

Abstract
Parkinson's disease (PD) is a major neurodegenerative disorder for which the etiology and pathogenesis remain as elusive as for Alzheimer's disease. PD appears to be caused by genetic and environmental factors, and pedigree and cohort studies have identified numerous susceptibility genes and loci related to PD. Autosomal recessive mutations in the genes Parkin, Pink1, DJ-1, ATP13A2, PLA2G6, and FBXO7 have been linked to PD susceptibility. Such mutations in ATP13A2, also named PARK9, were first identified in 2006 in a Chilean family and are associated with a juvenile-onset, levodopa-responsive type of Parkinsonism called Kufor-Rakeb syndrome (KRS). KRS involves pyramidal degeneration, supranuclear palsy, and cognitive impairment. Here we review current knowledge about the ATP13A2 gene, clinical characteristics of patients with PD-associated ATP13A2 mutations, and models of how the ATP13A2 protein may help prevent neurodegeneration by inhibiting α-synuclein aggregation and supporting normal lysosomal and mitochondrial function. We also discuss another ATP13A2 mutation that is associated with the family of neurodegenerative disorders called neuronal ceroid lipofuscinoses (NCLs), and we propose a single pathway whereby ATP13A2 mutations may contribute to NCLs and Parkinsonism. Finally, we highlight how studies of mutations in this gene may provide new insights into PD pathogenesis and identify potential therapeutic targets.
1. Introduction
Parkinson's disease (PD) is a neurodegenerative disorder for which the etiology and pathogenesis remain elusive, although it is known to be a multifactorial disease involving both genetic and environmental factors. Pedigree and cohort studies of patients with inherited forms of PD, which account for only 5–10% of cases [1], have identified numerous genes and loci associated with PD susceptibility [2, 3]. Autosomal recessive mutations in six of these genes have been linked to the disease: Parkin (PARK2) [4], DJ-1 (PARK7) [5], PINK1 (PARK6) [6], ATP13A2 (PARK9) [7], PLA2G6 (PARK14) [8], and FBXO7 (PARK15) [9].
Autosomal recessive mutations in the ATP13A2 gene were first discovered in 2006 in a single Chilean pedigree [7]. Several members of the family showed a rare, juvenile-onset, levodopa-responsive type of Parkinsonism named Kufor-Rakeb syndrome (KRS), involving pyramidal degeneration, supranuclear palsy, and cognitive impairment. Subsequent studies in several other countries linked other mutations to KRS and early-onset Parkinsonism. At the same time, ATP13A2 mutations have been associated with the occurrence of neurodegenerative disorders called neuronal ceroid lipofuscinoses (NCLs) in patients with Parkinsonism [10]. Some of the NCL-associated mutations overlap with PD-associated ones, suggesting a common pathway in the two types of neurological disease.
Here, we review recent advances in the emerging association of ATP13A2 mutations with Parkinsonism and NCLs. These findings point to the gene and/or protein as a potential therapeutic target.
2. ATP13A2 Mutations and PD
In the first study linking ATP13A2 mutations to PD, pedigree analysis of one Chilean family with several members with KRS led to the identification of two loss-of-function mutations: c.1306+5G>A in exon 13 and 3057delC/1019GfsX1021 in exon 26 [7]. In the same study, the authors also performed pedigree analysis of a Jordanian family with several members with KRS, leading to the identification of a 22-bp duplication in exon 16 (1632_1653dup22 or 552LfsX788). This duplication causes a frameshift, resulting in 236 extraneous amino acids followed by a stop codon. All these mutations were absent in a control group of 480 healthy individuals.
Sequencing the complete ATP13A2 coding region of 46 patients with juvenile- or young-onset PD led to the identification of three additional disease-associated mutations [11]: c.1510G>C/p.Gly504Arg in a Brazilian patient with sporadic PD, c.35C>T/p.Thr12Met (exon 2) in an Italian patient, and c.1597G>A/p.Gly533Arg (exon 16) in another Italian patient. This was the first study to identify any mutation associated with sporadic early-onset PD [11]. Subsequent studies in several countries identified additional novel ATP13A2 mutations in patients with early-onset disease (Table 1), including studies on individuals from Japan [12, 13], China [14–17], Europe [18], Iran [18, 19], Pakistan [20], Afghanistan [21], Lithuania [22], Inuit communities in Greenland [23], and Italy [24].
Table 1.
Review of the literature on ATP13A2 mutations associated with Parkinson's disease.
| Ref. | Author | Year | Country of patient origin | Mutation | Notes |
|---|---|---|---|---|---|
| [7] | Ramirez et al. | 2006 | Chile, Jordan | c.3057delC (p.1019GfsX1021) c.1306+5G>A (p.G399_L435del) c.1632_1653dup22 (p.Leu552fsX788) |
|
|
| |||||
| [11] | Di Fonzo et al. | 2007 | Brazil, Italy | c.1510G>C (p.Gly504Arg) c.35C>T (p.Thr12Met) c.1597G>A (p.Gly533Arg) |
|
|
| |||||
| [12] | Ning et al. | 2008 | Japan | c.546C>A (p.Phe182Leu) | |
|
| |||||
| [14] | Lin et al. | 2008 | Taiwan, Singapore | c.2236G>A (p.Ala746Thr) | Ethnic Chinese |
|
| |||||
| [18] | Djarmati et al. | 2009 | Various European countries | c.746C>T (p.Ala249Val) c.844A>T (p.Ser282Cys) c.2939G>A (p.Arg980His) |
|
| Iran | c.1346G>A (p.Arg449Gln) | ||||
|
| |||||
| [20] | Schneider et al. | 2010 | Pakistan | c.1103_1104insGA (p.Thr367fsX29) | |
|
| |||||
| [25] | Fei et al. | 2010 | China (mainland) | c.2236G>A (p.Ala746Thr) | |
|
| |||||
| [26] | Mao et al. | 2010 | China (mainland) | c.2236G>A (p.Ala746Thr) | |
|
| |||||
| [13] | Funayama et al. | 2010 | Japan | c.2236G>A (p.Ala746Thr) | |
|
| |||||
| [15] | Chen et al. | 2011 | Taiwan | c.3274A>G (p.Gly1014Ser) | Ethnic Chinese |
|
| |||||
| [21] | Fong et al. | 2011 | Lithuania | c.1108_1120del13 (p.Arg370fsX390) | |
|
| |||||
| [16] | Park et al. | 2011 | Various Asian countries | c.3176T>G (p.Leu1059Arg) c.3253delC (p.L1085wfsX1088) |
|
|
| |||||
| [22] | Crosiers et al. | 2011 | Afghanistan | c.2742_2743delTT (p.F851CfsX856) | |
|
| |||||
| [23] | Eiberg et al. | 2012 | Greenland | c.2473C>AA (p.Leu825fs) | Ethnic Inuits |
|
| |||||
| [17] | Zhu et al. | 2012 | China (mainland) | c.1754G>T (p. Ala585Asp) | Ethnic Chinese |
|
| |||||
| [24] | Santoro et al. | 2011 | Italy | c.2629G>A (p.Gly877Arg) | |
|
| |||||
| [27] | Chan et al. | 2013 | China (Hong Kong) | c.2236G>A (p.Ala746Thr) | Ethnic Chinese |
|
| |||||
| [28] | Darvish et al. | 2013 | Iran | Deletion of exon 2 | |
|
| |||||
| [19] | Malakouti-Nejad et al. | 2014 | Iran | c.2762C>T (p.Gln858∗) | |
The ATP13A2 mutation c.2236G>A/p.Ala746Thr (exon 20) was identified in three ethnic Chinese individuals from Taiwan and Singapore, two of whom had late-onset PD [14].
However, two subsequent studies failed to detect this mutation in patients with early- or late-onset PD from mainland China and Hong Kong [25–27]. A third study of 65 Chinese patients with early-onset PD detected the Ala746Thr mutation in two patients and four healthy controls [15]. The same study also discovered a novel mutation associated with early-onset disease (c.3274 A>G, Gly1014Ser, exon 26). These studies highlight the need for more research, particularly on Chinese individuals, to identify additional mutations associated with disease and to resolve conflicting results about the Ala746Thr mutation.
Studies using multiplex ligation-dependent probe amplification (MLPA) to measure exon dosage in Iranian patients found deletion of ATP13A2 exon 2 to be associated with KRS [28]. Three of the 232 affected individuals in the study came from the same family and showed an average age of disease onset of 12 years. Genomic rearrangements were not detected among patients with sporadic or familial PD. In fact, several studies have failed to identify associations of ATP13A2 mutations with sporadic PD or non-KRS familial PD [25, 29] or with late-onset PD [30]. These findings highlight the need to examine ATP13A2 mutations in patients with sporadic or familial PD from a broad range of ethnicities, in order to clarify whether the mutations are associated only with juvenile- or young-onset Parkinsonism or perhaps only with KRS.
3. Clinical Characteristics of PD Patients Carrying ATP13A2 Mutations
KRS was initially described in a family with Parkinsonism in the Kufor-Rakeb district in Jordan; affected individuals show a juvenile-onset, levodopa-responsive form of PD involving pyramidal signs, dementia, and supranuclear gaze palsy [31]. These symptoms are quite similar to those of pallidopyramidal syndrome, though KRS differs in that it involves dystonia, which is attributable to pyramidal dysfunction, as well as cognitive dysfunction and supranuclear upgaze paresis [31, 32].
From the literature, we extracted general clinical characteristics of 34 PD patients with ATP13A2 mutations (Table 2). Most patients had KRS or early-onset disease, either sporadic or familial; two patients had late-onset PD. Slightly more patients were male (21, 56.7%) than female (16, 43.3%); the average age of onset was 23.7 ± 13.8 years. The youngest patient was a 12-year-old Lithuanian boy who had had the disease for 6 years before his case was published; the oldest patient was a 63-year-old Taiwanese woman. Initial symptoms were diverse and included bradykinesia, dystonia, gait disturbance, mental retardation, anxiety, postural instability, and rest tremor. Clinical symptoms were varied and followed the following distribution from the most to the least frequent: rigidity (n = 37), bradykinesia (33), postural instability (29), supranuclear upgaze paresis (22), cognitive impairment (19), dystonia (17), resting tremor (17), hallucination (16), and myoclonus (16). A uni- or bilateral Babinski sign was present in 27 of 37 patients.
Table 2.
Clinical features of patients with Parkinson's disease and mutations in the ATP13A2 gene.
| Ref. | Internal code | Mutation | Country of origin | AO (years) | G | FH | IS | MS | MC | SUP | DYS | CD | H | BS | Response to levodopa | Imaging findings |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [26] | V44 | 1632_1653dup22 (552LeufsX788) | Jordan | 12 | M | + | B, MR, R | B, R, PI | + | + | − | + | + | + | + | Diffuse atrophy (MRI) |
| V48 | Jordan | 15 | M | + | B, R | B, R, PI | + | + | + | + | + | + | + | Diffuse atrophy (MRI) | ||
| V49 | Jordan | 13 | M | + | MR, R | B, R, PI | + | − | + | + | + | + | + | Diffuse atrophy (MRI) | ||
| V53 | Jordan | 12 | F | + | B | B, R, PI | + | − | + | + | + | + | + | NR | ||
|
| ||||||||||||||||
| [1] | II-8 | c.3057delC (p.1019GfsX1021) c.1306+5G>A (p.G399_L435del) |
Chile | 18 | M | + | BR, F | T, B, R | + | + | NR | + | + | + | Never tried | Enlarged sulci (CT) |
| II-9 | Chile | 17 | M | + | BR, B, R | T, B, R, PI | + | − | NR | + | + | − | − | Mild, diffuse atrophy; caudate hypointensity (MRI) | ||
| II-10 | Chile | 15 | F | + | B, F, BR | T, B, R, PI | + | + | NR | + | − | + | − | NR | ||
| II-11 | Chile | 12 | M | + | F, BR | T, B, R, PI | + | + | NR | + | + | + | − | Diffuse atrophy (CT) | ||
|
| ||||||||||||||||
| [11] | BR-3042 | c.1510G>C (Gly504Arg) | Brazil | 12 | M | − | B | B, R, PI | + | + | NR | + | + | + | + | Diffuse atrophy (CT) |
| VE29 | c.35C>T (Thr12Met) | Italy | 30 | M | + | NA | T, B, R, PI | NR | + | + | − | − | + | + | NR | |
| PK-69-1 | c.1597C>A (Gly533Arg) | Italy | 40 | M | + | NA | B, R, PI | NR | + | + | − | + | + | + | NR | |
|
| ||||||||||||||||
| [18] | L-1349 | c.746C>T (Ala249Val) | Germany | 31 | F | − | T | T, B, R, PI | NR | + | − | NR | + | − | + | Normal (MRI) |
| L-1928 | c.844A>T (Ser282Cys) | Norway | 20 | M | − | PI | R, PI | NR | + | − | NR | − | + | + | Normal (MRI) | |
| L-324 | c.1346G>A (Arg449Gln) | Iran | 36 | M | − | T | B, R, PI | NR | + | − | NR | + | + | + | Cerebral atrophy (CT) | |
| P-55 | c.2939G>A (Arg980His) | Serbia | 35 | F | − | PT | T, B, R, PI | NR | + | − | NR | + | + | + | Normal | |
|
| ||||||||||||||||
| [20] | NR | c.1103_1104insGA (p.Thr367fsX29) | Pakistan | 16 | M | − | B, MR | B, R, PI | − | + | + | + | + | + | + | Diffuse atrophy (MRI) |
|
| ||||||||||||||||
| [22] | II-3 | c.2742_2743delTT (p.F851CfsX856) | Afghanistan | 10 | M | − | B, MR | B, R, PI | + | + | + | + | − | + | + | Diffuse atrophy, bilateral hypointensity in putamina and caudate nuclei (MRI) |
|
| ||||||||||||||||
| [21] | NR | c.1108_1120del13 (p.Arg370fsX390) | Lithuania | 6 | M | − | Dysarthria, DYS | T, B, R, PI | NR | − | + | − | NR | − | + | Normal (MRI) |
|
| ||||||||||||||||
| [23] | VI-1 | c.2473C>AA, (p.Leu825AsnfsX32) | Greenland | 27 | F | + | FA | NR | NR | NR | NR | + | + | + | NR | Diffuse atrophy (MRI) |
| VI-6 | Greenland | 24 | M | + | Weakness | NR | NR | NR | + | + | + | + | NR | Diffuse atrophy (MRI) | ||
| V-1 | Greenland | 12 | M | + | T | B, R | + | + | + | + | + | + | NR | Normal (MRI) | ||
| V-3 | Greenland | 10 | F | + | CD | T, B, R | − | − | − | − | + | − | NR | NR | ||
| V-5 | Greenland | 29 | F | + | GD | PI | + | + | − | + | − | + | NR | Diffuse atrophy (MRI) | ||
| V-9 | Greenland | 15 | F | + | B, MR | T, B, R | + | NR | NR | + | − | NR | NR | NR | ||
|
| ||||||||||||||||
| [19] | X4015 | c.2762C>T (p.Gln858∗) | Iran | 14 | F | + | Motor defect | T, B, R, PI | NR | + | + | + | − | + | + | Diffuse atrophy (MRI) |
| X4041 | Iran | 10 | M | + | B | T, B, R, PI | NR | + | + | + | − | + | + | Diffuse atrophy (MRI) | ||
| R1042 | Iran | 30 | M | + | NR | T, B, R, PI | NR | NR | NR | NR | NR | NR | + | NR | ||
|
| ||||||||||||||||
| [24] | NAPO6 | c.2629G>A (Gly877Arg) | Italy | 10 | M | − | GD | B, R, PI | + | + | + | + | − | + | + | Diffuse atrophy (MRI) |
|
| ||||||||||||||||
| [12] | A | c.546C>A (Phe182Leu) | Japan | 22 | F | − | GD | T, B, R, PI | + | + | + | + | + | + | + | Diffuse atrophy (MRI) |
|
| ||||||||||||||||
| [14] | F37 | c.2236G>A (Ala746Thr) | China | 53 | F | − | NA | T, B, R | − | − | + | − | NR | + | + | Normal (MRI) |
| EK1 | China | 50 | M | − | NA | T, B, R, PI | − | − | + | − | NR | + | + | Normal (MRI) | ||
| Y56 | China | 39 | M | − | NA | T, B, R, PI | − | − | + | − | NR | + | + | Normal (MRI) | ||
|
| ||||||||||||||||
| [15] | H1288 | c.3274A>G (p.Gly1014Ser) | China | 48 | F | − | NA | T, B, R, PI | NR | NR | NR | NR | NR | NR | + | Normal (MRI) |
| H496 | c.2236G>A (Ala746Thr) | China | 49 | M | − | NA | T, B, R, PI | NR | NR | NR | NR | NR | NR | + | NR | |
| H2120 | c.2236G>A (Ala746Thr) | China | 51 | F | − | NA | T, B, R, PI | NR | NR | NR | NR | NR | NR | + | NR | |
|
| ||||||||||||||||
| [16] | NR | c.3176C>G (Leu1059Arg), c.3253delC (L1085wfsX1088) |
China | 17 | M | + | A | B, R | + | + | + | NR | − | + | + | Normal (MRI) |
| NR | China | 17 | F | + | A, D | B, R | + | + | + | NR | − | + | + | Normal (MRI) | ||
A: anxiety; AO: age of onset; B: bradykinesia; BS: Babinski sign; CD: cognitive dysfunction; D: depression; DYS: dystonia; F: female; FA: fatigue; FH: family history; G: gender; GD: gait disturbance; IS: initial symptom; M: male; MC: myoclonus; MS: motor symptom; NR: not reported; PI: postural instability; PT: postural tremor; R: rigidity; SUP: supranuclear upgaze palsy; T: tremor.
Most patients were examined by computed tomography (CT) or magnetic resonance imaging (MRI); the most frequent significant features were an enlarged subarachnoid space and diffuse atrophy ranging from mild to severe. Only two patients, an adolescent from Pakistan [20] and an adolescent from Chile [33], showed abnormal bilateral hypointensity in the putaminal and caudate nuclei on T2∗ diffuse MRI images. The clinicians attending the Pakistani patient were able to exclude manganese deposition as the cause of hypointensity, since the patient did not experience manganese exposure or show chronic liver failure; copper deposition, since the patient showed normal serum levels of copper and ceruloplasmin, and the slit lamp test showed no K-F ring; and calcium deposition, since the patient showed normal CT results. In the end, the clinicians attributed the abnormal MRI hypointensity to iron deposition. The clinicians attending the Chilean patient also attributed the hypointensity to ferritin deposits based on the absence of hypointensity on brain CT images, though they did not perform tests to exclude the possibility of deposition of other metals [33].
By single-photon emission CT (SPECT), patient NAPO6, an Italian with ATP13A2 mutation c.G2629A, showed specific-to-nondisplaceable V′′3 binding ratios that were 75% lower in the caudate and 85% lower in the putamen than those of healthy individuals [24]. His younger brother, designated NAPO7, carried the same ATP13A2 mutation and showed mild mental retardation but no clinically obvious Parkinsonism. His V′′3 ratio was 40% lower than normal in the caudate and 65% lower in the putamen, consistent with the fact that mild retardation can be an initial symptom of PD [9, 32]. These results suggest that combining genotyping of PD susceptibility genes with positron emission tomography or SPECT may improve diagnosis of early-stage PD, especially in subclinical patients.
4. Physiological Role of ATP13A2 and Link to PD
4.1. ATP13A2 and Function of Lysosomes and Mitochondria
ATP13A2 encodes a lysosomal transmembrane protein belonging to the 5P-type ATPase subfamily [34]. Wild-type ATP13A2 localizes to the lysosome, while all mutant forms associated with PD localize to the endoplasmic reticulum (ER) [9, 16, 35, 36]. In contrast to genes for other 5P-type ATPases, ATP13A2 in mice is expressed mainly in the brain, suggesting a brain-specific function. ATP13A2 levels in the substantia nigra are substantially lower in postmortem tissue biopsies of patients with sporadic PD than in the corresponding samples from healthy controls [37, 38], but they are higher in survival dopaminergic (DA) neurons of patients than in those of controls [37]. ATP13A2 levels are particularly high in the cytosol of nigral dopaminergic neurons, where the protein accumulates in Lewy bodies [37].
These circumstantial data implicate ATP13A2 in the pathogenesis and/or progression of PD, but more direct evidence requires insights into the function of the ATP13A2 protein. Studies with cultures of fibroblast cells and DA cells taken from PD patients with ATP13A2 mutations showed that inhibiting ATP13A2 function decreased the ability of lysosomes to degrade proteins and mediate clearance of autophagosomes [37]. These cellular functions returned to near-normal levels after ATP13A2 activity was restored. These results suggest that ATP13A2 is required for normal lysosome function, which is in turn required for preventing α-synuclein aggregation in neurons (Figure 1(a)). This aggregation is a pathological hallmark of both sporadic and familial PD [39].
Figure 1.
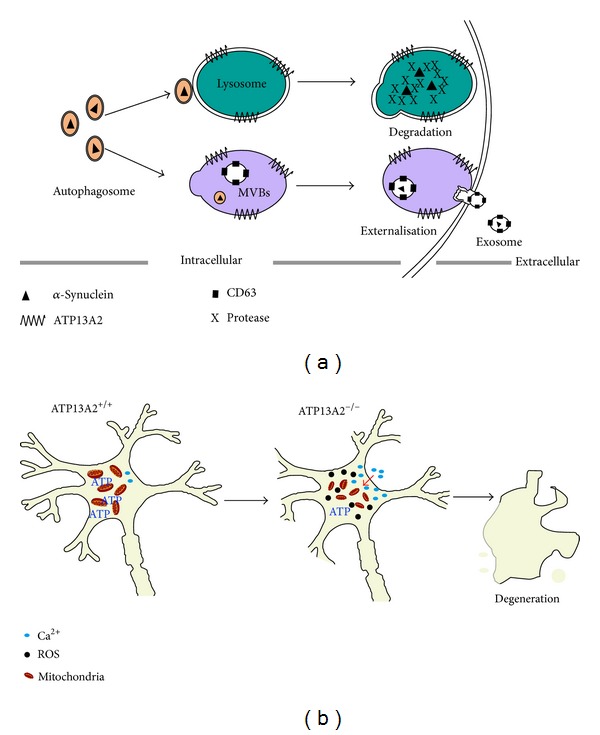
Model of how ATP13A2 expression may affect lysosomes and mitochondria to prevent neurodegeneration. (a) After α-synuclein has been internalized by autophagosomes, it can be immediately degraded in lysosomes containing ATP13A2 or secreted out of the cell via multivesicular bodies (MVBs) also containing ATP13A2. Both routes prevent intracellular accumulation of α-synuclein. (b) Knocking out ATP13A2 expression in neurons leads to mitochondrial defects, resulting in higher intracellular levels of reactive oxygen species (ROS) and Ca2+, both of which contribute to neurodegeneration.
Several additional studies provide further evidence that ATP13A2 prevents α-synuclein aggregation. SH-SY5Y cultures overexpressing ATP13A2 showed lower intracellular levels of α-synuclein, perhaps because of increased α-synuclein export via multivesicular bodies (MVBs) (Figure 1(a)) [40]. In both whole-animal and neuronal culture models of PD, coexpressing ATP13A2 with α-synuclein led to lower synuclein levels in DA neurons than expressing synuclein alone [41]. Neuronal cultures lacking the ATP13A2 gene showed significantly higher endogenous levels of α-synuclein than did the corresponding wild-type neurons [42]. Intriguingly the ATP13A2-knockout neurons did not show elevated levels or aggregates of tau protein, which may play an important role in the pathogenesis of Alzheimer's disease (AD). This raises the possibility that ATP13A2 interacts preferentially with α-synuclein, consistent with a recent study showing that ATP13A2 colocalized with α-synuclein in Lewy bodies but not with β-amyloid [38].
In addition to ensuring proper lysosomal function, ATP13A2 may work in mitochondria, such that the reduced activity of ATP13A2 mutants may lead to mitochondrial defects that contribute to neurodegeneration (Figure 1(b)) [43]. Fibroblasts from patients with KRS showed lower mitochondrial membrane potential and ATP synthesis rates than fibroblasts from healthy individuals [33]. Cell cultures deficient in ATP13A2 showed lower levels of autophagy than healthy cells, leading to higher levels of reactive oxygen species and concomitant oxidative stress [44]. Overexpressing ATP13A2 in neurons inhibited cadmium-induced mitochondrial fragmentation, while silencing ATP13A2 expression induced mitochondrial fragmentation [45] (Figure 1(b)). That same study further showed that increasing or decreasing ATP13A2 expression substantially shortened the neurites of primary midbrain DA neurons, without affecting neurites of cortical neurons. This may mean that the morphological and functional integrity of DA neurons depends on well-controlled ATP13A2 expression [45].
The available evidence suggests that ATP13A2, by supporting lysosomal and mitochondrial function, helps prevent the α-synuclein aggregation associated with Parkinsonism [46–49]. The implication is that the ATP13A2 mutations linked to KRS and other forms of PD are loss-of-function mutations that reduce ATP13A2 activity sufficiently to induce neurodegeneration. Future studies should examine in detail the activity, localization, and binding partners of these mutant proteins.
4.2. ATP13A2 and Cation Accumulation
ATP13A2 plays a critical role in the transmembrane transport of manganese and zinc and perhaps of iron and cadmium as well [15]; abnormal accumulation of any of these cations can cause neurodegeneration [41, 50–52]. Thus, patients with PD have been reported to show elevated levels of manganese and zinc in serum and cerebrospinal fluid [53–55], and manganese and zinc exposure are significant environmental risk factors for PD [56, 57]. ATP13A2 helps protect cells from this toxicity by regulating the homeostasis of manganese and zinc in neurons [41, 44, 58, 59]. It may be that dysregulation of ATP13A2 expression disrupts the homeostasis of manganese and zinc in the brain, leading to neurodegeneration.
This possibility is consistent with the interpretation of the abnormal hypointensity in the putamina and caudate nuclei of patients with KRS in T2∗ diffuse MRI images as iron deposits (see Section 3). This finding led those authors to propose KRS with iron deposits as a distinct condition called neurodegeneration with brain iron accumulation (NBIA) [20]. Indeed, iron accumulation was reported in the substantia nigra of PD patients [60], where it was particularly abundant in DA neurons [61]. Administering the iron chelator deferiprone to an animal model of PD induced by oxidative stress improved motor function and increased dopamine levels in the striatum [62]. In a pilot randomized clinical trial, double-blind and placebo-controlled, deferiprone showed some ability to delay or reverse the progression of PD [62].
How mutations in ATP13A2 may affect cation deposition is unclear. We speculate that loss-of-function mutations in ATP13A2 may work similarly to silencing of the PANK2 gene, which disrupts normal cation transfer and leads to mitochondrial and lysosomal dysfunction and ultimately to cation accumulation in the brain [63, 64]. In this way, ATP13A2 mutants may trigger deposition of the cations zinc, manganese, and iron, leading to metal-induced oxidative damage and ultimately causing decreases in glutathione peroxidase activity, glutathione (GSH) levels, and mitochondrial Complex I activity, as well as increases in levels of basal lipid peroxidation, free radicals, and glutamate [65–67]. The net result is significant neuronal loss that is the distinguishing pathological feature of PD.
This proposed mechanism implies that regulating or restoring the homeostasis of neurotoxic cations may be a neuroprotective therapy for patients with PD. However, only two of the 37 PD patients with ATP13A2 mutations that we reviewed showed cation accumulation on T2∗ diffuse MRI images (Table 2), and direct postmortem pathological evidence for metal accumulation in PD is lacking [20, 33]. Further studies are urgently needed to clarify whether ATP13A2 mutations contribute to PD by increasing susceptibility to cation toxicity.
5. ATP13A2 Mutations: A Link between Parkinsonism and NCLs
ATP13A2 mutations have been identified not only in patients with Parkinsonism, but also in patients with neuronal ceroid lipofuscinoses (NCLs) [10]. NCLs are a group of neurodegenerative disorders that are also lysosomal storage diseases. Clinical manifestations are seizures, progressive cognitive and motor decline, and failing vision. The pathological hallmark of NCLs is accumulation of autofluorescent lipopigment within neuronal lysosomes [68].
Recently, the mutation c.2429C>G in exon 22 of ATP13A2, predicted to result in the amino acid substitution p.Met810Arg, was identified in a Belgian family with NCLs [10]. Affected individuals showed not only typical NCL symptoms but also extrapyramidal involvement. Postmortem pathological examination revealed extensive lipofuscin deposits in the cortex, basal nuclei, cerebellum, and retina—but not the white matter—and electron microscopy showed whorled lamellar inclusions typical of NCLs [10]. A link between ATP13A2 mutations and NCL pathogenesis is further supported by studies in animal models [69, 70]. In fact, mice deficient in ATP13A2 exhibited neuronal ceroid lipofuscinosis, α-synuclein accumulation, and age-dependent sensorimotor deficits, suggesting that PD and NCLs share a pathogenic mechanism [71].
A shared disease pathway may help explain earlier reports of individuals who demonstrate an “overlapping” neurodegenerative syndrome combining Parkinsonism and NCLs [72–76]. ATP13A2 is a lysosomal transport protein that helps maintain optimal pH in lysosomes [46], and ceramide is metabolized in lysosomes [77]. The apoptosis that appears to cause NCLs is associated with increased levels of ceramide [78, 79], which have also been linked to α-synuclein deposition, which may contribute to PD pathogenesis [80]. It may be that ATP13A2 helps regulate ceramide metabolism, such that significant changes in ATP13A2 activity may contribute to the pathogenesis of both PD and NCLs. This model is similar to that of the lysosomal storage disorder called Gaucher disease. The homozygous mutations in the β-glucocerebrosidase gene that cause Gaucher disease also increase risk of PD [81]. Both diseases arise because lysosomal dysfunction leads to excessive aggregation of substrates that normally are degraded. Analogously, lysosomal dysfunction may underlie the clinically different neurodegenerative disorders of PD and NCLs.
6. Summary
Much has been learned about the physiological functions of ATP13A2 since mutations in the ATP13A2 gene were first linked to autosomal recessive familial KRS [7]. Patients with such mutations show onset at earlier ages than patients with other forms of PD, as well as some atypical clinical symptoms such as pyramidal degeneration, supranuclear palsy, cognitive impairment, and dystonia. Studies in animal models of PD and in cultures of cells taken from patients with KRS and other types of PD suggest that ATP13A2 is important for proper functioning of lysosomes and mitochondria and perhaps for clearance of divalent metals; defects in any of these three processes are tightly associated with neurodegeneration. Nevertheless, more studies are needed that directly examine how PD-associated mutations in ATP13A2 affect the activity and localization of the protein and ultimately the integrity of these three processes. ATP13A2 mutations that affect one of these processes, lysosomal functioning, may simultaneously increase the risk of PD and NCLs. In other words, these quite clinically different diseases may share a mechanism of lysosomal dysfunction. If further studies validate the literature, the ATP13A2 gene and/or protein may become a suitable therapeutic target for treating both PD and NCLs.
Acknowledgments
Work by the authors related to this review was supported by the Sichuan Key Project of Science and Technology (no. 2010SZ0086) and the National Natural Science Foundation of China (no. 30700243).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Lesage S, Brice A. Parkinson's disease: from monogenic forms to genetic susceptibility factors. Human Molecular Genetics. 2009;18(R1):R48–R59. doi: 10.1093/hmg/ddp012. [DOI] [PubMed] [Google Scholar]
- 2.Bonifati V. Genetics of Parkinson's disease—state of the art. Parkinsonism & Related Disorders. 2014;20(supplement 1):S23–S28. doi: 10.1016/S1353-8020(13)70009-9. [DOI] [PubMed] [Google Scholar]
- 3.Coppedè F. Genetics and epigenetics of Parkinson's disease. Scientific World Journal. 2012;2012:12 pages. doi: 10.1100/2012/489830.489830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lesage S, Lohmann E, Tison F, Durif F, Dürr A, Brice A. Gene symbol: PARK2. Disease: parkinsonism, juvenile, autosomal recessive. Human genetics. 2008;123(1, article 114) [PubMed] [Google Scholar]
- 5.Bonifati V, Rizzu P, Van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299(5604):256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 6.Li Y, Tomiyama H, Sato K, et al. Clinicogenetic study of PINK1 mutations in autosomal recessive early-onset parkinsonism. Neurology. 2005;64(11):1955–1957. doi: 10.1212/01.WNL.0000164009.36740.4E. [DOI] [PubMed] [Google Scholar]
- 7.Ramirez A, Heimbach A, Gründemann J, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nature Genetics. 2006;38(10):1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 8.Tomiyama H, Yoshino H, Ogaki K, et al. PLA2G6 variant in Parkinson’s disease. Journal of Human Genetics. 2011;56(5):401–403. doi: 10.1038/jhg.2011.22. [DOI] [PubMed] [Google Scholar]
- 9.Di Fonzo A, Dekker MCJ, Montagna P, et al. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology. 2009;72(3):240–245. doi: 10.1212/01.wnl.0000338144.10967.2b. [DOI] [PubMed] [Google Scholar]
- 10.Bras J, Verloes A, Schneider SA, Mole SE, Guerreiro RJ. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Human Molecular Genetics. 2012;21(12):2646–2650. doi: 10.1093/hmg/dds089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Fonzo A, Chien HF, Socal M, et al. ATP13A2 missense mutations in juvenile Parkinsonism and young onset Parkinson disease. Neurology. 2007;68(19):1557–1562. doi: 10.1212/01.wnl.0000260963.08711.08. [DOI] [PubMed] [Google Scholar]
- 12.Ning YP, Kanai K, Tomiyama H, et al. PARK9-linked parkinsonism in eastern Asia: mutation detection in ATP13A2 and clinical phenotype. Neurology. 2008;70(16, part 2):1491–1493. doi: 10.1212/01.wnl.0000310427.72236.68. [DOI] [PubMed] [Google Scholar]
- 13.Funayama M, Tomiyama H, Wu RM, et al. Rapid screening of ATP13A2 variant with highresolution melting analysis. Movement Disorders. 2010;25(14):2434–2437. doi: 10.1002/mds.23106. [DOI] [PubMed] [Google Scholar]
- 14.Lin CH, Tan EK, Chen ML, et al. Novel ATP13A2 variant associated with Parkinson disease in Taiwan and Singapore. Neurology. 2008;71(21):1727–1732. doi: 10.1212/01.wnl.0000335167.72412.68. [DOI] [PubMed] [Google Scholar]
- 15.Chen C-M, Lin C-H, Juan H-F, et al. ATP13A2 variability in Taiwanese Parkinson's disease. American Journal of Medical Genetics B: Neuropsychiatric Genetics. 2011;156(6):720–729. doi: 10.1002/ajmg.b.31214. [DOI] [PubMed] [Google Scholar]
- 16.Park J, Mehta P, Cooper AA, et al. Pathogenic effects of novel mutations in the P-type ATPase ATP13A2 (PARK9) causing Kufor-Rakeb syndrome, a form of early-onset parkinsonism. Human Mutation. 2011;32(8):956–964. doi: 10.1002/humu.21527. [DOI] [PubMed] [Google Scholar]
- 17.Zhu LH, Luo XG, Zhou YS, et al. Lack of association between three single nucleotide polymorphisms in the PARK9, PARK15, and BST1 genes and Parkinson's disease in the northern Han Chinese population. Chinese Medical Journal. 2012;125(4):588–592. [PubMed] [Google Scholar]
- 18.Djarmati A, Hagenah J, Reetz K, et al. ATP13A2 variants in early-onset Parkinson's disease patients and controls. Movement Disorders. 2009;24(14):2104–2111. doi: 10.1002/mds.22728. [DOI] [PubMed] [Google Scholar]
- 19.Malakouti-Nejad M, Shahidi GA, Rohani M, et al. Identification of p.Gln858∗ in ATP13A2 in two EOPD patients and presentation of their clinical features. Neuroscience Letters. 2014;577:106–111. doi: 10.1016/j.neulet.2014.06.023. [DOI] [PubMed] [Google Scholar]
- 20.Schneider SA, Paisan-Ruiz C, Quinn NP, et al. ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Movement Disorders. 2010;25(8):979–984. doi: 10.1002/mds.22947. [DOI] [PubMed] [Google Scholar]
- 21.Fong CY, Rolfs A, Schwarzbraun T, Klein C, O'Callaghan FJK. Juvenile parkinsonism associated with heterozygous frameshift ATP13A2 gene mutation. European Journal of Paediatric Neurology. 2011;15(3):271–275. doi: 10.1016/j.ejpn.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Crosiers D, Ceulemans B, Meeus B, et al. Juvenile dystonia-parkinsonism and dementia caused by a novel ATP13A2 frameshift mutation. Parkinsonism and Related Disorders. 2011;17(2):135–138. doi: 10.1016/j.parkreldis.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 23.Eiberg H, Hansen L, Korbo L, et al. Novel mutation in ATP13A2 widens the spectrum of Kufor-Rakeb syndrome (PARK9) Clinical Genetics. 2012;82(3):256–263. doi: 10.1111/j.1399-0004.2011.01745.x. [DOI] [PubMed] [Google Scholar]
- 24.Santoro L, Breedveld GJ, Manganelli F, et al. Novel ATP13A2 (PARK9) homozygous mutation in a family with marked phenotype variability. Neurogenetics. 2011;12(1):33–39. doi: 10.1007/s10048-010-0259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fei QZ, Cao L, Xiao Q, et al. Lack of association between ATP13A2 Ala746Thr variant and Parkinson’s disease in Han population of mainland China. Neuroscience Letters. 2010;475(2):61–63. doi: 10.1016/j.neulet.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 26.Mao XY, Burgunder JM, Zhang ZJ, et al. ATP13A2 G2236A variant is rare in patients with early-onset Parkinson's disease and familial Parkinson's disease from mainland China. Parkinsonism and Related Disorders. 2010;16(3):235–236. doi: 10.1016/j.parkreldis.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 27.Chan AYY, Baum L, Tang NLS, et al. The role of the Ala746Thr variant in the ATP13A2 gene among Chinese patients with Parkinson's disease. Journal of Clinical Neuroscience. 2013;20(5):761–762. doi: 10.1016/j.jocn.2012.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Darvish H, Movafagh A, Omrani MD, et al. Detection of copy number changes in genes associated with Parkinson's disease in Iranian patients. Neuroscience Letters. 2013;551:75–78. doi: 10.1016/j.neulet.2013.07.013. [DOI] [PubMed] [Google Scholar]
- 29.Vilariño-Güell C, Soto AI, Lincoln SJ, et al. ATP13A2 variability in Parkinson disease. Human Mutation. 2009;30(3):406–410. doi: 10.1002/humu.20877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rakovic A, Stiller B, Djarmati A, et al. Genetic association study of the P-type ATPase ATP13A2 in late-onset Parkinson's disease. Movement Disorders. 2009;24(3):429–433. doi: 10.1002/mds.22399. [DOI] [PubMed] [Google Scholar]
- 31.Najim Al-Din AS, Wriekat A, Mubaidin A, Dasouki M, Hiari M. Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurologica Scandinavica. 1994;89(5):347–352. doi: 10.1111/j.1600-0404.1994.tb02645.x. [DOI] [PubMed] [Google Scholar]
- 32.Williams DR, Hadeed A, Najim al-Din AS, Wreikat A, Lees AJ. Kufor Rakeb disease: autosomal recessive, levodopa-responsive Parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia. Movement Disorders. 2005;20(10):1264–1271. doi: 10.1002/mds.20511. [DOI] [PubMed] [Google Scholar]
- 33.Behrens MI, Brüggemann N, Chana P, et al. Clinical spectrum of Kufor-Rakeb syndrome in the Chilean kindred with ATP13A2 mutations. Movement Disorders. 2010;25(12):1929–1937. doi: 10.1002/mds.22996. [DOI] [PubMed] [Google Scholar]
- 34.Schultheis PJ, Hagen TT, O’Toole KK, et al. Characterization of the P5 subfamily of P-type transport ATPases in mice. Biochemical and Biophysical Research Communications. 2004;323(3):731–738. doi: 10.1016/j.bbrc.2004.08.156. [DOI] [PubMed] [Google Scholar]
- 35.Schröder B, Wrocklage C, Pan C, et al. Integral and associated lysosomal membrane proteins. Traffic. 2007;8(12):1676–1686. doi: 10.1111/j.1600-0854.2007.00643.x. [DOI] [PubMed] [Google Scholar]
- 36.Matsui H, Sato F, Sato S, et al. ATP13A2 deficiency induces a decrease in cathepsin D activity, fingerprint-like inclusion body formation, and selective degeneration of dopaminergic neurons. FEBS Letters. 2013;587(9):1316–1325. doi: 10.1016/j.febslet.2013.02.046. [DOI] [PubMed] [Google Scholar]
- 37.Dehay B, Ramirez A, Martinez-Vicente M, et al. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(24):9611–9616. doi: 10.1073/pnas.1112368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy KE, Cottle L, Gysbers AM, Cooper AA, Halliday GM. ATP13A2 (PARK9) protein levels are reduced in brain tissue of cases with Lewy bodies. Acta Neuropathologica Communications. 2013;1, article 11 doi: 10.1186/2051-5960-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tofaris GK. Lysosome-dependent pathways as a unifying theme in Parkinson's disease. Movement Disorders. 2012;27(11):1364–1369. doi: 10.1002/mds.25136. [DOI] [PubMed] [Google Scholar]
- 40.Kong SM, Chan BK, Park JS, et al. Parkinson's disease-linked human PARK9/ ATP13A2 maintains zinc homeostasis and promotes α-Synuclein externalization via exosomes. Human Molecular Genetics. 2014;23(11):2816–2833. doi: 10.1093/hmg/ddu099. [DOI] [PubMed] [Google Scholar]
- 41.Gitler AD, Chesi A, Geddie ML, et al. α-Synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nature Genetics. 2009;41(3):308–315. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. The Journal of Neuroscience. 2012;32(12):4240–4246. doi: 10.1523/JNEUROSCI.5575-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park JS, Koentjoro B, Veivers D, Mackay-Sim A, Sue CM. Parkinson's disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis andmitochondrial dysfunction. Human Molecular Genetics. 2014;23(11):2802–2815. doi: 10.1093/hmg/ddt623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gusdon AM, Zhu J, van Houten B, Chu CT. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiology of Disease. 2012;45(3):962–972. doi: 10.1016/j.nbd.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramonet D, Podhajska A, Stafa K, et al. PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Human Molecular Genetics. 2012;21(8):1725–1743. doi: 10.1093/hmg/ddr606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nixon RA, Yang DS, Lee JH. Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy. 2008;4(5):590–599. doi: 10.4161/auto.6259. [DOI] [PubMed] [Google Scholar]
- 47.Dehay B, Martinez-Vicente M, Caldwell GA, et al. Lysosomal impairment in Parkinson's disease. Movement Disorders. 2013;28(6):725–732. doi: 10.1002/mds.25462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olanow CW, Brundin P. Parkinson's disease and alpha synuclein: is Parkinson's disease a prion-like disorder? Movement Disorders. 2013;28(1):31–40. doi: 10.1002/mds.25373. [DOI] [PubMed] [Google Scholar]
- 49.Bové J, Martínez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nature Reviews Neuroscience. 2011;12(8):437–452. doi: 10.1038/nrn3068. [DOI] [PubMed] [Google Scholar]
- 50.Schmidt K, Wolfe DM, Stiller B, Pearce DA. Cd2+, Mn2+, Ni2+ and Se2+ toxicity to Saccharomyces cerevisiae lacking YPK9p the orthologue of human ATP13A2. Biochemical and Biophysical Research Communications. 2009;383(2):198–202. doi: 10.1016/j.bbrc.2009.03.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mastroberardino PG, Hoffman EK, Horowitz MP, et al. A novel transferrin/TfR2-mediated mitochondrial iron transport system is disrupted in Parkinson’s disease. Neurobiology of Disease. 2009;34(3):417–431. doi: 10.1016/j.nbd.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sheline CT, Zhu J, Zhang W, Shi C, Cai A. Mitochondrial inhibitor models of Huntington's disease and Parkinson's disease induce zinc accumulation and are attenuated by inhibition of zinc neurotoxicity in vitro or in vivo. Neurodegenerative Diseases. 2013;11(1):49–58. doi: 10.1159/000336558. [DOI] [PubMed] [Google Scholar]
- 53.Fukushima T, Tan X, Luo Y, Kanda H. Serum vitamins and heavy metals in blood and urine, and the correlations among them in parkinson's disease patients in China. Neuroepidemiology. 2011;36(4):240–244. doi: 10.1159/000328253. [DOI] [PubMed] [Google Scholar]
- 54.Hozumi I, Hasegawa T, Honda A, et al. Patterns of levels of biological metals in CSF differ among neurodegenerative diseases. Journal of the Neurological Sciences. 2011;303(1-2):95–99. doi: 10.1016/j.jns.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 55.Jiménez-Jiménez FJ, Fernández-Calle P, Martínez-Vanaclocha M, et al. Serum levels of zinc and copper in patients with Parkinson's disease. Journal of the Neurological Sciences. 1992;112(1-2):30–33. doi: 10.1016/0022-510x(92)90127-7. [DOI] [PubMed] [Google Scholar]
- 56.Guilarte TR. Manganese and Parkinson's disease: a critical review and new findings. Environmental Health Perspectives. 2010;118(8):1071–1080. doi: 10.1289/ehp.0901748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pals P, van Everbroeck B, Grubben B, et al. Case-control study of environmental risk factors for Parkinson's disease in Belgium. European Journal of Epidemiology. 2003;18(12):1133–1142. doi: 10.1023/b:ejep.0000006639.05690.92. [DOI] [PubMed] [Google Scholar]
- 58.Rentschler G, Covolo L, Ahmadi Haddad A, Lucchini RG, Zoni S, Broberg K. ATP13A2 (PARK9) polymorphisms influence the neurotoxic effects of manganese. NeuroToxicology. 2012;33(4):697–702. doi: 10.1016/j.neuro.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tan J, Zhang T, Jiang L, et al. Regulation of intracellular manganese homeostasis by Kufor-Rakeb syndrome-associated ATP13A2 protein. Journal of Biological Chemistry. 2011;286(34):29654–29662. doi: 10.1074/jbc.M111.233874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berg D, Hochstrasser H. Iron metabolism in parkinsonian syndromes. Movement Disorders. 2006;21(9):1299–1310. doi: 10.1002/mds.21020. [DOI] [PubMed] [Google Scholar]
- 61.Oakley AE, Collingwood JF, Dobson J, et al. Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology. 2007;68(21):1820–1825. doi: 10.1212/01.wnl.0000262033.01945.9a. [DOI] [PubMed] [Google Scholar]
- 62.Devos D, Moreau C, Devedjian JC. Targeting chelatable iron as a therapeutic modality in Parkinson’sdisease. Antioxidants and Redox Signaling. 2014;21(2):195–210. doi: 10.1089/ars.2013.5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Poli M, Derosas M, Luscieti S, et al. Pantothenate kinase-2 (Pank2) silencing causes cell growth reduction, cell-specific ferroportin upregulation and iron deregulation. Neurobiology of Disease. 2010;39(2):204–210. doi: 10.1016/j.nbd.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 64.Tsunemi T, Krainc D. Zn2+ dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Human Molecular Genetics. 2014;23(11):2791–2801. doi: 10.1093/hmg/ddt572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu J, Marzetti E, Seo AY, Kim J, Prolla TA, Leeuwenburgh C. The emerging role of iron dyshomeostasis in the mitochondrial decay of aging. Mechanisms of Ageing and Development. 2010;131(7-8):487–493. doi: 10.1016/j.mad.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Youdim MBH, Riederer P. The role of iron in senescence of dopaminergic neurons in Parkinson's disease. Journal of Neural Transmission, Supplement. 1993;(40):57–67. [PubMed] [Google Scholar]
- 67.Karki P, Lee E, Aschner M. Manganese neurotoxicity: a focus on glutamate transporters. Annals of Occupational and Environmental Medicine. 2013;25(1, article 4) doi: 10.1186/2052-4374-25-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goebel HH, Gerhard L, Kominami E, Haltia M. Neuronal ceroid-lipofuscinosis—late-infantile or Jansky-Bielschowsky type—revisited. Brain Pathology. 1996;6(3):225–228. doi: 10.1111/j.1750-3639.1996.tb00850.x. [DOI] [PubMed] [Google Scholar]
- 69.Wöhlke A, Philipp U, Bock P, et al. A one base pair deletion in the canine ATP13A2 gene causes exon skipping and late-onset neuronal ceroid lipofuscinosis in the Tibetan terrier. PLoS Genetics. 2011;7(10) doi: 10.1371/journal.pgen.1002304.e1002304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Farias FHG, Zeng R, Johnson GS, et al. A truncating mutation in ATP13A2 is responsible for adult-onset neuronal ceroid lipofuscinosis in Tibetan terriers. Neurobiology of Disease. 2011;42(3):468–474. doi: 10.1016/j.nbd.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 71.Schultheis PJ, Fleming SM, Clippinger AK, et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited α-synuclein accumulation and age-dependent sensorimotor deficits. Human Molecular Genetics. 2013;22(10):2067–2082. doi: 10.1093/hmg/ddt057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Burneo JG, Arnold T, Palmer CA, Kuzniecky RI, Oh SJ, Faught E. Adult-onset neuronal ceroid lipofuscinosis (Kufs disease) with autosomal dominant inheritance in Alabama. Epilepsia. 2003;44(6):841–846. doi: 10.1046/j.1528-1157.2003.39802.x. [DOI] [PubMed] [Google Scholar]
- 73.Berkovic SF, Carpenter S, Andermann F, Andermann E, Wolfe LS. Kufs' disease: a critical reappraisal. Brain. 1988;111(1, part 1):27–62. doi: 10.1093/brain/111.1.27. [DOI] [PubMed] [Google Scholar]
- 74.Nijssen PCG, Brusse E, Leyten ACM, Martin JJ, Teepen JLJM, Roos RAC. Autosomal dominant adult neuronal ceroid lipofuscinosis: parkinsonism due to both striatal and nigral dysfunction. Movement Disorders. 2002;17(3):482–487. doi: 10.1002/mds.10104. [DOI] [PubMed] [Google Scholar]
- 75.Taschner PEM, de Vos N, Thompson AD, et al. Chromosome 16 microdeletion in a patient with juvenile neuronal ceroid lipofuscinosis (Batten disease) The American Journal of Human Genetics. 1995;56(3):663–668. [PMC free article] [PubMed] [Google Scholar]
- 76.Lavrov AY, Ilyna ES, Zakharova EY, Boukina AM, Tishkanina SV. The first three Russian cases of classical, late-infantile, neuronal ceroid lipofuscinosis. European Journal of Paediatric Neurology. 2002;6(3):161–164. doi: 10.1053/ejpn.2002.0584. [DOI] [PubMed] [Google Scholar]
- 77.Bras J, Singleton A, Cookson MR, Hardy J. Emerging pathways in genetic Parkinson's disease: potential role of ceramide metabolism in Lewy body disease. FEBS Journal. 2008;275(23):5767–5773. doi: 10.1111/j.1742-4658.2008.06709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.El Haddad S, Khoury M, Daoud M, et al. CLN5 and CLN8 protein association with ceramide synthase: biochemical and proteomic approaches. Electrophoresis. 2012;33(24):3798–3809. doi: 10.1002/elps.201200472. [DOI] [PubMed] [Google Scholar]
- 79.Persaud-Sawin D, Mousallem T, Wang C, Zucker A, Kominami E, Boustany RN. Neuronal ceroid lipofuscinosis: a common pathway? Pediatric Research. 2007;61(2):146–152. doi: 10.1203/pdr.0b013e31802d8a4a. [DOI] [PubMed] [Google Scholar]
- 80.van Ham TJ, Thijssen KL, Breitling R, Hofstra RMW, Plasterk RHA, Nollen EAA. C. elegans model identifies genetic modifiers of α-synuclein inclusion formation during aging. PLoS Genetics. 2008;4(3) doi: 10.1371/journal.pgen.1000027.e1000027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Halperin A, Elstein D, Zimran A. Increased incidence of Parkinson disease among relatives of patients with Gaucher disease. Blood Cells, Molecules, and Diseases. 2006;36(3):426–428. doi: 10.1016/j.bcmd.2006.02.004. [DOI] [PubMed] [Google Scholar]