ALD.
Figure and structure alteration of liver mitochondria induced by ethanol
Mitochondria have long been known to be very important organelles of the eukaryocyte. They produce ATP to supply energy directly for life activity of the mammal cells. Moreover, mitochondria are considered the key organelle in the initiation of apoptosis since mitochondrial disintegration not only leads to a loss of their function but also causes the release of proapoptotic factors. Emerging evidence suggests that mitochondrial dysfunction caused by oxidative stress is an early event that plays an important role in the pathogenesis of ethanol induced apoptosis.
In this study, we found that the mitochondria of the model group were obviously damaged, and membrane and cristae were broken or disappeared under electron microscopy. We also observed U-form mitochondria (deformed mitochondria) in different sizes and megamito-chondria. In hepatocytes, there are some myelin figures, chromatins distribute abnormally and some can be edge-gathering, indicating the existence of apoptosis. All of these further confirmed the damage in the structure and function of mitochondria. Meanwhile, the structural and functional damage of mitochondria play a very important role in accelerating hepatocyte aging and apoptosis induced by ethanol.
Mitochondria PTP opening influenced by ethanol
In recent years, the knowledge of apoptosis has shifted gradually from the mode of nucleolus-central to mitochondria-central adjustment[2]. In apoptosis, the basic change in mitochondria is mitochondrial permeability transition (MPT) and decreased transmembrane potential and other transformation of apoptosis, leading finally to cell apoptosis.
PTP is a nonselective passage with a high conductance which stretches across the outer and inner membrane of mitochondria. Physiologically, the diameter of PPT is 0.2-0.3 μm, large enough to allow micromolecule solute to pass[3]. By the effect of Ca2+, ADP, ATP and so on, PTP switches alternately. This condition may play a role in material exchange between mitochondria and cytoplasm. But pathologically, the opening diameter is obviously large
(1.8-2.6 μm) so that the solute smaller than 1.5 kDa can pass through PPT to the cytoplasm, causing mitochondrial swelling and transmembrane potential disappearance[4]. PTP opening has a self-enlarging phenomenon that after reveiving the death signal, can perform positive feedback in the same cell. It means that once PTP maintains opening, it will no longer depend on the previous stimulant signal, and the cell will die irreversibly. Our experiment indicated that long-term alcohol abuse of ethanol can make PTP open, the mechanism is possibly because in hepatocytes, anti-oxidation materials such as materials containing sulfydryl are reduced, and/or lipid peroxidation intensifies, causing injury to the mitochondrial membrane. Thus, the permeability of the membrane increases, mitochondria swell, and finally the mitochondria are injured.
Relationship between ethanol and mitochondria transmembrane potential
The permeability of the mitochondrial outer membrane is comparatively large, material smaller than 15 kDa can pass through it freely, so substances of the outer compartment of the mitochondria are similar to cytoplasm. The permeability of the mitochondrial inner membrane is small enough to allow the solute smaller than 1.5 kDa to pass through. Oxadative phosphorylation to supply energy takes place in mitochondria. In this process, energy produced by the tricarboxylic acid cycle is transmitted to electrons, which shuttles along respiratory enzymes of the inner mitochondrial membrane, thus manifesting the generation of ATP. The proton pump is localized on the inner membrane, it pumps protons from groundmass to the intermembrane in order to form the transmembrane potential between outer and inner membrane of the mitochondria Δψm[5]. When protons return, they pass energy to ADP and Pi to generate ATP. So Δψm plays a crucial role in keeping the function of mitochondria. Further studies of apoptosis indicate that there is mitochondrial polarization and Δψm disruption in injured cells induced by apoptosis repulsion, moreover these changes take place before alteration due to apoptosis in cells. All of these indicate that Δψm alteration is the early stage of apoptosis[6].
In our experiment, we measured mitochondrial transmembrane potential (Δψm) changes during culture using Rhodamine123 (specific fluorescence mark) staining and flow cytometry. Results reveal that long-term alcohol consumption can cause transmembrane potential significantly lower than the control group, indicating that mitochondrial depolarization and Δψm alteration must be an early-onset, and even a crucial event in the cell death induced by ethanol.
Mechanism of mitochondrial quality degression induced by ethanol
The mitochondrial quality is the amount of all the intra-cellular mitochondria and their content, and it is an important index of mitochondrial injury. Nonyl-Acridine Orange (NAO) is fluorescent can bind specifically to unoxidizable phospholipids. The inner membrane of mitochondria, and the fluorescence intensity is proportional to the quality of phospholipids in the mitochondrial inner membrane[7]. In our experiment, we used NAO flow cytometry to measure the quality of ALD mitochondria. The biological values are to show the quantity of mitochondria with normal respiratory efficiency, and to indicate the degree of injury to the mitochondria. The results show that mitochondrial quality of the model group is obviously lower than that of the control group. This indicates that in the process of ALD, the mitochondrial quality is lowered.
Effect of Ca2+ in hepatocyte apoptosis and ALD injury
Physiologically, hepatocytes are surrounded with a high concentration of Ca2+, and the concentration difference between outer and inner cell is 3-4 orders of magnitudes. Distribution of Ca2+ is uneven intracellularly, the content of Ca2+ is higher in mitochondria and endoplasmic reticulum than in cytoplasm and caryon[8]. Such a concen-tration gradient between organelle and cytoplasm is a precondition that Ca2+ can be an intracellular messenger. Slow accumulation of Ca2+ in mitochondria causes over-loading of Ca2+ so as to make PTP open, leading to a prompt decrease of the membrane potential (Δψm), swelling of mitochondria, and finally apoptosis[9].
In our study, we found that the Ca2+ level of the model group is significantly higher than that of the control group. It indicates that Ca2+ plays an important role in the process of ALD. The Ca2+ signal may induce mitochondrial damage through several possible ways as follows: (1) the elevation of Ca2+ density in hepatocytes makes PTP open, causing Ca2+ to transfer from cytoplasm to mitochondria. This damage is reversible; (2) the over-loading of Ca2+ in mitochondria maintains PTP open, leading to a decrease of Δψm, swelling of mitochondria, and finally apoptosis. So our study indicats that the Ca2+ signal transduction system plays an crucial role in the pathogenesis of ALD.
In summary, our study demonstrates that in the process of hepatocyte injury, ethanol metabolism induce mitochondrial membrane depolarization and permeability changes in cultured hepatocytes. It indicates that mitochondrial damage must be of pathophysiological importance in the process of ALD. The pathway is possibly Ca2+ transferring from cytoplasm to mitochondria, increasing mitochondrial permeability, decreasing mitochondrial membrane potential, finally leading to hepatocyte injury.
Footnotes
Supported by Natural Science Foundation of Shandong Province, No. 032050113
S- Editor Wang J L- Editor Ma JY E- Editor Chen GJ
References
- 1.Alpini G, Phillips JO, Vroman B, LaRusso NF. Recent advances in the isolation of liver cells. Hepatology. 1994;20:494–514. [PubMed] [Google Scholar]
- 2.Loeffler M, Kroemer G. The mitochondrion in cell death control: certainties and incognita. Exp Cell Res. 2000;256:19–26. doi: 10.1006/excr.2000.4833. [DOI] [PubMed] [Google Scholar]
- 3.Crompton M. Mitochondrial intermembrane junctional complexes and their role in cell death. J Physiol. 2000;529 Pt 1:11–21. doi: 10.1111/j.1469-7793.2000.00011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chelli B, Falleni A, Salvetti F, Gremigni V, Lucacchini A, Martini C. Peripheral-type benzodiazepine receptor ligands: mitochondrial permeability transition induction in rat cardiac tissue. Biochem Pharmacol. 2001;61:695–705. doi: 10.1016/s0006-2952(00)00588-8. [DOI] [PubMed] [Google Scholar]
- 5.Zamzami N, Susin SA, Marchetti P, Hirsch T, Gómez-Monterrey I, Castedo M, Kroemer G. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding WX, Shen HM, Ong CN. Critical role of reactive oxygen species and mitochondrial permeability transition in microcystin-induced rapid apoptosis in rat hepatocytes. Hepatology. 2000;32:547–555. doi: 10.1053/jhep.2000.16183. [DOI] [PubMed] [Google Scholar]
- 7.Gohil VM, Gvozdenovic-Jeremic J, Schlame M, Greenberg ML. Binding of 10-N-nonyl acridine orange to cardiolipin-deficient yeast cells: implications for assay of cardiolipin. Anal Biochem. 2005;343:350–352. doi: 10.1016/j.ab.2005.04.039. [DOI] [PubMed] [Google Scholar]
- 8.Woo SH, Park IC, Park MJ, Lee HC, Lee SJ, Chun YJ, Lee SH, Hong SI, Rhee CH. Arsenic trioxide induces apoptosis through a reactive oxygen species-dependent pathway and loss of mitochondrial membrane potential in HeLa cells. Int J Oncol. 2002;21:57–63. [PubMed] [Google Scholar]
- 9.Anuradha CD, Kanno S, Hirano S. Oxidative damage to mitochondria is a preliminary step to caspase-3 activation in fluoride-induced apoptosis in HL-60 cells. Free Radic Biol Med. 2001;31:367–373. doi: 10.1016/s0891-5849(01)00591-3. [DOI] [PubMed] [Google Scholar]