

Abstract
We performed IGH clonotypic sequence analysis in WM in order to determine whether a preferential IGH gene rearrangement was observed and to assess IGHV mutational status in blood and/or bone marrow samples from 36 WM patients. In addition we investigated the presence of MYD88 L265P somatic mutation. After IGH VDJ locus amplification, monoclonal VDJ rearranged fragments were sequenced and analyzed. MYD88 L265P mutation was detected by AS-PCR. The most frequent family usage was IGHV3 (74%); IGHV3-23 and IGHV3-74 segments were used in 26% and 17%, respectively. Somatic hypermutation was seen in 91% of cases. MYD88 L265P mutation was found in 65,5% of patients and absent in the 3 unmutated. These findings did not correlate with clinical findings and outcome. Conclusion. IGH genes' repertoire differed in WM from those observed in other B-cell disorders with a recurrent IGHV3-23 and IGHV3-74 usage; monoclonal IGHV was mutated in most cases, and a high but not omnipresent prevalence of MYD88 L265P mutation was observed. In addition, the identification of 3 patients with unmutated IGHV gene segments, negative for the MYD88 L265P mutation, could support the hypothesis that an extra-germinal B-cell may represent the originating malignant cell in this minority of WM patients.
1. Introduction
Waldenström macroglobulinemia (WM) is an uncommon B-cell lymphoproliferative disorder characterized by bone marrow lymphoplasmacytic infiltration and by the presence of a monoclonal IgM immunoglobulin in the serum [1]. It belongs to the lymphoplasmacytic lymphoma type [2]. Clinical manifestations of WM include lymphoma-related lymphadenopathy, organomegaly, fatigue, disease related fever, or symptoms related to bone marrow failure (cytopenias) and IgM-related cryoglobulinemia, cold agglutinin syndrome, demyelinating neuropathy, and symptomatic hyperviscosity [3]. The monoclonal IgM is produced by malignant B-cells harboring a unique clonotypic rearrangement of immunoglobulin heavy chain variable genes (IGH), the VDJH rearrangement, associated with a specific constant region [4, 5]. On immunophenotype WM lymphoplasmacytes are usually CD5−, CD10−, CD23−, CD19+, CD20+ but numerous variations can be observed and in addition there is no characteristic pathognomonic genetic alteration; thus, differential diagnosis with other entities that can secrete IgM, share the same immunophenotype, and present lymphoplasmacytic differentiation may be sometimes difficult [6].
Immunoglobulin heavy chain gene (IGH) sequence analysis can provide useful clues in the investigation of B-cell lymphoproliferative disorders. It provides evidence regarding the maturation status of specific B-cell entities. Disorders characterized by germline immunoglobulin genes are likely to be derived from naive B cells, which have not encountered antigen. Most of B-cell lymphoproliferative disorders, however, exhibit somatic hypermutation (SHM) of immunoglobulin variable genes and are, therefore, derived from cells that have encountered antigen in germinal center. In addition, in chronic lymphocytic leukemia (CLL), marginal zone lymphoma (MZL), and mantle cell lymphoma (MCL), biased usage of IGHV genes and stereotyped clusters of immunoglobulin receptor support the role of antigen-driven mechanisms in their pathogenesis [7, 8]. The WM IGHV gene repertoire is completely different from other B-cell lymphoproliferative disorders like CLL and MZLs, as it is characterized by an overrepresentation of IGHV3-23 genes with high IGHV mutation rates [8–13]. These features indicate that the transformation leading to WM occurred in postgerminal center B-cells that bear SHM and have been submitted to T-dependant antigen selection.
Recently, whole genome sequencing in WM patients revealed a highly recurrent somatic mutation (MYD88 L265P) in these patients [8, 14–20]. It was furthermore suggested that MYD88 L265P mutation possibly constitutes the initiating event, responsible for disease transformation [21, 22]. Furthermore its detection could constitute a valuable differential diagnosis tool.
In the present study we characterized IGH genes rearrangements and somatic hypermutations (SHM) in a cohort of WM patients and we investigated any eventual correlation with patients' clinical features. The frequency of the MYD88 L265P mutation was also investigated and correlated with the IGH genes rearrangements in an attempt to reveal new insights in WM pathogenesis and the nature of WM B-cell.
2. Materials and Methods
2.1. Patients
A cohort of 36 WM patients was studied retrospectively. Diagnostic workout included physical examination, hematological and laboratory parameters, chest radiographs, and computed tomography scans of the thorax, abdomen, and pelvis. Bone marrow smears and biopsy as well as immunophenotype were performed in all patients, and lymph node histology was additionally performed in the cases with lymphadenopathy. International diagnostic criteria were used for the diagnosis of WM. Patients' characteristics are shown in Table 1.
Table 1.
Clinical and laboratory findings for the study's WM patients.
| Mean value (median value) | Range | |
|---|---|---|
| Age (years) | 65,5 (64) | 42–84 |
| Gender (male/female) | ||
| IPSS stage | 19/17 | |
| 1 | 45% | |
| 2 | 39% | |
| 3 | 16% | |
| Bone marrow involvement | 46,7% (40%) | 5–100% |
| Lymphadenopathy | 21% | |
| Splenomegaly | 19% | |
| Hepatomegaly | 9% | |
| Extranodal sites | 3% | |
| IgM (mg/dL) | 2777,2 (2500) | 138–7870 |
| Hb (g/dL) | 10,9 (11,1) | 6–14,3 |
| Platelets (×109/L) | 233,2 (234) | 60–472 |
| WBC (×109/L) | 7,1 (6,7) | 2,1–16,8 |
| B2M (mg/dL) | 4,1 (3,4) | 1,9–10,4 |
| Abnormal (high) LDH | 27% | |
Forty-five percent, 39% and 16%, of patients were staged 1, 2, and 3, respectively, according to IPSS [23]. Seventy-two percent were symptomatic and required treatment; median time to treatment and overall survival were 13 and 61 months, respectively.
The study was approved by the local ethical committee.
2.2. Specimens and DNA Extraction
We analyzed genomic DNA extracted from patients' blood and/or bone marrow samples (bone marrow mononuclear cells, bone marrow smears, and bone marrow biopsies).
Genomic DNA was extracted by standard protocols using QIAmp DNA Mini kit (QIAGEN) according to the manufacturer's recommendations.
2.3. Sequencing of IGHV Gene Sequences and Analysis of IGHV Sequences
Immunoglobulin heavy chain VDJ locus was amplified by PCR using the Biomed-2 strategy with FR1 primers as previously described [24]. It was possible to confirm monoclonality of the PCR product in 26/36 samples studied, by using capillary electrophoresis in Agilent 2100 Bioanalyzer using Agilent DNA 1000 kit (Agilent Technologies) according to the manufacturer's recommendations. PCR products were directly sequenced on both strands with the same primers using Sanger's chain-termination method and fluorescent dideoxynucleotides with GenomeLab DTCS Quick start kit in Beckman-Coulter CEQ 8000 sequencer platform.
In order to confirm monoclonality in the remaining 10/36 samples, cloning techniques were used as follows: (1) ligation of the PCR product to pCRII-TOPO vector (Invitrogen) using TOPO TA Cloning Kit (Invitrogen) according to manufacturer's recommendations, (2) transformation of One Shot TOP10 Chemically Competent E. coli cells (Invitrogen) by insertion of plasmids, (3) selection of 8–10 colonies of transformed E. coli cells followed by liquid culture (in LB medium for 12–14 hours at 37°C), and (4) plasmid DNA purification using PureLink HiPure Plasmid DNA Purification Kit (Invitrogen) according to manufacturer's recommendations. Plasmid DNA was sequenced, as described above, and sequences (8–10) were aligned using DNASTAR SeqMan Pro software in order to confirm monoclonality by detecting the same IGH-VDJ rearrangement in at least 3 out of 8 sequences.
Each clonal DNA IGHV sequence was aligned with the closest germline sequence using the international immunogenetics information system (IMGT, http://www.imgt.org/). Sequences were translated into amino acids in order to identify the functional IGHV gene rearrangement. The percentage of homology between the functional IGHV segment used in the tumor and the germline sequence was then calculated (excluding primer sequences). Somatic hypermutation was defined as a >2% deviation from germline (as per convention) [25]. The length of CDR3 regions was determined according to IMGT numbering.
2.4. Screening for MYD88 L265P Mutations
Samples from 31 patients were also investigated for detection of MYD88 L265P mutation by allele specific PCR (AS-PCR). Two PCRs were performed for each sample, one for wild type MYD88 and one for possible detection of mutated MYD88 by using primers as previously described [18]. PCRs were carried out by using a HotStarTaq DNA Plus Master Mix kit (QIAGEN). PCR consisted of an initial denaturation step of 15 minutes at 95°C, followed by 35 cycles of 95°C for 30 seconds, 58°C for 30 seconds, and 72°C for 30 seconds, with a final extension step of 5 minutes at 72°C. Agarose gel (1,5%) electrophoresis was performed to visualize the PCR products (220 bp).
2.5. Statistical Analysis
The SPSS software v.15 was used. Correlations between molecular findings and clinical parameters were assessed by the Mann-Whitney or by the chi-square test. Pictorial representation of survival curves was done by the Kaplan-Mayer method and their comparison by the log-rank test.
3. Results and Discussion
3.1. IGHV Usage and Mutation Analysis
Thirty-six WM patients were studied. Two out of the 36 patients were excluded from the analysis, as in these two patients genomic DNA was extracted from peripheral blood and not bone marrow. Although these two patients had not lymphoma cells in blood (by morphology or immunophenotype analysis), monoclonality of IGHV-PCR product was confirmed in both samples (Figure 1 illustrates the electropherogram of IGHV6-PCR product in one of these two patients after capillary electrophoresis). Mutated IGHV genes were detected in these two patients (one IGHV3-74 and one IGHV6-1).
Figure 1.
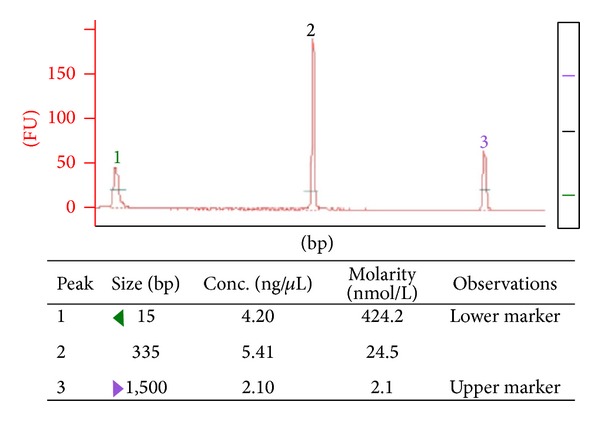
Electropherogram—after capillary electrophoresis in Agilent 2100 Bioanalyzer using Agilent DNA 1000 kit (Agilent Technologies)—of IGHV6-PCR product in one of the two patients of whom genomic DNA was extracted from blood sample and not bone marrow. Monoclonality of IGHV-PCR product is obvious (peak number 2) and was further confirmed by direct sequencing.
We observed an IGHV3 overrepresentation (74,3%), as high as described in previous studies [8, 12] but lower than reported by others [11, 26]. The distinctive IGHV gene segments usage in our patients is presented in Table 2. It should be mentioned that in one patient two different productive IGH VDJ sequences were identified (two different B-cell clones). There was an overrrepresentation of IGHV3-23 gene usage (25,7%), as expected according to previous studies; while IGHV3-74 gene -another member of the IGHV3 family- was also overrepresented (17,1%), which is not reported in other studies [12, 26]. The repertoire of IGHV genes in WM, as presented in this study and previous studies, has some similarities with IgM-MGUS IGHV genes' repertoire [12, 13], but it is absolutely different from the ones observed in CLL/SLL [7], MCL [27], and MZL [8, 28, 29]. This is important because the aforementioned B-cell malignancies can secrete a monoclonal IgM and be in some cases misdiagnosed as WM or vice versa.
Table 2.
Distinctive IGHV gene segments usage in present study.
| Segment | Number of patients | % |
|---|---|---|
| IGHV1-2 | 1 | 2,86 |
| IGHV1-8 | 1 | 2,86 |
| IGHV2-5 | 1 | 2,86 |
| IGHV3-7 | 3 | 8,57 |
| IGHV3-23 | 9 | 25,71 |
| IGHV3-30 | 3 | 8,57 |
| IGHV3-33 | 2 | 5,71 |
| IGHV3-48 | 1 | 2,86 |
| IGHV3-64 | 1 | 2,86 |
| IGHV3-73 | 1 | 2,86 |
| IGHV3-74 | 6 | 17,14 |
| IGHV4-34 | 3 | 8,57 |
| IGHV4-59 | 2 | 5,71 |
| IGHV5-51 | 1 | 2,86 |
SHM was seen in all but three cases (91,4%). Mean percentages of mutations in all cases, IGHV3 family, IGHV3-23, and IGHV3-74 segments were 7,5%, 8%, 9,4%, and 7,5%, respectively (Table 3). These findings are in agreement with previous studies [11, 12, 26] and suggest that WM cells are derived from postgerminal center memory B cells that have been submitted to T-dependant antigen selection. It should be mentioned that in this study unmutated IGHV genes (≤2% deviation from germline homolog gene) were detected in 3 cases, as it was described in previous studies [10–12, 26], while in some other studies [8, 9, 13] all (100%) were mutated. In detail, 3 (8,6%) unmutated IGHV genes were detected (two IGHV3-33 and one IGHV5-51), and one of the three was 100% homolog to germline gene. It is remarkable that none of these three genes belonged to the highly represented IGHV segments in WM (IGHV3-23 and IGHV3-74). The existence of unmutated IGHV genes could mean that the transformation leading to WM does not target exclusively postgerminal center B-cells that bear SHM and have been submitted to T-dependent antigen selection. Even higher percentages of unmutated IGHV genes have been observed in resembling diseases such as splenic MZL (SMZL) [30–32]. Indeed an erroneous diagnosis can never be excluded in this disease although the 3 patients presented typical WM features. CDR3 length was short (≤17 amino acids) in 80% of all cases as previously described [8, 10–12, 33], although in the three unmutated cases the mean of CDR3 length was 22,3 amino acids.
Table 3.
Mean (median) of somatic mutations' percentage in different groups.
| Mean (median) somatic mutations' percentage | Range (%) | |
|---|---|---|
| In all 35∗ cases | 7,5% (7,3%) | 0–16,1 |
| In 32 cases with mutated genes (<98% homology) | 8,1% (7,6%) | 2,83–16,1 |
| In IGHV3 cases' group (27 cases) | 8% (8,3%) | 0–14,46 |
| In IGHV3-23 cases' group (9 cases) | 9,4% (9,7%) | 2,83–14,46 |
| In IGHV3-74 cases' group (6 cases) | 7,5% (8,1%) | 4,02–9,65 |
∗34 patients, 1 with two clones; IGHV3-74 and IGHV4-59.
The above-mentioned findings were compared with patients' physical and routine laboratory workup results and no correlations were found nor was it the case with time to treatment or survival. Although in CLL the IGHV genes mutational status is one of the most important independent prognostic factors [34–37], this was not the case for our WM patients.
3.2. MYD88 Mutation Analysis
Since MYD88 has been reported to be mutated (L265P) in the large majority of WM patients, we next looked for this mutation. Two out of the 31 patients investigated for detection of MYD88 L265P mutation were excluded from the analysis, as in these two patients genomic DNA was extracted from blood and not bone marrow. However, in these samples monoclonality of IGHV-PCR product was confirmed. These two samples were negative for MYD88 L265P mutation. Nineteen out of 29 patients (65,5%) were positive for the MYD88 L265P mutation. This percentage is high and in accordance with the study of Gachard et al. [8]; however it is lower compared to other studies reporting a MYD88 L265P mutation prevalence of 79% to 100% [14–16, 18, 20]. These variations could reflect differences in methodology followed by each study. The use, in our study, of BM unselected tissue for MYD88 L265P AS-PCR assay could have contributed to the seemingly lower detection rate as has been raised by others. Cases not exhibiting MYD88 L265P mutation had a statistically significant lower bone marrow infiltration by lymphoplasmacytes (P < 0.005).
The findings were also compared with patients' physical and routine laboratory workup results and no correlations were found nor was it the case with time to treatment or survival, as also described by Jiménez et al. [16]. We have also seen a difference in MYD88 L265P detection based on bone marrow involvement, with a higher BM involvement in MYD88 L265P positive patients.
Finally it should be mentioned that the mean of SHM levels of IGHV genes in MYD88 L265P positive patients was 8,3%, while in MYD88 L265P negative patients it was 5,4%. Five of seven patients with IGHV3-23 who were tested for MYD88 L265P and five out of five patient with IGHV3-74 were positive for this mutation, which suggests that IGHV3-23 and IGHV3-74 are represented more in MYD88 L265P positive patients. This may reinforce the concept that there are biological differences between the patients with and without the MYD88 L265P mutation. This is further supported by an additional observation in this study: all three cases of unmutated IGHV genes were negative for MYD88 L265P. These findings imply that a different (extra-germinal) B-cell represents the origin of the malignant cell in a minority of patients. Such hypothesis of the origin of the malignant cell in some WM patients is also described by Sahota et al. [38]. Larger studies may support further this concept.
In addition, the landscape is still unclear in this field as MYD88 L265P mutation was recently found in other lymphoma entities [16, 18] while it was negative in lymphoplasmacytic lymphoma not secreting IgM [39]. Further studies are needed.
4. Conclusions
WM IGH genes repertoire, as expected, differs from that observed in normal B-cells and other B-cell diseases such as MZL, MCL, and B-CLL/SLL.
In addition to the known hyperrepresentation of the IGHV3-23 gene, another member of the IGHV3 family, the IGHV3-74 gene is also overrepresented in WM, as shown in the present study. The high IGHV mutation rate supports a derivation of WM cells from postgerminal center memory B cells in the majority (91,4%) of WM patients. However, the identification of a minority of patients (3 of 34) with unmutated IGHV gene segments, negative for the MYD88 L265P mutation, supports the hypothesis that they represent a subgroup of WM not arising from postgerminal B cells with a different disease pathogenesis. Finally, consensus and guidelines for MYD88 L265P detection's methodology are needed, as it is quite obvious that this mutation could be both helpful in the diagnosis of WM and a potential therapeutic target in WM patients.
Acknowledgments
The authors thank D. Palaiologou, E. Staikou, and A. Stefanou for excellent technical assistance.
Conflict of Interests
The authors declare no conflict of interests.
References
- 1.Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom's macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom's Macroglobulinemia. Seminars in Oncology. 2003;30(2):110–115. doi: 10.1053/sonc.2003.50082. [DOI] [PubMed] [Google Scholar]
- 2.Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematipoetic and Lymphoid Tissues. Lyon, France: IARC Press; 2008. [Google Scholar]
- 3.Treon SP, Hunter ZR, Aggarwal A, et al. Characterization of familial Waldenström's macroglobulinemia. Annals of Oncology. 2006;17(3):488–494. doi: 10.1093/annonc/mdj111. [DOI] [PubMed] [Google Scholar]
- 4.Wagner SD, Martinelli V, Luzzatto L. Similar patterns of Vκ gene usage but different degrees of somatic mutation in hairy cell leukemia, prolymphocytic leukemia, Waldenstrom's macroglobulinemia, and myeloma. Blood. 1994;83(12):3647–3653. [PubMed] [Google Scholar]
- 5.Aoki H, Takishita M, Kosaka M, Saito S. Frequent somatic mutations in D and/or JH segments of Ig gene in Waldenstrom's macroglobulinemia and chronic lymphocytic leukemia (CLL) with Richter's syndrome but not in common CLL. Blood. 1995;85(7):1913–1919. [PubMed] [Google Scholar]
- 6.Pangalis GA, Kyrtsonis M, Kontopidou FN, et al. Differential diagnosis of Waldenstrom's macroglobulinemia from other low-grade B-cell lymphoproliferative disorders. Seminars in Oncology. 2003;30(2):201–205. doi: 10.1053/sonc.2003.50046. [DOI] [PubMed] [Google Scholar]
- 7.Stamatopoulos K, Belessi C, Moreno C, et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: pathogenetic implications and clinical correlations. Blood. 2007;109(1):259–270. doi: 10.1182/blood-2006-03-012948. [DOI] [PubMed] [Google Scholar]
- 8.Gachard N, Parrens M, Soubeyran I, et al. IGHV gene features and MYD88 L265P mutation separate the three marginal zone lymphoma entities and Waldenström macroglobulinemia/lymphoplasmacytic lymphomas. Leukemia. 2013;27(1):183–189. doi: 10.1038/leu.2012.257. [DOI] [PubMed] [Google Scholar]
- 9.Sahota SS, Forconi F, Ottensmeier CH, et al. Typical waldenstrom macroglobulinemia is derived from a B-cell arrested after cessation of somatic mutation but prior to isotype switch events. Blood. 2002;100(4):1505–1507. [PubMed] [Google Scholar]
- 10.Walsh SH, Laurell A, Sundström G, Roos G, Sundström C, Rosenquist R. Lymphoplasmacytic lymphoma/Waldenström's macroglobulinemia derives from an extensively hypermutated B cell that lacks ongoing somatic hypermutation. Leukemia Research. 2005;29(7):729–734. doi: 10.1016/j.leukres.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Kriangkum J, Taylor BJ, Treon SP, Mant MJ, Belch AR, Pilarski LM. Clonotypic IgM V/D/J sequence analysis in Waldenstrom macroglobulinemia suggests an unusual B-cell origin and an expansion of polyclonal B cells in peripheral blood. Blood. 2004;104(7):2134–2142. doi: 10.1182/blood-2003-11-4024. [DOI] [PubMed] [Google Scholar]
- 12.Martín-Jiménez P, García-Sanz R, Balanzategui A, et al. Molecular characterization of heavy chain immunoglobulin gene rearrangements in Waldenström's macroglobulinemia and IgM monoclonal gammopathy of undetermined significance. Haematologica. 2007;92(5):635–642. doi: 10.3324/haematol.10755. [DOI] [PubMed] [Google Scholar]
- 13.Rollett RA, Wilkinson EJ, Gonzalez D, et al. Immunoglobulin heavy chain sequence analysis in Waldenström's macroglobulinemia and immunoglobulin M monoclonal gammopathy of undetermined significance. Clinical Lymphoma and Myeloma. 2006;7(1):70–72. doi: 10.3816/CLM.2006.n.042. [DOI] [PubMed] [Google Scholar]
- 14.Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström's macroglobulinemia. The New England Journal of Medicine. 2012;367(9):826–833. doi: 10.1056/NEJMoa1200710. [DOI] [PubMed] [Google Scholar]
- 15.Xu L, Hunter ZR, Yang G, et al. MYD88 L265P in Waldenström macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood. 2013;121(11):2051–2058. doi: 10.1182/blood-2012-09-454355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiménez C, Sebastián E, Chillón MC, et al. MYD88 L265P is a marker highly characteristic of, but not restricted to, Waldenström's macroglobulinemia. Leukemia. 2013;27(8):1722–1728. doi: 10.1038/leu.2013.62. [DOI] [PubMed] [Google Scholar]
- 17.Poulain S, Roumier C, Decambron A, et al. MYD88 L265P mutation in Waldenstrom macroglobulinemia. Blood. 2013;121(22):4504–4511. doi: 10.1182/blood-2012-06-436329. [DOI] [PubMed] [Google Scholar]
- 18.Varettoni M, Arcaini L, Zibellini S, et al. Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenstrom's macroglobulinemia and related lymphoid neoplasms. Blood. 2013;121(13):2522–2528. doi: 10.1182/blood-2012-09-457101. [DOI] [PubMed] [Google Scholar]
- 19.Willenbacher W, Willenbacher E, Brunner A, Manzl C. Improved accuracy of discrimination between IgM Multiple Myeloma and Waldenström Macroglobulinaemia by testing for MYD88 L265P mutations. British Journal of Haematology. 2013;161(6):902–904. doi: 10.1111/bjh.12313. [DOI] [PubMed] [Google Scholar]
- 20.Ondrejka SL, Lin JJ, Warden DW, Durkin L, Cook JR, Hsi ED. MYD88 L265P somatic mutation: its usefulness in the differential diagnosis of bone marrow involvement by B-cell lymphoproliferative disorders. The American Journal of Clinical Pathology. 2013;140(3):387–394. doi: 10.1309/AJCP10ZCLFZGYZIP. [DOI] [PubMed] [Google Scholar]
- 21.Treon SP, Hunter ZR. A new era for Waldenstrom macroglobulinemia: MYD88 L265P. Blood. 2013;121(22):4434–4436. doi: 10.1182/blood-2013-04-494849. [DOI] [PubMed] [Google Scholar]
- 22.Yang G, Zhou Y, Liu X, et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenstrom macroglobulinemia. Blood. 2013;122(7):1222–1232. doi: 10.1182/blood-2012-12-475111. [DOI] [PubMed] [Google Scholar]
- 23.Morel P, Duhamel A, Gobbi P, et al. International prognostic scoring system for Waldenström macroglobulinemia. Blood. 2009;113(18):4163–4170. doi: 10.1182/blood-2008-08-174961. [DOI] [PubMed] [Google Scholar]
- 24.van Dongen JJM, Langerak AW, Brüggemann M, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 concerted action BMH4-CT98-3936. Leukemia. 2003;17(12):2257–2317. doi: 10.1038/sj.leu.2403202. [DOI] [PubMed] [Google Scholar]
- 25.Matsuda F, Shin EK, Nagaoka H, et al. Structure and physical map of 64 variable segments in the 3' 0.8-megabase region of the human immunoglobulin heavy-chain locus. Nature Genetics. 1993;3(1):88–94. doi: 10.1038/ng0193-88. [DOI] [PubMed] [Google Scholar]
- 26.Varettoni M, Zibellini S, Capello D, et al. Clues to pathogenesis of Waldenström macroglobulinemia and immunoglobulin M monoclonal gammopathy of undetermined significance provided by analysis of immunoglobulin heavy chain gene rearrangement and clustering of B-cell receptors. 2013;54:2485–2489. doi: 10.3109/10428194.2013.779689. [DOI] [PubMed] [Google Scholar]
- 27.Hadzidimitriou A, Agathangelidis A, Darzentas N, et al. Is there a role for antigen selection in mantle cell lymphoma? Immunogenetic support from a series of 807 cases. Blood. 2011;118(11):3088–3095. doi: 10.1182/blood-2011-03-343434. [DOI] [PubMed] [Google Scholar]
- 28.Warsame AA, Aasheim H, Nustad K, et al. Splenic marginal zone lymphoma with VH1-02 gene rearrangement expresses poly- and self-reactive antibodies with similar reactivity. Blood. 2011;118(12):3331–3339. doi: 10.1182/blood-2011-03-341651. [DOI] [PubMed] [Google Scholar]
- 29.Zibellini S, Capello D, Forconi F, et al. Stereotyped patterns of B-cell receptor in splenic marginal zone lymphoma. Haematologica. 2010;95(10):1792–1796. doi: 10.3324/haematol.2010.025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Algara P, Mateo MS, Sanchez-Beato M, et al. Analysis of the IgVH somatic mutations in splenic marginal zone lymphoma defines a group of unmutated cases with frequent 7q deletion and adverse clinical course. Blood. 2002;99(4):1299–1304. doi: 10.1182/blood.v99.4.1299. [DOI] [PubMed] [Google Scholar]
- 31.Arcaini L, Zibellini S, Passamonti F, et al. Splenic marginal zone lymphoma: clinical clustering of immunoglobulin heavy chain repertoires. Blood Cells, Molecules, and Diseases. 2009;42(3):286–291. doi: 10.1016/j.bcmd.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 32.Kalpadakis C, Pangalis GA, Dimitriadou E, et al. Mutation analysis of IgVH genes in splenic marginal zone lymphomas: correlation with clinical characteristics and outcome. Anticancer Research. 2009;29(5):1811–1816. [PubMed] [Google Scholar]
- 33.Kriangkum J, Taylor BJ, Mant MJ, Treon SP, Belch AR, Pilarski LM. The malignant clone in Waldenstrom's macroglobulinemia. Seminars in Oncology. 2003;30(2):132–135. doi: 10.1053/sonc.2003.50061. [DOI] [PubMed] [Google Scholar]
- 34.Damle RN, Wasil T, Fais F, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94(6):1840–1847. [PubMed] [Google Scholar]
- 35.Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848–1854. [PubMed] [Google Scholar]
- 36.Krober A, Seiler T, Benner A, et al. VH mutation status, CD38 expression level, genomic aberrations, and survival in chronic lymphocytic leukemia. Blood. 2002;100(4):1410–1416. [PubMed] [Google Scholar]
- 37.Stilgenbauer S, Bullinger L, Lichter P, Döhner H. Genetics of chronic lymphocytic leukemia: genomic aberrations and VH gene mutation status in pathogenesis and clinical course. Leukemia. 2002;16(6):993–1007. doi: 10.1038/sj.leu.2402537. [DOI] [PubMed] [Google Scholar]
- 38.Sahota S, Babbage G, Weston-Bell N. CD27 in defining memory B-cell origins in Waldenström's macroglobulinemia. Clinical Lymphoma and Myeloma. 2009;9(1):33–35. doi: 10.3816/CLM.2009.n.007. [DOI] [PubMed] [Google Scholar]
- 39.Manasanch EE, Braylan R, Stetler-Stevenson M, et al. Lack of MYD88 L265P in non-immunoglobulin M lymphoplasmacytic lymphoma. Leukemia and Lymphoma. 2013;55:1402–1403. doi: 10.3109/10428194.2013.831091. [DOI] [PMC free article] [PubMed] [Google Scholar]