

Abstract
Farnesoid X receptor (FXR, NRIH4) plays a major role in the control of cholesterol metabolism. This suggests that antagonizing the transcriptional activity of FXR is a potential means to treat cholestasis and related metabolic disorders. Here we describe the synthesis, biological evaluation, and structure-activity relationship (SAR) studies of trisubstituted-pyrazol carboxamides as novel and potent FXR antagonists. One of these novel FXR antagonists, 4j has an IC50 of 7.5 nM in an FXR binding assay and 468.5 nM in a cell-based FXR antagonistic assay. Compound 4j has no detectable FXR agonistic activity or cytotoxicity. Notably, 4j is the most potent FXR antagonist identified to date; it has a promising in vitro profile and could serve as an excellent chemical tool to elucidate the biological function of FXR.
1. INTRODUCTION
The activation of Farnesoid X receptor (FXR, NRIH4),1 in particular, gives bile acids the ability to modulate genomic signaling pathways. FXR is a ligand-dependent transcription factor that regulates gene networks involved in regulating lipid and cholesterol homeostasis.2 As such, FXR is expressed primarily in tissues exposed to high concentrations of bile acids, such as the intestine, kidney, adrenal gland, and liver.3 Bile acids are the purported endogenous agonists for FXR.3–4 As the bile acid sensor, FXR regulates the expression of transporters and biosynthetic enzymes crucial for the physiological maintenance of bile acid homeostasis. Because of FXR’s role in bile acid homeostasis, modulating FXR may be beneficial for treating all aspects of the metabolic syndrome, a complex disease cluster that includes risk factors such as dyslipidemia, insulin-resistance, increased blood pressure, visceral obesity and hypercoagubility.5 Recent findings also suggest that FXR acts as a key metabolic regulator in the liver to maintain the homeostasis of liver metabolites.6 FXR ligands have been investigated in preclinical studies for targeted therapy of metabolic diseases, but have shown limitations.7 There is, therefore, a need for novel, potent and selective modulators as agonists or antagonists of FXR, both for potential clinical applications, as well as studies to better understand FXR’s biological functions.
Several potent, selective FXR agonists have been reported recently, such as GW40648 and 6α-ethylchenodeoxycholic acid (6-ECDCA).9 However, preclinical development of FXR agonists is limited by the complex response, including possible undesirable effects, triggered by activation of FXR in the liver. For example, FXR activation inhibits bile acid synthesis and basolateral efflux of bile through indirect downregulation of cholesterol 7 alpha-hydroxylase, or cytochrome P450 7A1 (CYP7A1), the rate-limiting enzyme of the bile acid synthesis pathway.10 In addition, although activation of FXR would reduce triglyceride levels in hypertriglyceridemic patients, it would also decrease the levels of high density lipoprotein and lead to accumulation of cholesterol as a consequence of the inhibited bile acid synthesis. Furthermore, FXR agonists interfere with the ability of constitutive androstane receptor to regulate basolateral transporters in hepatocytes.11 These effects might worsen liver injury in a subset of patients who have obstructive cholestasis (a severe liver disease that impairs bile flow and causes irreversible liver damage), thereby limiting the preclinical development and possible clinical use of FXR agonists. In contrast, FXR antagonists might be useful for understanding the function of FXR and ultimately for treating liver disorders, if they targeted a specific cluster of genes and thus avoided the detrimental side effects mediated by FXR agonists. Treatment with FXR antagonists could potentially upregulate CYP7A1 expression and decrease overall cholesterol concentrations. This activity would occur through FXR’s regulation of the expression of small heterodimer partner.2 In addition, FXR can promote expression of an intestinal bile acid-binding protein (I-BABP) gene.12 When expression of I-BABP is repressed by an antagonist of FXR, re-absorption of bile acids in the ileum is repressed, which may reduce the amount of bile acid that returns to the liver, and, thereby promote the expression of CYP7A1. Thus, an FXR antagonist could potentially induce repression of I-BABP expression in the ileum, leading to reduced levels of serum cholesterol, and suggesting potential as a therapeutic agent for hypercholesterolemia in humans. Therefore, an FXR-specific antagonist could be used to validate the clinical relevance of antagonizing FXR.
Despite the potential of FXR as a therapeutic target for metabolic diseases, few selective FXR antagonists (synthetic small molecules or natural products) are described in the literature. Recently, the natural product guggulsterone was identified as the first putative FXR antagonist.13–14 However, the mechanism by which it antagonizes FXR is unclear. It appears that rather than being a true antagonist of FXR, guggulsterone is a unique FXR ligand that has antagonistic activity in coactivator association assays and can enhance the action of FXR agonists in vivo.15 In addition to guggulsterone, several marine products, such as sulfated polyhydroxy sterols isolated from marine invertebrates, have been identified as putative FXR modulators.16 This is exemplified by sulfated sterol. Theonellasterol has also been reported as an FXR antagonist. In rodent models, theonellasterol attenuated both liver injury and the extent of liver necrosis caused by bile duct ligation.17 In addition, several scalarane sesterterpenes, isolated from a marine sponge, inhibited FXR with IC50 values between 2.4 and 24 µM.18 Most recently, suvanine, a marine sponge sesterterpene, was reported by Di Leva et al. as an FXR antagonist.19 Using suvanine as a template, Di Leva et al. developed suvanine derivative as a novel FXR antagonist (Figure 1).
Figure 1.

Chemical structures of examples of FXR ligands.
The nonsteroidal antagonist AGN34 displayed an interesting profile of antagonist properties of the RXR/FXR heterodimer acting at RXR. AGN43 antagonizes FXR in vitro reporter assays but acts in a gene-selective manner in vivo.20 Interestingly, the nonsteroidal compound1,3,4-trisubstituted-pyrazolone derivative was recently identified as an FXR antagonist (Figure 1).21 This compound strongly suppresses the regulating effects of CDCA on FXR target genes. These findings further suggest the feasibility of identifying FXR antagonists that regulate a specific subset of FXR responsive genes in a gene-specific fashion.22
We recently reported the discovery of an FXR antagonist through the use of a time-resolved fluorescence resonance energy transfer (TR-FRET) assay,23 as well as several chemotypes identified as inhibitors of FXR. These chemotypes are represented by heterocyclic amide compounds, including substituted pyrazole compounds.24 a–b The pyrazole heterocycle has long been considered a privileged scaffold in drug discovery on the basis of its wide-ranging biological activities, including modulating G-protein coupled receptor modulators, inhibiting cyclooxygenase-2, and p38 MAP kinase.25 A challenge in investigating pyrazole heterocycles as potential FXR antagonists is selecting a structural motif to serve as the scaffold of a focused library, given the diversity of structures that can be optimized to develop more potent and selective FXR antagonists. Therefore we started by assessing the antagonistic effect of a collection of known substituted pyrazole carboxamides (Enamine Small Molecule Compound Libraries) against FXR, and identified E16, a 1,3-disubstituted -pyrazole-4-carboxamide as a potent FXR antagonist. We then used E16 as the template for further structural optimization, which led to several novel compounds. These compounds were exemplified by 1-(3-Methoxybenzyl)-3-(3-methoxyphenyl)-N-(4-methyl-3-(morpholinosulfonyl) phenyl)-1H-pyrazole-4-carboxamide) 4j, which had significantly improved FXR antagonistic activities as compared to E16. To our knowledge, 4j is the most potent FXR antagonist to date. Our efforts in designing structurally novel, nonsteroidal FXR antagonists by modifying pyrazole carboxamides may represent an important direction in developing novel FXR antagonists.
2. RESULTS and DISCUSSION
2.1. Identification of a Substituted Pyrazole-4-carboxamide E16 as a Template for Designing Novel FXR Antagonists
We began by evaluating the FXR antagonistic activities of known or commercially available substituted pyrazole carboxamides. We first evaluated 16 commercially available substituted pyrazole carboxamides (Enamine Small Molecule Compound Libraries) by FXR biochemical TR-FRET binding, FXR cell-based agonistic and antagonistic, and cytotoxicity assays (Tables 1 and 2). Among the carboxamide analogs, E16 (Table 1), a 1,3,4-trisubstituted-pyrazole carboxamide analog, had the highest FXR binding affinity (IC50 = 47 nM) and was a potent FXR antagonist (IC50 = 2.62 µM) (Table 2 and Figure 2). None of the substituted pyrazole carboxamides tested displayed FXR agonistic activity (data not shown) or significant cytotoxicity (< 5% of control) (Table 2).
Table 1.
Structures of the 16 substituted pyrazole-4-carboxamides (from Enamine Small Molecule Compound Libraries).
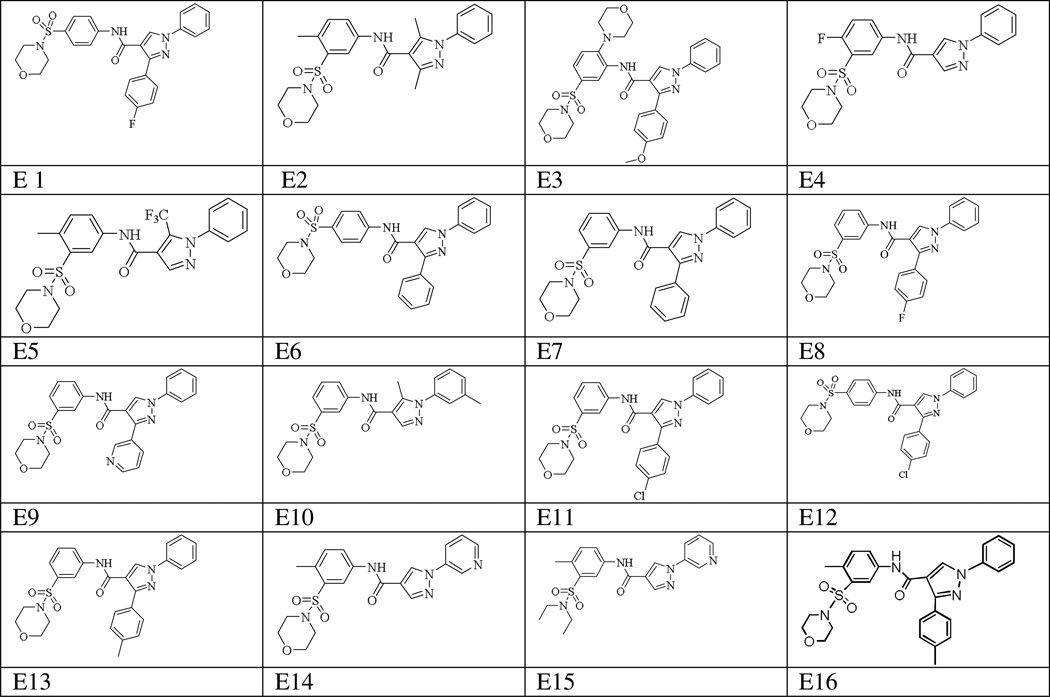 |
Table 2.
Biological activities of the 16 substituted pyrazole-4-carboxamides as determined by FXR TR-FRET binding, cell-based FXR antagonistic and cytotoxic assays. None of the 16 compounds showed FXR agonistic activity (data not shown).
| Compound | FXR TR-FRET Binding | GB FXR Cell Anta. | GB FXR Cell Cyto | |||
|---|---|---|---|---|---|---|
| %Inh. | IC50 | %Inh. | IC50 | %Inh. | IC50 | |
| E1 | 21.2±0.6 (164 nM) | NA | 34.7±1.2 (40 μM) | NA | NT | NA |
| E2 | 70.6±4.2 (13.3 µM) | 3.3±0.6 µM | 99.0±3.7 (40 µM) | 11.8±0.6 µM | 47.4±1.4 (40 µM) | NA |
| E3 | 11.6±0.2 (54.8 nM) | NA | 27.0±0.3 (40 µM) | NA | NT | NA |
| E4 | 83.8±1.4 (13.3 µM) | 8.3±0.9 µM | 80.3±0.1 (40 µM) | 9.4±0.9 µM | 27.4±1.2 (40 µM) | NA |
| E5 | 99.8±6.1 (13.3 µM) | 996±67 nM | 101.1±2.7 (40 µM) | 11.8±1.1 µM | 51.3±1.7 (40 µM) | NA |
| E6 | 12.7±0.9 (493 nM) | NA | 18.6±0.4 (40 µM) | NA | 7.4±0.3 (40 µM) | NA |
| E7 | 44.4±1.9 (493 nM) | NA | 54.9±2.1 (40 µM) | NA | 7.1±0.3 (40 µM) | NA |
| E8 | 53.3±0.6 (493 nM) | NA | 42.6±0.9 (40 µM) | NA | 16.3±0.2 (40 µM) | NA |
| E9 | 90.4±4.0 (4.44 µM) | 1.3±0.2µM | 82.3±2.6 (40 µM) | 10.8±0.4 µM | NT | NA |
| E10 | 85.1±0.9 (13.3 µM) | 6.6±0.7µM | 66.9±3.0 (40 µM) | 29.3±1.7 µM | NT | NA |
| E11 | 36.9±1.5 (54.8 nM) | NA | 38.8±1.7 (40 µM) | NA | NT | NA |
| E12 | 17.8±0.2 (54.8 nM) | NA | 20.6±0.3 (40 µM) | NA | NT | NA |
| E13 | 27.5±0.9 (493 nM) | NA | 32.8±1.1 (40 µM) | NA | 5.4±0.2 (40 µM) | NA |
| E14 | 18.8±1.2 (13.3 µM) | NA | 16.1±0.5 (40 µM) | NA | 27.9±0.7 (40 µM) | NA |
| E15 | 23.0±0.5 (1.48 µM) | NA | 24.0±0.9 (40 µM) | NA | 10.3±0.3 (40 µM) | NA |
| E16 | 100.2±5.2 (164 nM) | 47.0±5.1 nM | 77.2±2.2 (40 µM) | 2.62±0.13 µM | 17.9±0.5 (40 µM) | NA |
NA: not applicable
NT: Not toxic (toxicity less than 5% of control)
GB FXR Cell Anta: GeneBLAzer FXR cell-based antagonistic assay
GB FXR Cell Cyto: Cytotoxicity assay performed in the GeneBLAzer FXR cells
IC50 values represent the means ± SE of at least three independent experiments derived as described in the EXPERIMENTAL SECTION
%Inh: %Inhibition is presented as the means ± SE of at least three independent experiments calculated as described in the EXPERIMENTAL SECTION. The value shown in the table represents the maximum percent inhibition observed, and its lowest corresponding compound concentration is noted in the parentheses.
Figure 2.

(A) FXR binding activities of GW4064, E16, and lithocholic acid in an FXR TR-FRET binding assay. (B) FXR agonistic activities (GW4064, E16, or lithocholic acid alone) and antagonistic activities (E16 and lithocholic acid in the presence of 400 nM GW4064) in a cell-based FXR transactivation assay. GW4064 and lithocholic acid were used as agonistic and antagonistic controls, respectively.
2.2. Design
With the novel substituted pyrazole-4-carboxamide framework in hand, we started to design a series of new trisubstituted-pyrazole carboxamide analogs to evaluate their receptor binding affinity and antagonistic potency in modulating the transcriptional activity of FXR. Recently, several crystal structures of the FXR-ligand binding domain (LBD) in complex with agonists were reported.26 However, no structural information is available on the binding mode of these FXR antagonists nor are co-crystal structures of a substituted pyrazole carboxamide framework in complex with the LBD of FXR available. Therefore, we could not directly identify the structural feature of the substituted pyrazole carboxamide framework that interacts with the ligand-dependent activation function 2 motif of the FXR LBD27. However, the flexible nature of the FXR side chains suggests the FXR LBD may have considerable flexibility to accommodate ligands of various shapes by changing its conformation in response to ligand binding.28 Huang et al. predicted the binding poses of the 1,3,4-substituted-pyrazolone analogs in the FXR ligand binding pocket by means of molecular docking to address the flexibility of the FXR LBD.21 It is recognized that the chemical structure of 1,3,4-substituted-pyrazolone is the most similar to the substituted 1,3,4-substituted-pyrazole, even though their functions are diverse.
To understand the structural basis for the SAR, we analyzed the published binding poses of the 1,3,4-substituted-pyrazolone derivative (Figure 1). The binding model showed that the 1,3-substituted alkyl rings of the pyrazolone core are crucial for the hydrophobic interactions between the two phenyl rings and the two aromatic residues (Phe329 and His294), even though the antagonistic potency of the pyrazolone derivative for FXR is still relatively low (IC50 = 8.96 ± 3.62 µM).21 The SAR analysis of the 1,3,4-substituted-pyrazolone derivative demonstrated that a subtle interplay between steric/hydrophobic effects of the 1,3- substituent of the pyrazolone core seemed to be critical for high FXR antagonistic potency. These observations suggest that varying the 1,3-substituents of the pyrazole core could be critical for high antagonistic potency.
On the basis of the detailed SAR studies for the 16 substituted pyrazole carboxamides (Tables 1 and 2), the substituted morpholinosulfonyl moiety of aniline on the 4-position appeared to be necessary to improve the antagonistic activity against FXR (E16 in Table 1). To increase the diversity of substituted moieties (R1, R2, and R3 in Figure 3) and optimize the 1,3,4-trisubstituted pyrazole-based molecules to improve their antagonistic activity against FXR, we investigated the alternative deconstructive hypothesis for fragment binding.29 Retrospectively, 1,3,4-trisubstituted-pyrazole carboxamides can be considered as being formed by two fragments: the substituted-N1-C3- pyrazole core (F1) and the substituted morpholinosulfonyl aniline moiety chain (F2) (Figure 3).
Figure 3.

The retro-analysis of 1,3,4-trisubstituted pyrazole carboxamides.
On the basis of our preliminary results (Tables 1 and 2) suggesting the substituted N1-C3- pyrazole moiety is sensitive to activity, various hydrophobic residues, such as alkyl or methoxy residues, substituted in the R1 or R2 positions of the phenyl-ring might significantly improve the antagonistic activity (Tables 1 and 2). We also determined whether the substituted N1-C3- pyrazole core (F1) and morpholinosulfonyl aniline (F2) could bind separately to FXR. We selected three commercially available products, 2-or 4-methyl -3-(morpholinosulfonyl)aniline and 3-(morpholinosulfonyl)aniline as F2 functionalities for this evaluation. A series of substituted F1 fragments was prepared (described in Experimental Section; compounds 3a–3e). These fragments failed to show an antagonistic effect against FXR. However, by connecting F1 moieties to F2 moieties through a linker of a single covalent bond by an amide bridge, the resulting carboxamide compounds had dramatically improved antagonistic activity against FXR, exemplified by compounds in series 4 (Figure 4). This suggested that 1,3,4-trisubstituted-pyrazole carboxamides can be created as potent antagonists of FXR by linking the F1 and F2 fragments.
Figure 4.

A focused two-component small array of pyrazoles designed for optimization.
Because the F2 fragments have a hydrophobic region centered on the double hydrogen bond acceptor (sulfone) and had very weak antagonistic activity against FXR, it may provide considerable conformational rigidity to the receptor recognition after joining with F1 fragments.30 Detailed SAR studies indicated that the position of the morpholinosulfonyl moiety of the F2 phenyl ring is important to modifications (Tables 1 and 2). Further analysis of the essential structural requirements for effective FXR antagonists showed that the meta-position-substituted morpholinosulfonyl moiety on the aniline amide moiety would influence the antagonistic activity. Therefore, we hypothesized that an appropriate 3-(morpholinosulfonyl)aniline moiety of the amide moiety needed to be fixed to act as a specific anchor during FXR binding recognition.
Variations of R3 on the F2 fragments could also affect the antagonistic potency. For example, introduction of a 4-methyl group on the F2 fragment resulted in greater antagonistic activity (Tables 1 and 2). Additionally, replacing the 4-methyl substituent on the aniline ring with a 2-methyl substituent conjugated with the same F1 resulted in lower antagonistic activities. Further, inclusion of an unmethylated 3-morpholinosulfonyl aniline substantially decreased the antagonistic potency (Tables 1 and 2). Taken together, this preliminary SAR analysis of a set of analogs provided important insight into the essential structural requirements that indicated 4-methyl-3-(morpholinosulfonyl)aniline in the F2 functionality would be an appropriate factor for effective FXR antagonists.
Guided by the SAR insights for the F2 fragments described above, we envisioned that F2 fragments might be a key pharmacophore region where an appropriate substituent, such as 4-methyl substituted aniline, would be crucial for hydrophobic interactions with FXR. We chose the 4-methyl -3-(morpholinosulfonyl)aniline as a fixed essential fragment and modified two regions of F1 fragments. We hypothesized that by subtly optimizing the steric/hydrophobic effects of the R1 substituents and fine-tuning the hydrogen-bonding capability of the R2 substituents, we could establish a focused library to develop novel FXR antagonists that could display high FXR binding affinity and potent cellular antagonistic effects against FXR.
2.3. Chemistry
Synthesis of 1,3,4-Trisubstituted Pyrazole Nonsteroidal FXR Antagonists
As indicated in Figure 4, the N1-and C3-pyrazole moieties on the pyrazole core could be explored with various electronic and hydrophobic substituted alkyls and phenyls. We hypothesized that potent analogs of 1,3-disubstituted-pyrazole-4-carboxamides may have structural features in common. From a parallel library synthesis perspective, compounds of this type are attractive because they contain only two vectors (denoted R1 and R2) that allow the introduction of considerable structural diversity. To explore novel carboxamide analogs of 1,3-disubstituted-pyrazole-4-carboxamides that could have high binding affinity and antagonistic effects against FXR, an efficient synthetic strategy was developed to optimize the N1-C3-pyrazole core structure through substitutions at R1 and R2 as a focused two-component small array of pyrazole. Combining structural considerations and chemical feasibility, our initial strategy comprised traditional medicinal chemistry involving variations in both the electronic and steric properties of the appendages of the central pyrazole nucleus, which could act as a template to put substituents into the correct positions to fit the receptor. A scaffold expansion based on two diverse points given by the substituents and their positions on the pyrazole ring, as well as the substituents (R1 and R2) on the N1,C3- pyrazole rings, was therefore designed (Figures 5 and 6).
Figure 5.

Initial exploration of pyrazole core via Vilsmeier-Haack reaction by the introduction of the first diversity element (R1).
Figure 6.

Alternative exploration of the pyrazole core to introduce R1 and R2 as substituted N1-C3-substituted pyrazoles of 4f–4s.
On the basis of the above design, two protocols for preparing the 1,3,4-trisubstituted-pyrazole carboxamides were developed to optimize the synthesis of compounds 4. These protocols included preparing various structural isomers to determine whether the positions of the R1 and R2 could affect compound activity. The key intermediates 3 and 7 (Figures 5 and 6) could be prepared by two possible routes to allow access to final functional analogs. In the first route, lead optimization was initiated by exploring the phenyl functionality located on the N1-position. A series of para- and meta-substitutions on the N1- pyrazole ring was investigated (Figure 5). In general procedures for preparing phenyl hydrazones, synthesis often begins with commercially available substituted acetophenone 12 and substituted phenylhydrazines 11. Condensation between acetophenone and substituted phenylhydrazines provides hydrazones 1. A Vilsmeier-Haack reaction, which is the key reaction for the cyclization of hydrazones to pyrazoles, assembles the pyrazole nucleus scaffolds.31 However, these conditions either do not form key aldehydes or have poor yields. We therefore modified the Vilsmeier-Haack reaction by adding the corresponding phenylhydrazones to an excess equivalence of POCl3 and DMF at 0 °C, then heated the reaction mixture to 60–70 °C with stirring over 18 h under N2 to afford the desired key cyclized aldehydes 2 at a 70–90% yield after hydrolysis. Oxidation of the substituted pyrazole-4-carboxaldehydes 2 with a Jones’ reagent32 yielded the corresponding substituted pyrazole-4-carboxylic acids 3. Finally, the corresponding acids 3 were coupled with commercially available 4-methyl-3-(morpholinosulfonyl)aniline 13 through a Mitsunobu reaction33 in the presence of cross-linking reagents (i.e., HOBt and EDCI) to produce the desired 4. The synthetic pathways used to obtain the 5 analogs of 1,3,4-trisubstituted-1H-pyrazoles 4 are illustrated in Figure 5.
An alternative strategy to explore the SAR by introducing selected R2 functional groups to investigate the structure requirements for FXR antagonistic activity is shown in Figure 6. The introduction of R2 groups was carried out by treating commercially available β-keto esters 14 with triethyl orthoformate in the presence of Ac2O as a catalyst to provide the corresponding ethoxymethylene intermediate 5. Cyclization of 5 with hydrazine in dry ethanol afforded the pyrazole core 6. The N1-alkylation reaction of 6 with excess appropriate bromoalkyl and bromobenzyl was performed in the presence of cesium carbonate as the base and acetonitrile as solvent. The ethyl esters 6 were hydrolyzed under basic conditions to afford the corresponding carboxylic acid 7. This was followed by a Mitsunobu coupling with 4-methyl-3-(morpholinosulfonyl)aniline in the presence of EDCI/DMAP as a fixed coupling reagent to produce the desired 14 products 4f–4s. Notably, in this alternative pathway, the combination of an activating coupling agent (i.e., HOBt and EDCI) did not work well, so the combination of EDCI/DMAP as an alternative coupling reagent was used.
To explore the linker between the amide bridge and the aromatic core, we focused our efforts on modifying the chemical space in the amide moiety to examine if the amide bridge was sensitive to activity. Three amides of series 4 were altered to generate compounds 8. We introduced a methyl moiety to assess the SAR. Compounds 4f, 4m, and 4o were chosen to react with iodomethane in the presence of sodium hydride to give N-methylation compounds 8 (Figure 7). Because the amide position was hypothesized to be a site of potential metabolism,34 further optimization involved reduction of amides. The amide groups in 4f, 4m, and 4o were reduced to corresponding amines with 1N borane-THF complex to yield compounds 9 (Figure 7).
Figure 7.

N-methylation and amide reduction of 4f, 4m, and 4o gave corresponding compounds 8 and 9.
2.2. Biological Evaluation
2.2.1. FXR TR-FRET Binding Assay
The FXR binding affinities of E16, the 23 newly synthesized novel 1,3-trisubstituted-pyrazol-4-carboxamide analogs shown in Figures 5–7, and control compounds GW4064 and lithocholic acid (LCA) were tested through an in vitro FXR TR-FRET binding assay using DY246 as the fluorescent probe.23 We chose TR-FRET assays because of their advantages of low background fluorescence interference and increased sensitivity as compared to other fluorescence-based assays, such as FI, FP and FRET assays.34 We chose DY246 because it is a derivative of GW4064, which is a potent FXR agonist. In addition, DY246 acts as a potent FXR agonist (EC50 of 550 nM) and has successfully been used as a fluorescent probe in a high throughput screening campaign to identify FXR antagonists.23 In a representative assay, DMSO (vehicle and negative control), 10 µM GW4064 (positive control), and titrations of GW4064, LCA or other chemicals were incubated for 20 min with an appropriate mixture of GST-FXR-LBD, terbium (Tb)-anti-GST and DY246, after which the TR-FRET signals were collected and activity for each chemical was normalized to DMSO (negative control; 0% inhibition) and 10 µM GW4064 (positive control; 100% inhibition). The activities of compounds that displayed dose-dependent inhibitory effects were fit into a one-site competitive binding equation to derive IC50 values. The TR-FRET binding activities are summarized in Tables 3, 4, and 5. Many of these compounds had significantly improved FXR binding affinity when compared to E16. Noticeably, 4j had an IC50 of 7.5 nM in the FXR TR-FRET binding assay, which was 5.26-fold more potent than E16 (IC50 of 47 nM) (Tables 3, 4, and 5; Figure 8A).
Table 3.
The effect of varying the substitution pattern of the 1,3,4-substituted-pyrazole-4-carboxamide derivatives on their biological activities.
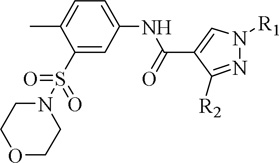 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cmpds | R1 | R2 | FXR TR-FRET Binding | GB FXR Cell Anta. | GB FXR Cell Cyto |
|||
| %Inh. | IC50a | %Inh. | IC50a | %Inh. | IC50 | |||
| GW4064 | 100±4.3 (10 μM) | 19.8±2.5 nM | NA | NA | NT | NA | ||
| Lithocholic Acid | 82.3±0.9 (40 µM) | 18.3±1.2 | 16.1±0.5 (40 µM) | NA | NT | NA | ||
| E16 | Ph | 4-CH3-Ph | 100.2±5.2 (164 nM) | 47.0±5.1 nM | 77.2±2.2 (40 µM) | 2.62±0.13 µM | 17.9±0.5 (40 µM) | NA |
| 4a | 4-F-Ph | 4-CH3-Ph | 95.0±1.2 (493 nM) | 24.3±0.6 nM | 72.0±2.2 (40 µM) | 2.49±0.0.06 µM | 18.5±1.0 (40 µM) | |
| 4b | 3-Cl-Ph | 4-CH3-Ph | 99.3±1.1 (164 nM) | 10.4±0.9 nM | 60.6±0.9 (40 µM) | 5.91±0.15 µM | NT | NA |
| 4c | 2,4-Di-Cl-Ph | 4-CH3-Ph | 119.8±7.8 (164 nM) | 14.1±0.3 nM | 89.9±1.9 (40 µM) | 1.24±0.12 µM | NT | NA |
| 4d | 3-CF3-Ph | 4-CH3-Ph | 77.5±4.3 (164 nM) | 23.9±2.1 nM | 54.8±1.3 (40 µM) | 22.29±1.14µM | 7.3±0.2 (40 µM) | NA |
| 4e |  |
4-CH3-Ph | 101.8±2.8 (164 nM) | 17.7±0.4 nM | 79.1±2.4 (40 µM) | 7.91±1.16 µM | 11.5±0.2 (13.3 µM) | NA |
| 4f | (CH3)2CH | 4-CH3-Ph | 130.6±3.8 (4.4 µM) | 49.1±9.9 nM | 99.1±4.4 (40 µM) | 1.69±0.18µM | NT | NA |
| 4g | CH3(CH2)3 | 4-CH3-Ph | 130.1±0.7 (1.5 µM) | 52.5±4.1 nM | 97.9±8.1 (20 µM) | 11.51±0.07 µM | 10.3±0.3 (4.4 µM) | NA |
| 4h |  |
4-CH3-Ph | 97.8±1.2 (164 nM) | 10.2±1.6 nM | 84.4±4.1 (40 µM) | 844.1±10.3 nM | 7.9±0.3 (4.4 µM) | NA |
| 4i | 4-CH3-Ph | 105.0±4.9 (1.5 µM) | 76.9±2.7 nM | 100.1±3.2 (40 µM) | 848.8±69.0 nM | NT | NA | |
| 4j | 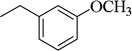 |
4-CH3-Ph | 130.1±5.4 (164 nM) | 7.5±0.8 nM | 98.9±4.3 (40 µM) | 468.5±8.4 nM | NT | NA |
| 4k | 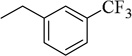 |
4-CH3-Ph | 99.2±6.4 (164 nM) | 18.4±1.2 nM | 93.4±3.9 (40 µM) | 515.0±25.4 nM | NT | NA |
| 4l | HO(CH2)4 | 4-CH3-Ph | 89.3±0.1 (493 nM) | 130.3±16 nM | 97.6±3.7 (40 µM) | 2.61 ±0.29 µM | 5.9±0.2 (40 µM) | NA |
| 4m | 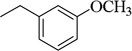 |
CH(CH3)2 | 100.6±5.6 (4.4 µM) | 199.4±21.2 nM | 99.2±2.7 (40 µM) | 3.11 ±0.58 µM | NT | NA |
| 4n | (CH3)2CH | 3-CH3O-Ph | 149.8±6.9 (13.3µM | 159.7±22.6 nM | 97.8±1.5 (40 µM) | 3.49 ±0.64 µM | 10.8±0.2 (4.4 µM | |
| 4o | 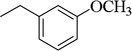 |
3-CH3O-Ph | 114.1±3.3 (493 nM) | 42.8±0.5 nM | 89.4±4.2 (40 µM) | 1.24 ±0.09 µM | NT | NA |
| 4p | (CH3)2CH | 4-CF3-Ph | 133.2±5.3 (4.4 µM) | 77.5±3.7 nM | 100.0±3.7 (40 µM) | 1.95 ±0.28 µM | 19.5±1.1 (40 µM) | NA |
| 4q | 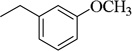 |
4-CF3-Ph | 109.1±3.6 (164 nM) | 30.9±3.6 nM | 97.6±0.9 (40 µM) | 592.7±3.0 nM | NT | NA |
| 4r | (CH3)2CH | CH3O(CH2)2 | 98.2±3.8 (40 µM) | 3.42±0.19 µM | 62.5±0.8 (40 µM) | 29.58 ±1.75 µM | NT | NA |
| 4s | 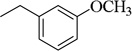 |
CH3O(CH2)2 | 113.5±0.8 (13.3µM) | 389.5±9.4 nM | 95.9±2.4 (40 µM) | 11.1 ±0.64 µM | NT | NA |
NA: not applicable
NT: Not toxic (toxicity less than 5%)
GB FXR Cell Anta: GeneBLAzer FXR cell-based antagonistic assay
GB FXR Cell Cyto: Cytotoxicity assay performed in the GeneBLAzer FXR cells
IC50 values represent the means ± SE of at least three independent experiments derived as described in the EXPERIMENTAL SECTION
%Inh: %Inhibition is presented as the means ± SE of at least three independent experiments calculated as described in the EXPERIMENTAL SECTION. The value shown in the table represents the maximum percent inhibition observed, and its lowest corresponding compound concentration is noted in the parentheses.
Table 4.
The effect of varying the substitution pattern of the 1,3,4-substituted-pyrazole-4-carboxamide derivatives on their biological activities (N-methylation).
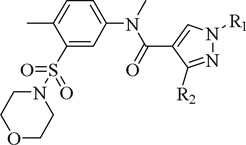 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cmpd | R1 | R2 | FXR TR-FRET Binding | GB FXR Cell Anta. | GB FXR Cell Cyto | |||
| %Inh. | IC50 | %Inh. | IC50 | %Inh. | IC50 | |||
| 8a | (CH3)2CH | 4-CH3-Ph | 132.9±6.2 (40 µM) | 489.4±21.2 nM | 86.4±2.8 (40 µM) | 16.16±0.28 µM | 15.0±0.7 (40 µM) | NA |
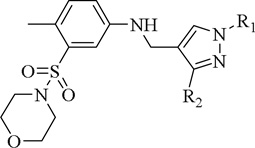
| 8b | 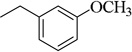 |
(CH3)2CH | 80.3±5.2 (40 µM) | 5.33±0.24 µM | 2.4±0.1 (20 µM) | NA | 13.5±0.4 (40 µM) | NA |
| 8c |  |
3-CH3O-Ph | 69.7±3.5 (40 µM) | 3.81±0.20 µM | 23.8±0.4 (40 µM) | NA | 19.7±0.4 (40 µM) | NA |
Table 5.
The effect of varying the substitution pattern of the 1,3,4-substituted-pyrazole-4-carboxamide derivatives on their biological activities (amide reduction).
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cmpd | R1 | R2 | FXR TR-FRET Binding | GB FXR Cell Anta. | GB FXR Cell Cyto | |||
| %Inh. | IC50 | %Inh. | IC50 | %Inh. | IC50 | |||
| 9a | (CH3)2CH | 4-CH3-Ph | 99.5±2.6 (13.3 µM) | 2.04±0.02 µM | 36.8±0.3 (40 µM) | NA | 10.5±0.3 (40 µM) | NA |
| 9b |  |
(CH3)2CH | 72.5±2.7 (4.4 µM) | 2.31±0.32 µM | 64.8±1.9 (40 µM) | 27.83 ±1.54 µM | 13.9±0.6 (40 µM) | NA |
| 9c |  |
3-CH3O-Ph | 39.8±1.8 (1.48 µM) | NA | 3.1±0.1 (40 µM) | NA | NT | NA |
Figure 8.

(A) FXR binding activities of E16 and 4j in a FXR TR-FRET binding assay. (B) Agonistic (E16 and 4j) and antagonistic activities (E16 and 4j in an FXR cell-based transactivation assay.
2.2.2. Cell-based FXR Transactivation Assay
To investigate the cellular activities of the novel 1,3-substituted-pyrazol-4-carboxamide analogs, we used a GeneBLAzer FXR cell-based assay, which detects the levels of a β-lactamase reporter controlled by a promoter containing the binding site of the DNA binding domain (DBD) of the GAL4 transcription factor; the Gal4 DBD was fused to the LBD of FXR in this assay.36 In typical test for agonists, DMSO (vehicle and negative control), 10 µM GW4064 (positive control), and titrations of GW4064, LCA or the 23 newly synthesized novel 1,3-trisubstituted-pyrazol-4-carboxamide analogs were mixed with GeneBLAzer FXR-UAS-bla HEK 293T cells. Reporter assays were performed after a16 h incubation. No agonistic activities were observed for any of the newly synthesized compounds (data not shown).
For the experiments to evaluate the antagonistic activity, titrations of compounds were tested in the presence of 400 nM GW4064. Assay controls included: DMSO alone (100% inhibition), 400 nM GW4064 alone (0% inhibition), and titrations of LCA in the presence of 400 nM GW4064 (as an assay reference). Activities of compounds that displayed activating or inhibitory activities in a dose-dependent manner were fit into a sigmoidal dose-response equation to derive individual EC50 or IC50 values. The tested chemicals’ FXR activities are summarized in Tables 3, 4, and 5. Many of these compounds had significantly improved antagonistic activity when compared to E16. Noticeably, in the FXR antagonistic assay, 4j had an IC50 of 468.5 nM, which was 4.59-fold more potent than that of E16 (IC50 of 2.62 µM) (Tables 3, 4, and 5; Figure 8B).
2.2.3. Cytotoxicity Assay
For the FXR cell-based antagonistic assays, if a tested compound caused a signal reduction, it was usually considered an antagonist. However, signal reduction could also be caused non-specifically if a chemical was cytotoxic, although GeneBLAzer assays are relatively insensitive to cytotoxicity because of their use of ratiometric reading. To examine if certain chemical’s antagonistic effect was caused by cytotoxicity, we used the same GeneBLAzer FXR cells in cytotoxicity assays. These assays were set up the same as for an FXR agonistic assay, with the exception of signal reading. Instead of detecting reporter activities, the CellTiter-Glo® assay was used to detect cell viability. Conditions including DMSO with or without cells were used as negative (0% inhibition) or positive (100% inhibition) controls, respectively. Cytotoxic activities are summarized in Tables 3, 4, and 5. All chemicals tested showed marginal toxicity (% inhibition < 5 %), therefore the antagonistic effect of the compounds was not caused by non-specific cytotoxicity.
2.3. SAR
We investigated the effect of introducing electron withdrawing groups, such as fluorine, chlorine, dichlorines, and trifluoromethyl atoms, at ortho, meta, or para positions of the N1-phenyl.37 All of these derivatives (4a, 4b, 4c, 4d, and 4e) improved FXR antagonistic activity by about 3- to 4-fold as compared to E16 (Table 3), indicating that the electron withdrawing groups were well tolerated for antagonistic activity. This observation can be attributed to the higher interaction strength of fluorine or chlorine atoms compared to hydrogen, which enables a tighter fit.
Based on this initial SAR, we next introduced longer benzyl chains or small alkyl groups to replace the N1-phenyl of the pyrazole moiety to see if it was a suitable position to find more potent antagonists for FXR. The alternative synthetic methodology for constructing the corresponding pyrazole-4-carboxylic acids worked well. Replacing the phenyl ring with small hydrophobic substituents such as isopropyl (4f) or n-butyl (4g) allowed slight retention of the antagonistic activity toward FXR. Substituting with a methylcyclopropane (4i) decreased the activity by 2-fold for FXR as compared to E16. Interestingly, incorporating a bulkier group, such as benzo[d]thiazole (4e), led to 3-fold higher potency for FXR. Other substitutions, including longer and bulkier benzyl aromatic substituents such as 2-naphthyl (4h) and benzyl with an electron donating group m-methoxy (4j), enhanced antagonistic activity. Among the N1-alkylated analogs, 4j exhibited the most potent FXR-antagonistic activity, having IC50 values of 7.5 nM and 468.5 nM in a FXR TR-FRET binding assay and a FXR cell-based antagonistic assay, respectively. In addition, 4j had no FXR agonistic activity and no cytotoxicity. These results suggest that an expanded hydrophobic moiety is necessary at the N1-position and the positive influence of the m-methoxybenzyl group may be generally preferred at the N1-position of the pyrazole ring.
Replacing the original C3-p-methyl phenyl of 4j with the stronger electron-donating m-methoxy phenyl group resulted in analog 4o, which had slightly decreased antagonistic activity (Table 3). This indicated that the binding pocket of FXR that hosted the m-methoxy phenyl group on the C3 position of pyrazole is not sufficiently wide to accept larger substituents for hydrogen bond interactions with FXR. Notably, replacing the original of C3-p-methyl phenyl (R2) of 4j with a smaller isopropyl group (analog 4m) significantly decreased the potency and affinity, demonstrating that the C3 moiety is very sensitive in terms of steric bulkiness and/or hydrophobicity. For instance, introducing a p-trifluoromethyl phenyl with electron withdrawing properties at the C3 position of the pyrazole ring gave products 4p and 4q, which had nanomolar IC50 values for in vitro potency. 4p and 4q appeared to have similar antagonistic activities as their parent compounds 4f and 4o. In contrast, replacing the p-trifluoromethyl phenyl at the C3 position of the pyrazole ring with a linear 2-methoxy-ethyl group to obtain compounds 4r and 4s, resulted in severe loss of potency relative to that of 4f and 4o, indicating that an alkyl methoxy in the C3 position of pyrazole ring is not beneficial. Therefore, we speculate that an aromatic phenyl ring at the C3-position of the pyrazole ring is required not only to optimally bind to a hydrophobic area of FXR, but is also critical for π−π interaction with the receptor to increase FXR antagonistic activity. These examples demonstrate the favorable interaction that can be gained from fine-tuning the hydrophobic shape complementarily.
We also modified compounds 4f, 4m, and 4o with amide moieties. The carboxamide nitrogens of 4f, 4m, and 4o were methylated to form N-methyl analogs 8a, 8b, and 8c (Figure 7). Unfortunately, N-methylation of the CONH function of amide compounds 8 decreased the antagonistic activity (Table 4), as compared to the activity of the original amides, 8a, 8b, and 8c were essentially inactive at 1 µM against FXR (data not shown). Similarly, compounds 9 had weak activity at 1 µM, at least on FXR (data not shown). Compound 9 had more than 300-fold less affinity and potency as compared with 4j. The discrepancies in activity of these analogs between in vitro and in vivo might be due to their stability, solubility and /or permeability.
3. CONCLUSIONS
FXR is a nuclear receptor that functions as a physiological receptor for bile acids. A highly potent and selective antagonist of FXR would represent a novel option that could prove useful for treating dyslipidemia and cholestasis. Thus, there is considerable need to identify lead structures and pharmacophore in this relatively unexplored area. We investigated the preliminary SAR-based optimization of 1,3,4-trisubstituted-pyrazole carboxamides as FXR antagonists. In an effort to develop specific FXR antagonists, a traditional medicinal chemistry parallel strategy was used. A series of 1,3,4-trisubstituted-pyrazole carboxamide analogues were developed and analyzed for physicochemical properties and SAR based on in vitro data. All of these modified compounds were evaluated for FXR binding affinities in biochemical assays and antagonistic activity in cell-based transactivation assays. All amide compounds reported here behaved as potent FXR antagonists, and did not have FXR agonistic activity (data not shown). Of interest, compound 4j had IC50 values of 7.5 nM and 468.5 nM in FXR TR-FRET binding assays and FXR cell-based antagonistic assays, respectively, suggesting it is a promising candidate FXR antagonist. In addition, compound 4j does not have any FXR agonistic activity and cytotoxicity, suggesting that expansion of R1 and R2 on the pyrazole scaffold increases hydrogen bond acceptor interactions with FXR. This work has resulted in the most potent antagonist to date for FXR, 4j, which may emerge as novel chemical tool to explore the effect of FXR inhibition in pharmacological settings. Studies aimed at further profiling 4j in vitro and expanding the SAR of these analogs with the goal of developing FXR antagonists that can be delivered orally and are active in vivo are under way.
4. EXPERIMENTAL SECTION
4.1. Chemistry
General procedures
Organic reagents were purchased from commercial suppliers unless otherwise noted and were used without further purification. All solvents were analytical or reagent grade. All reactions were carried out in flame-dried glassware under argon or nitrogen. Melting points were determined and reported automatically by an optoelectronic sensor in open capillary tubes and were uncorrected. 1H NMR and 13C NMR spectra were measured at 500 MHz and 125 MHz, respectively, using CDCl3 or CD3OD as solvents and tetramethylsilane (Me4Si) as the internal standard. Flash column chromatography was performed using Sigma-Aldrich silica gel 60 (200–400 mesh), carried out under moderate pressure with columns of an appropriate size packed and eluted with appropriate eluents. All reactions were monitored by thin layer chromatography (TLC) on precoated plates (silica gel HLF). TLC spots were visualized either by exposure to iodine vapor or by irradiation with UV light. Organic solvents were removed under vacuum by a rotary evaporator. Elemental analyses were performed by Columbia Analytical Services Inc, Tucson, Arizona.
4.1.1. (E)-1-(4-fluorophenyl)-2-(1-p-tolylethylidene)hydrazine (1a)
To a solution of 4’-methylacetophenone (0.5 g, 3.73 mmol) in 8 mL of acetic acid and 5 mL of MeOH, 4-fluorophenylhydrazine hydrochloride (0.6 g, 3.73 mmol) was added. The reaction mixture was stirred at room temperature for 18 h. The reaction mixture was diluted with an additional 30 mL of water and subsequently filtered. The solid was dried in vacuo to afford 0.63 g of beige solid 1a (70% yield): mp 101.8 °C. 1H NMR (CDCl3) δ 7.68 (d, 2H), 7.19 (d, 3H), 7.07 (t, 2H), 6.98 (d, 2H), 2.37 (s, 3H), 2.23 (s, 3H).
4.1.2. (E)-1-(3-Chlorophenyl)-2-(1-p-tolylethylidene)hydrazine (1b)
To a solution of 4’-methylacetophenone (0.5 g, 3.73 mmol) in 8 mL of acetic acid and 5 mL of water/MeOH, 3-chlorophenylhydrazine hydrochloride (0.67 g, 3.73 mmol) was added. The reaction mixture was stirred at room temperature for 18 h. The reaction mixture was diluted with an additional 30 mL of water and subsequently filtered. The solid was dried in vacuo to afford 0.58 g of beige solid 1b (60% yield): mp 100.5 °C. 1H NMR (CDCl3) δ 7.68 (t, 2H), 7.20 (t, 5H), 6.98 (s, 1H), 6.83 (d, 1H), 2.38 (s, 3H), 2.28 (s, 3H).
4.1.3. (E)-1-(2,4-dichlorophenyl)-2-(1-p-tolylethylidene)hydrazine (1c)
To a solution of 4’-methylacetophenone (0.5 g, 3.73 mmol) in 8 mL of acetic acid and 5 mL of water/MeOH, 2,4-dichlorophenylhydrazine hydrochloride (0.8 g, 3.73 mmol) was added. The reaction mixture was stirred at room temperature for 18 h. The reaction mixture was diluted with an additional 30 mL of water and subsequently filtered. The solid was dried in vacuo to afford 0.98 g of beige solid 1c (90% yield): mp 122.7 °C. 1H NMR (CDCl3) δ 7.71 (t, 2H), 7.61 (d, 2H), 7.29 (s, 1H), 7.021(d, 3H), 2.38 (s, 3H), 2.28 (s, 3H).
4.1.4. (E)-1-(3-Trifluromethylphenyl)-2-(1-p-tolylethylidene)hydrazine (1d)
To a solution of 4’-methylacetophenone (0.5 g, 3.73 mmol) in 8 mL of acetic acid and 5 mL of water/MeOH, 3-trifluoromethylphenylhydrazine hydrochloride (0.66 g, 3.73 mmol) was added. The reaction mixture was stirred at room temperature for 18 h. The reaction mixture was diluted with an additional 30 mL of water and subsequently filtered. The solid was dried in vacuo to afford 1.01 g of beige solid 1d (93% yield): mp 103.7 °C. 1H NMR (CDCl3) δ 7.72 (d, 2H), 7.44 (m, 4H), 7.28 (d, 2H), 7.14 (s, 1H), 2.41 (s, 3H), 2.27 (s, 3H).
4.1.5. (E)-2-(2-(1-(p-tolyl)ethylidene)hydrazinyl)benzo[d]thiazole (1e)
To a solution of 4’-methylacetophenone (0.5 g, 3.73 mmol) in 8 mL of acetic acid and 1 mL of water, 2-hydrazinobenthiazole (0.62 g, 3.73 mmol) was added. The reaction mixture was stirred at room temperature for 18 h. The reaction mixture was diluted with an additional 20 mL of water and subsequently filtered. The solid was dried in vacuo to afford 1 g of beige solid 1e (99% yield): mp 112.6 °C. 1H NMR (CDCl3) δ 7.74 (d, 2H), 7.68 (d, 1H), 7.53 (d, 1H), 7.36 (d, 1H), 7.25 (d, 2H), 7.16 (d, 1H), 2.41 (s, 3H), 2.17 (s, 3H).
4.1.6. 1-(4-fluorophenyl)-3-p-tolyl-1H-pyrazole-4-carbaldehyde (2a)
A solution of DMF (3 mL) and POCl3 (1.5 mL, 16.4 mmol) was made in an ice bath under argon and stirred for 10 min. 1a (0.21 g, 0.87 mmol) dissolved in dichloroethane (5 mL) was added to this solution; the mixture was warmed to 70 °C and stirred for an additional 18 h. The reaction mixture was then cooled to room temperature, slowly poured into saturated aqueous NaHCO3 (200 mL) under ice cold conditions, and then warmed to room temperature and stirred for 3 h. The aqueous layer was extracted from the reaction mixture with EtOAc. The organic layers were washed with water and brine, dried over Na2SO4 and concentrated in vacuo to yield 0.21 g (88%) of the title compound: mp 143.8 °C, which was used without further purification. 1H NMR (CDCl3) δ 10.05 (s, 1H), 8.47 (s, 1H), 7.78 (m, 1H), 7.71 (d, 2H), 7.33 (d, 2H), 7.19 (m, 3H), 2.44 (s, 3H). 13C NMR (CDCl3) δ 187.0, 156.5, 132.7, 130.9, 130.2, 129.8, 123.9, 123.1, 118.1, 117.8, 23.3.
4.1.7. 1-(3-Chlorophenyl)-3-p-tolyl-1H-pyrazole-4-carbaldehyde (2b)
A solution of DMF (3 mL) and POCl3 (1.5 mL, 16.4 mmol) was made in an ice bath under argon and stirred for 10 min. 1b (0.58 g, 2.24 mmol) dissolved in dichloroethane (5 mL) was added to this solution; the mixture was heated to 70 °C and stirred for an additional 18 h. The reaction mixture was then cooled to room temperature and slowly poured into saturated aqueous NaHCO3 (200 mL) under ice cold conditions, then warmed to room temperature and stirred for 3 h. The aqueous layer was extracted with EtOAc. The organic layers were washed with water, brine, dried over Na2SO4 and concentrated in vacuo to yield 0.64 g (88%) of the title compound: mp 118.7 °C, which was used without further purification. 1H NMR (CDCl3) δ 10.05 (s, 1H), 8.53 (s, 1H), 7.88 (s, 1H), 7.67 (d, 3H), 7.46 (t, 1H), 7.31 (d, 3H), 2.44 (s, 3H). 13C NMR (CDCl3) δ 186.5, 159.3, 141.1, 132.1, 130.9, 130.2, 129.3, 121.6, 118.8, 23.0.
4.1.8. 1-(2,4-Dichlorophenyl)-3-p-tolyl-1H-pyrazole-4-carbaldehyde (2c)
A solution of DMF (3 mL) and POCl3 (1.5 mL, 16.4 mmol) was made in an ice bath under argon and stirred for 10 min. 1c (0.61 g, 2.08 mmol) dissolved in dichloroethane (5 mL) was added to this solution; the mixture was heated to 70 °C and stirred for an additional 18 h. The reaction mixture was then cooled to room temperature and slowly poured into saturated aqueous NaHCO3 (200 mL) under ice cold conditions and then warmed to room temperature and stirred for 3 h. The aqueous layer was extracted with EtOAc. The organic layers were washed with water, brine, dried over Na2SO4 and concentrated in vacuo to yield 0.53 g (78%) of the title compound: mp 85.8 °C, which was used without further purification. 1H NMR (CDCl3) δ 10.05 (s, 1H), 8.48 (s, 1H), 7.66 (m, 3H), 7.54 (s, 1H), 7.42 (d, 1H), 7.37 (d, 2H), 2.38 (s, 3H). 13C NMR (CDCl3) δ 186.6, 156.59 140.9, 137.4, 136.8, 134.1, 132.0, 131.7, 130.8, 129.4, 127.2, 105.7, 22.7.
4.1.9. 3-p-Tolyl-1-(3-(trifluoromethyl)phenyl)-1H-pyrazole-4-carbaldehyde (2d)
A solution of DMF (3 mL) and POCl3 (1.5 mL, 16.4 mmol) was made in an ice bath under argon and stirred for 10 min. 1d (0.75 g, 2.56 mmol) dissolved in dichloroethane (7 mL) was added to this solution; the mixture was heated to 70 °C and stirred for an additional 18 h. The reaction mixture was then cooled to room temperature and slowly poured into saturated aqueous NaHCO3 (200 mL) under ice cold conditions and then warmed to room temperature and stirred for 3 h. The aqueous layer was extracted with EtOAc. The organic layers were washed with water and brine, dried over Na2SO4 and concentrated in vacuo to yield 0.79 g (93%) of the title compound (mp 155.1 °C), which was used without further purification. 1H NMR (CDCl3) δ 10.08 (s, 1H), 8.60 (s, 1H), 8.13 (s, 1H), 7.99 (q, 1H), 7.74 (d, 2H), 7.67 (d, 2H), 7.35 (d, 2H), 2.45 (s, 3H). 13C NMR (CDCl3) δ 186.6, 157.3, 141.7, 140.8, 132.3, 131.8, 130.9, 130.3, 126.6, 126.2, 123.8, 22.8.
4.1.10. 1-(2,3-Dihydro-1H-inden-1-yl)-3-(p-tolyl)-1H-pyrazole-4-carbaldehyde (2f)
A solution of DMF (3 mL) and POCl3 (1.5 mL, 16.4 mmol) was made in an ice bath under argon and stirred for 10 min. 1f (1.0 g, 3.55 mmol) dissolved in dichloroethane (5 mL) was added to the solution; the mixture was heated to 60 °C and stirred for an additional 18 h. The reaction mixture was then cooled to room temperature, slowly poured into saturated aqueous NaHCO3 (200 mL) under ice cold conditions and then warmed to room temperature and stirred for an additional 5 h. The aqueous layer was extracted with EtOAc. The organic layers were washed with water, brine, dried over Na2SO4 and concentrated in vacuo to yield 0.74 g (70%) of the title compound: mp 145.3 °C, which was used without further purification. 1H NMR (CDCl3) δ 10.11 (s, 1H), 9.08 (s, 1H), 7.98 (d, 1H), 7.89 (d, 1H), 7.79 (d, 2H), 7.55 (t, 1H), 7.44 (t, 1H), 7.35 (d, 2H), 2.41 (s, 3H). 13C NMR (CDCl3) δ 186.0, 157.1, 152.0, 141.5, 134.0, 130.9, 130.3, 128.3, 124.4, 123.2, 23.4.
4.1.11. 1-(4-fluorophenyl)-3-p-tolyl-1H-pyrazole-4-carboxylic acid (3a)
To a solution of 2a (0.21 g, 0.75 mmol) dissolved in 16 mL of acetone, 5 mL of Jones’ reagent was added. The reaction mixture was stirred at room temperature and monitored by TLC. The reaction mixture was poured into water and extracted with EtOAc. The combined organic layers were washed with water and brine, dried over Na2SO4, and concentrated in vacuo to afford 0.16 g (72%) of the title acid: mp 202.0 °C. 1H NMR (CDCl3) δ 8.51 (s, 1H), 7.75 (m, 3H), 7.32 (d, 2H), 7.18 (t, 4H), 2.41 (s, 3H). 13C NMR (CDCl3) δ 170.7, 164.5, 155.9, 140.6, 135.1, 132.6, 130.9, 130.6, 130.2, 130.1, 122.8, 118.2, 22.7. Anal. Calcd for C17H13FN2O2: C, 68.91; H, 4.41; N, 9.45. Found: C, 68.13; H, 4.43; N, 8.94.
4.1.12. 1-(3-Chlorophenyl)-3-p-tolyl-1H-pyrazole-4-carboxylic acid (3b)
To a solution of 2b (0.51 g, 1.72 mmol) dissolved in 16 mL of acetone, 5 mL of Jones’ reagent was added. The reaction mixture was stirred at room temperature and monitored by TLC. The reaction mixture was poured into water and extracted with EtOAc. The combined organic layers were washed with water and brine, dried over Na2SO4, and concentrated in vacuo to afford 0.51 g (95%) of the title acid: mp 194.4 °C. 1H NMR (CDCl3) δ 8.57 (s, 1H), 7.86 (d, 1H), 7.77 (t, 2H), 7.66 (d, 1H), 7.44 (t, 1H), 7.34 (d, 1H), 7.28 (m, 2H), 2.41 (s, 3H). 13C NMR (CDCl3) δ 168.8, 156.9, 147.7, 141.6, 140.3, 137.2, 134.7, 132.1, 130.6, 129.1, 121.3, 118.7, 22.8. Anal. Calcd for C17H13FClN2O2: C, 65.29; H, 4.19; N, 8.96. Found: C, 64.65; H, 3.37; N, 8.80.
4.1.13. 1-(2,4-Dichlorophenyl)-3-p-tolyl-1H-pyrazole-4-carboxylic acid (3c)
To a solution of 2c (0.53 g, 1.6 mmol) dissolved in 16 mL of acetone, 5 mL of Jones’ reagent was added. The reaction mixture was stirred at room temperature and monitored by TLC. The reaction mixture was poured into water and extracted with EtOAc. The combined organic layers were washed with water and brine, dried over Na2SO4, and concentrated in vacuo to afford 0.51 g (91%) of the title acid: mp 211.7 °C. 1H NMR (CDCl3) δ 8.52 (s, 1H), 7.75 (d, 2H), 7.66 (d, 1H), 7.58 (s, 1H), 7.41 (d, 1H), 7.24 (d, 2H), 2.43 (s, 3H). 13C NMR (CDCl3) δ 171.6, 155.7, 139.3, 131.9, 130.6, 130.2, 134.7, 132.1, 130.6, 129.8, 22.7. Anal. Calcd for C17H12Cl2N2O2: C, 58.81; H, 3.48, N, 8.07. Found: C, 58.46; H, 3.40, N, 7.68.
4.1.14. 3-(p-Tolyl)-1-(3-(trifluoromethyl)phenyl)-1H-pyrazole-4-carboxylic acid (3d)
To a solution of 2d (0.79 g, 2.39 mmol) dissolved in 16 mL of acetone, 5 mL of Jones’ reagent was added. The reaction mixture was stirred at room temperature and monitored by TLC. The reaction mixture was poured into water and extracted with EtOAc. The combined organic layers were washed with water and brine, dried over Na2SO4, and concentrated in vacuo to afford 0.81 g (98%) of the title acid: mp 181.8 °C. 1H NMR (CDCl3) δ 8.64 (s, 1H), 8.09 (s, 1H), 7.98 (d, 1H), 7.79 (d, 2H), 7.63 (d, 2H), 7.31 (t, 2H), 2.45 (s, 3H). 13C NMR (CDCl3) δ 168.2, 155.7, 140.8, 140.5, 134.8, 131.7, 130.7, 130.2, 129.8, 125.5, 123.7, 117.9, 114.5, 22.8. Anal. Calcd for C18H13F3N2O2: C, 62.43; H, 3.78; N, 8.09. Found: C, 61.95; H, 3.74; N, 7.62.
4.1.15. 1-(Benzo[d]thiazol-2-yl)-3-(p-tolyl)-1H-pyrazole-4-carboxylic acid (3e)
To a solution of 2e (0.71 g, 2.31 mmol) dissolved in 16 mL of acetone, 5 mL of Jones’ reagent was added. The reaction mixture was stirred at room temperature and monitored by TLC. The reaction mixture was poured into water and extracted with EtOAc. The combined organic layers were washed with water and brine, dried over Na2SO4, and concentrated in vacuo to afford 0.41 g (57%) of the title acid: mp 208.0 °C. 1H NMR (CDCl3) δ 9.09 (s, 1H), 7.98 (d, 1H), 7.90 (d, 1H), 7.81 (d, 2H), 7.54 (d, 1H), 7.43 (d, 1H), 7.35 (d, 2H), 2.45 (s, 3H). 13C NMR (CDCl3) δ 171.2, 160.1, 151.6, 141.3, 134.0, 131.3, 130.3, 129.0, 126.9, 124.2, 123.5, 23.0.
General Procedure of Mitsunobu Reaction
EDCI and HOBT were added to a solution of 3 in CH2Cl2. The reaction mixture was stirred for 1–2 h. 4-methyl-3-(morpholinosulfonyl)aniline was added to the reaction and the reaction mixture was stirred at room temperature overnight and quenched with water. Layers were separated and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4 and then evaporated under a vacuum. The resulting yellow solids were purified by flash column chromatography to afford 4a–4e.
4.1.16. 1-(4-fluorophenyl)-N-(4-methyl-3-(morpholinosulfonyl)phenyl)-3-p-tolyl-1H-pyrazole-4-carboxamide (4a)
To a solution of 3a (0.2 g, 0.67 mmol) in CH2Cl2 (10 mL) was added EDCI (0.22 g, 1.16 mmol) and HOBT (0.16 g, 1.16 mmol). The reaction mixture was stirred for 1 h. To the reaction was added 4-methyl-3-(morpholinosulfonyl)aniline (0.17 g, 0.68 mmol). The reaction mixture was stirred at room temperature for 12 h and quenched with water. Layers were separated, and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4 and then evaporated under a vacuum. The resulting yellow solid was purified by flash column chromatography (Hexanes/EtOAc 1:1) to afford 4a as a white solid (0.09 g, 26% yield), mp 95.9 °C. 1H NMR (CDCl3) δ 8.52 (s, 1H), 7.73 (s, 1H), 7.72 (d, 2H), 7.64 (t, 2H), 7.54 (d, 2H), 7.36 (d, 2H), 7.27 (d, 3H), 3.72 (d, 4H), 3.17 (d, 4H), 2.57 (s, 3H), 2.47 (s, 3H). 13C NMR (CDCl3) δ 162.1, 152.6, 141.4, 137.5, 137.4, 134.8, 134.6, 132.7, 131.2, 130.7, 130.2, 125.2, 122.7, 122.3, 119.5, 117.9, 117.7, 67.7, 46.8, 23.9, 22.6. Anal. Calcd for C28H27FN4O4S: C, 62.91; H, 5.09; N, 10.48; F, 3.55. Found: C, 62.75; H, 4.57; N, 10.46; F, 3.4.
4.1.17. 1-(3-Chlorophenyl)-N-(4-methyl-3-(morpholinosulfonyl)phenyl)-3-(p-tolyl)-1H-pyrazole-4-carboxamide (4b)
To a solution of 3b (0.2 g, 0.67 mmol) in CH2Cl2 (10 mL) was added EDCI (0.22 g, 1.16 mmol) and HOBT (0.15 g, 1.16 mmol). The reaction mixture was stirred for 1 h. To the reaction was added 4-methyl-3-(morpholinosulfonyl)aniline (0.17 g, 0.68 mmol). The reaction mixture was stirred at room temperature for 12 h and quenched with water. The organic layer was collected and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4, evaporated under a vacuum. The resulting yellow solid was purified by flash column chromatography (H/EtOAc 1:1) to afford 4b as a white solid (0.07 g, 19% yield), mp 204.9 °C. 1H NMR (CDCl3) δ 8.60 (s, 1H), 7.88 (s, 1H), 7.72 (d, 1H), 7.62(m, 3H), 7.45 (d, 2H), 7.41 (m, 3H), 7.24 (d, 2H), 3.75 (d, 4H), 3.21 (d, 4H), 2.59 (s, 3H), 2.50 (s, 3H). 13C NMR (CDCl3) δ 162.1, 152.7, 147.6, 141.7, 137.9, 134.8, 132.8, 132.0, 131.3, 130.7, 128.9, 125.1, 122.3, 121.3, 118.5, 67.7, 64.9, 23.0, 21.5, Anal. Calcd for C28H27ClN4O4S: C, 61.03; H, 4.94; N, 10.17; Cl, 6.43. Found: C, 60.78; H, 4.61; N, 9.94; Cl, 6.7.
4.1.18. 1-(2,4-Dichlorophenyl)-N-(4-methyl-3-(morpholinosulfonyl)phenyl)-3-(p-tolyl)-1H-pyrazole-4-carboxamide (4c)
To a solution of 3c (0.2 g, 0.58 mmol) in CH2Cl2 (5 mL) was added EDCI (0.22 g, 1.16 mmol) and HOBT (0.16 g, 1.16 mmol). The reaction mixture was stirred for 1 h. To the reaction was added 4-methyl-3-(morpholinosulfonyl)aniline (0.15 g, 0.58 mmol). The reaction mixture was stirred at room temperature for 12 h and quenched with water. The organic layer was collected and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4, evaporated under a vacuum. The resulting yellow solid was purified by flash column chromatography (H/EtOAc 1:1) to afford 4c as a white solid (0.06 g, 20% yield), mp 99.8 °C. 1H NMR (CDCl3) δ 8.52 (s, 1H), 7.73 (s, 1H), 7.62 (m, 3H), 7.45 (d, 2H), 7.38 (m, 3H), 7.24 (d, 2H), 3.73 (d, 4H), 3.19 (d, 4H), 2.57 (s, 3H), 2.46 (s, 3H). 13C NMR (CDCl3) δ 162.0, 152.6, 141.9, 139.9, 137.7, 134.7, 134.3, 132.0, 131.3, 130.8, 129.5, 127.4, 125.2, 122.0, 118.3, 116.6, 67.7, 46.8, 22.9, 21.8. Anal. Calcd for C28H26Cl2N4O4S: C, 57.44; H, 4.48; N, 9.57; Cl, 12.22. Found: C, 57.11; H, 4.27; N, 8.91; Cl, 12.2.
4.1.19. N-(4-Methyl-3-(morpholinosulfonyl)phenyl)-3-(p-tolyl)-1-(3-(trifluoromethyl) phenyl)-1H-pyrazole-4-carboxamide (4d)
To a solution of 3d (0.2 g, 0.58 mmol) in CH2Cl2 (10 mL) was added EDCI (0.34 g, 1.76 mmol) and HOBT (0.24 g, 1.76 mmol). The reaction mixture was stirred for 1 h. To the reaction was added 4-methyl-3-(morpholinosulfonyl)aniline (0.2 g, 0.78 mmol). The reaction mixture was stirred at room temperature for 16 h and quenched with water. The organic layer was collected and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4, evaporated under a vacuum. The resulting yellow solid was purified by flash column chromatography (H/EtOAc 1:1) to afford 4d as a white solid (0.1 g, 31% yield), mp 192.7 °C. 1H NMR (CDCl3) δ 8.67 (s, 1H), 8.12 (s, 1H), 7.92 (d, 1H), 7.72 (s, 1H), 7.64 (d, 4H), 7.51 (t, 3H), 7.41 (d, 2H), 3.74 (d, 4H), 3.19 (d, 4H), 2.58 (s, 3H), 2.50 (s, 3H). 13C NMR (CDCl3) δ 165.0, 151.6, 141.9, 139.9, 137.7, 134.7, 134.3, 132.0, 131.3, 130.8, 129.5, 127.4, 125.2, 124.1, 118.3, 116.6, 67.8, 46.9, 23.9, 23.6. Anal. Calcd for C29H27F3N4O4S: C, 59.58; H, 4.66; N, 9.58; F, 9.75. Found: C, 60.07; H, 4.38; N, 9.53; F, 10.
4.1.20. 1-(Benzo[d]thiazol-2-yl)-N-(4-methyl-3-(morpholinosulfonyl)phenyl)-3-(p-tolyl)-1H-pyrazole- 4-carboxamide (4e)
To a solution of 3e (0.2 g, 0.59 mmol) in CH2Cl2 (5 mL) was added EDCI (0.14 g, 0.71 mmol) and HOBT (0.1 g, 0.7 mmol). The reaction mixture was stirred for 1 h. To the reaction was added 4-methyl-3-(morpholinosulfonyl)aniline (0.15 g, 0.59 mmol). The reaction mixture was stirred at room temperature for 12 h and quenched with water. The organic layer was collected and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4, evaporated under a vacuum. The resulting yellow oil was purified by flash column chromatography (H/EtOAc 1:1) to afford 4e as a pale-yellow solid (0.07 g, 21% yield), mp 153.1 °C. 1H NMR (CDCl3) δ 8.51 (s, 1H), 8.02 (s, 1H), 7.97 (d, 2H), 7.79 (d, 1H), 7.71 (t, 2H), 7.55 (t, 1H), 7.40 (dd, 4H), 3.74 (d, 4H), 3.19 (d, 4H), 2.63 (s, 3H), 2.44 (s, 3H). 13C NMR (CDCl3) δ 165.0, 159.6, 144.5, 135.4, 137.7, 131.3, 131.0, 130.7, 130.2, 128.5, 126.1, 123.1, 123.0, 67.7, 47.7, 23.0, 21.7. Anal. Calcd for C29H27N5O4S2: C, 60.71; H, 4.74; N, 12.21. Found: C, 60.57; H, 4.2; N, 12.6.
General Procedure of Mitsunobu Reaction for producing 4f–4s
EDCI and DMAP were added to a solution of 7 in CH2Cl2. To the reaction was added 4-methyl-3-(morpholinosulfonyl)aniline. The reaction mixture was stirred at room temperature for 6 h and quenched with water and then treated with 10% HCl. The organic layer was collected, and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4, evaporated under a vacuum. The resulting yellow oil was purified by flash column chromatography to afford 4f–4s.
Selected examples
4.1.21. 1-Isopropyl-N-(4-methyl-3-(morpholinosulfonyl)phenyl)-3-(p-tolyl)-1H-pyrazole-4-carboxamide (4f)
To a solution of 7a (0.11 g, 0.43 mmol) in CH2Cl2 (5 mL) was added EDCI (0.14 g, 0.73 mmol) and DMAP (0.09 g, 0.73 mmol). To the reaction was added 4-methyl-3-(morpholinosulfonyl)aniline (0.11 g, 0.43 mmol). The reaction mixture was stirred at room temperature for 6 h, quenched with water and then treated with 10% HCl. The organic layer was collected, and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4, evaporated under a vacuum. The resulting yellow oil was purified by flash column chromatography (H/EtOAc 1:1) to afford 4f as a white solid (0.11 g, 50% yield), mp 198.2 °C. 1H NMR (CDCl3) δ 8.13 (s, 1H), 7.69 (s, 1H), 7.54 (d, 2H), 7.44 (d, 2H), 7.35 (d, 2H), 7.26 (d, 1H), 4.55 (m, 1H), 3.74 (d, 4H), 3.17 (d, 4H), 2.56 (s, 3H), 2.46 (s, 3H), 1.59 (d, 6H). 13C NMR (CDCl3) δ 163.2, 145.3, 140.8, 135.2, 134.7, 130.3, 130.1, 126.1, 121.8, 120.9, 117.9, 109.9, 67.7, 56.5, 46.7, 24.1, 22.9, 21.1. Anal. Calcd for C25H30N4O4S. ½ H2O: C, 61.07; H, 6.15; N, 11.39. Found: C, 61.26; H, 6.31; N, 11.29.
4.1.22. 1-Butyl-N-(4-methyl-3-(morpholinosulfonyl)phenyl)-3-(p-tolyl)-1H-pyrazole-4-carboxamide (4g)
To a solution of 7b (0.2 g, 0.77 mmol) in CH2Cl2 (10 mL) was added EDCI (0.18 g, 0.92 mmol) and HOBT (0.12 g, 0.92 mmol). The reaction mixture was stirred for 1 h. To the reaction was added 4-methyl-3-(morpholinosulfonyl)aniline (0.2 g, 0.77 mmol). The reaction mixture was stirred at room temperature for 12 h and quenched with water. The organic layer was collected, and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4, evaporated under a vacuum. The resulting yellow oil was purified by flash column chromatography (H/EtOAc 1:1) to afford 4g as a white solid (0.2 g, 53% yield), mp 177.4 °C. 1H NMR (CDCl3) δ 8.07 (s, 1H), 7.69 (s, 1H), 7.54 (d, 2H), 7.44 (d, 2H), 7.35 (d, 2H), 7.23 (s, 1H), 4.18 (m, 2H), 3.74 (d, 4H), 3.17 (d, 4H), 2.56 (s, 3H), 2.46 (s, 3H), 1.92 (t, 2H), 1.41 (t, 2H), 0.98 (t, 3H). 13C NMR (CDCl3) δ 162.7, 151.3, 140.9, 137.6, 134.8, 131.7, 131.5, 130.7, 125.2, 122.1, 120.9, 116.9, 67.7, 53.9, 46.8, 33.5, 22.8, 21.4, 21.1, 14.9. Anal. Calcd for C26H32N4O4S: C, 62.88; H, 6.49; N, 11.28. Found: C, 62.65; H, 6.27; N, 11.08.
4.1.23. N-(4-Methyl-3-(morpholinosulfonyl)phenyl)-1-(naphthalen-2-ylmethyl)-3-(p-tolyl)-1H-pyrazole-4-carboxamide (4h)
To a solution of 7c (0.2 g, 0.78 mmol) in CH2Cl2 (10 mL) was added EDCI (0.18 g, 0.94 mmol) and DMAP (0.11 g, 0.94 mmol). To the reaction was added 4-methyl-3-(morpholinosulfonyl)aniline (0.2 g, 0.78 mmol). The reaction mixture was stirred at room temperature for 6 h and then treated with 10% HCl. The organic layer was collected, and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4, evaporated under a vacuum. The resulting yellow oil was purified by flash column chromatography (H/EtOAc 1:1) to afford 4h as a white solid (0.2 g, 50% yield), mp 144.9 °C. 1H NMR (CDCl3) δ 8.13 (s, 1H), 7.85 (m, 4H), 7.68 (s, 1H), 7.53 (m, 4H), 7.45 (m, 2H), 7.35 (d, 2H), 7.20 (d, 2H), 5.51 (s, 2H), 3.72 (t, 4H), 3.15 (t, 4H), 2.51 (s, 3H), 2.46 (s, 3H). 13C NMR (CDCl3) δ 162.2, 151.2, 141.2, 137.4, 136.9, 135.6, 134.8, 133.7, 131.3, 130.8, 130.4, 129.4, 129.2, 129.1, 128.1, 127.1, 125.1, 122.1, 67.8, 58.4, 46.9, 22.9, 21.6. Anal. Calcd for C33H32N4O4S: C, 68.25; H, 5.55; N, 9.65. Found: C, 68.00; H, 6.04; N, 9.48.
4.1.24. 1-(Cyclopropylmethyl)-N-(4-methyl-3-(morpholinosulfonyl)phenyl)-3-(p-tolyl)-1H-pyrazole-4-carboxamide (4i)
To a solution of 7d (0.2 g, 0.78 mmol) in CH2Cl2 (10 mL) was added EDCI (0.18 g, 0.94 mmol) and DMAP (0.11 g, 0.94 mmol). To the reaction was added 4-methyl-3-(morpholinosulfonyl)aniline (0.2 g, 0.78 mmol). The reaction mixture was stirred at room temperature for 6 h and then treated with 10% HCl. The organic layer was collected, and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4, evaporated under a vacuum. The resulting yellow solid was purified by crystallization from EtOAc gave 4i as a white solid (0.3 g, 78% yield), mp 210.0 °C. 1H NMR (CDCl3) δ 8.23 (s, 1H), 7.71 (d, 1H), 7.54 (d, 2H), 7.46 (d, 2H), 7.35 (d, 2H), 7.21 (d, 1H), 4.05 (d, 2H), 3.74 (t, 4H), 3.17 (t, 4H), 2.51 (s, 3H), 1.36 (brs, 1H), 0.74 (q, 2H), 0.45 (t, 2H). 13C NMR (CDCl3) δ 162.5, 150.8, 141.0, 137.6, 137.1, 131.3, 130.8, 125.0, 122.1, 67.8, 58.9, 46.9, 23.2, 21.9, 12.0, 5.6. Anal. Calcd for C26H30N4O4S. ¼ H2O: C, 61.98; H, 6.00; N, 11.02. Found: C, 61.63; H, 5.82; N, 10.84.
4.1.25. 1-(3-Methoxybenzyl)-N-(4-methyl-3-(morpholinosulfonyl)phenyl)-3-(p-tolyl)-1H-pyrazole-4-carboxamide (4j, DY268)
To a solution of 7e (0.1 g, 0.31 mmol) in CH2Cl2 (8 mL) was added EDCI (0.071 g, 0.37 mmol) and DMAP (0.045 g, 0.37 mmol). To the reaction was added 4-methyl-3-(morpholinosulfonyl)aniline (0.08 g, 0.31 mmol). The reaction mixture was stirred at room temperature for 6 h and then treated with 10% HCl. The organic layer was collected, and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4, evaporated under a vacuum. The resulting yellow oil was purified by flash column chromatography (H/EtOAc 1:1) to afford 4j as a white solid (0.08 g, 47% yield), mp 183.3 °C. 1H NMR (CDCl3) δ 8.01 (s, 1H), 7.69 (s, 1H), 7.55 (d, 2H), 7.44 (d, 2H), 7.34 (m, 3H), 7.22 (d, 2H), 6.93 (d, 1H), 6.87 (s, 1H), 5.32 (s, 2H), 3.85 (s, 3H), 3.74 (t, 4H), 3.17 (t, 4H), 2.56 (s, 3H), 2.46 (s, 3H). 13C NMR (CDCl3) δ 162.6, 160.0, 141.4, 137.1, 135.5, 134.8, 131.5, 131.3, 130.8, 125.1, 122.1, 115.5, 115.4, 67.7, 58.0, 56.7, 46.8, 22.8, 21.6. Anal. Calcd for C30H32N4O5S: C, 64.27; H, 5.75; N, 9.99. Found: C, 64.01; H, 5.77; N, 9.63.
4.1.26. N-(4-methyl-3-(morpholinosulfonyl)phenyl)-3-(p-tolyl)-1-(3-(trifluoromethyl)benzyl)-1H-pyrazole-4-carboxamide (4k)
To a solution of 7f (0.11 g, 0.34 mmol) in CH2Cl2 (5 mL) was added EDCI (0.078 g, 0.41 mmol) and DMAP (0.05 g, 0.41 mmol). To the reaction was added 4-methyl-3-(morpholinosulfonyl)aniline (0.087 g, 0.34 mmol). The reaction mixture was stirred at room temperature for 6 h and then treated with 10% HCl. The organic layer was collected and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4, evaporated under a vacuum. The resulting yellow solid was purified by crystallization from EtOAc gave 4k as a white solid (0.1 g, 56% yield), mp 179.3 °C. 1H NMR (CDCl3) δ 8.07 (s, 1H), 7.68 (s, 1H), 7.59 (s, 1H), 7.51 (t, 4H), 7.45 (t, 2H), 7.43 (d, 2H), 7.21 (d, 2H), 5.41 (s, 2H), 3.72 (t, 4H), 3.15 (t, 4H), 2.51 (s, 3H), 2.46 (s, 3H). 13C NMR (CDCl3) δ 162.4, 151.6, 141.4, 137.3, 135.7, 134.8, 134.6, 132.8, 131.3, 130.8, 126.2, 125.1, 122.2, 118.4, 67.7, 57.4, 46.8, 22.8, 21.6. Anal. Calcd for C30H29F3N4O4S: C, 60.19; H, 4.88; N, 9.36, F, 9.52. Found: C, 60.45; H, 4.70; N, 9.15, F, 9.20.
4.1.27. 1-(4-Hydroxybutyl)-N-(4-methyl-3-(morpholinosulfonyl)phenyl)-3-(p-tolyl)-1H-pyrazole-4-carboxamide (4l)
To a solution of 7g (0.09 g, 0.34 mmol) in CH2Cl2 (5 mL) was added EDCI (0.078 g, 0.41 mmol) and DMAP (0.05 g, 0.41 mmol). To the reaction was added 4-methyl-3-(morpholinosulfonyl)aniline (0.087 g, 0.34 mmol). The reaction mixture was stirred at room temperature for 6 h and then treated with 10% HCl. The organic layer was collected, and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4, evaporated under a vacuum. The resulting yellow oil was purified by flash column chromatography (CHCl3/MeOH 95:5) to afford 4l as a white solid (0.05 g, 27% yield), mp 57.8 °C. 1H NMR (CDCl3) δ 8.01 (s, 1H), 7.68 (s, 1H), 7.50 (d, 2H), 7.49 (t, 1H), 7.15 (t, 4H), 4.21 (d, 2H), 4.20 (d, 2H), 3.72 (t, 4H), 3.11 (t, 4H), 2.51 (s, 3H), 2.46 (s, 3H), 1.94 (t, 2H), 1.26 (t, 2H). 13C NMR (CDCl3) δ 162.2, 154.9, 141.3, 139.8, 137.6, 135.2, 134.8, 131.8, 131.3, 130.6, 129.9, 125.2, 122.3, 67.7, 63.3, 53.8, 46.8, 30.7, 27.2, 21.6. Anal. Calcd for C26H32N4O4S: C, 60.71; H, 4.74; N, 12.21. Found: C, 60.57; H, 4.2; N, 12.26.
4.1.28. (Z)-Ethyl 3-ethoxy-2-(4-methylbenzoyl)acrylate (5a)
Ethyl 3-oxo-3-(p-tolyl) propanoate (1.0 g, 4.85 mmol) and triethylorthoformate (1.15 g, 7.76 mmol) were heated to reflux and stirred for 30 min. Acetic anhydride (1.5 g, 14.55 mmol) was then added, and refluxing continued for 12 h. Then the mixture was cooled and diluted with EtOAc, and water was added to the solution, which was then stirred for 10 min to decompose excess triethylorthoformate. The aqueous layer was extracted with EtOAc. The organic layers were washed with water and brine, dried over Na2SO4 and concentrated in vacuo to yield 1.21 g (94%) of the title compound, which was used without further purification. 1H NMR (CDCl3) δ 7.82 (d, 2H), 7.69 (s, 1H), 7.27 (d, 2H), 4.17 (q, 2H), 4.11 (q, 2H), 2.40 (s, 3H), 1.28 (t, 3H), 1.17 (t, 3H). 13C NMR (CDCl3) δ 192.9, 162.7, 145.5, 136.2, 130.8, 130.5, 130.3, 113.7, 73.1, 61.9, 23.1, 16.6, 15.6.
4.1.29. (Z)-Ethyl 2-(ethoxymethylene)-4-methyl-3-oxopentanoate (5b)
Ethyl isobutyryl acetate (2.0 g, 12.64 mmol) and triethylorthoformate (3.0 g, 20.22 mmol) were heated to reflux for 30 min. Acetic anhydride (3.8 g, 37.92 mmol) was then added, and refluxing continued for about 12 h. The reaction mixture was cooled and diluted with EtOAc, and water was added to the solution, which was then stirred for 10 min to decompose excess triethylorthoformate. The aqueous layer was extracted with EtOAc. The organic layers were washed with water and brine, dried over Na2SO4 and concentrated in vacuo to yield 2.49 g (92%) of the title compound, which was used without further purification. 1H NMR (CDCl3) δ 7.52 (s, 1H), 4.29 (q, 2H), 4.19 (q, 2H), 3.14 (m, 1H), 1.37 (t, 3H), 1.32 (t, 3H), 1.16 (s, 3H), 1.11 (s, 3H). 13C NMR (CDCl3) δ 203.4, 165.8, 163.1, 114.7, 73.4, 62.2, 42.1, 20.2, 19.3, 16.6, 15.6.
4.1.30. (Z)-Ethyl 3-ethoxy-2-(3-methoxybenzoyl)acrylate (5c)
Ethyl (3-methoxybenzoyl) acetate (1.0 g, 4.5 mmol) and triethylorthoformate (1.07 g, 7.2 mmol) were heated to reflux for 30 min. Acetic anhydride (1.37 g, 13.5 mmol) was then added, and refluxing continued for about 12 h. The reaction mixture was cooled and diluted with EtOAc, and water was added to the solution, which was then stirred for 10 min to decompose excess triethylorthoformate. The aqueous layer was extracted with EtOAc. The organic layers were washed with water and brine, dried over Na2SO4 and concentrated in vacuo to yield 1.1 g (88%) of the title compound, which was used without further purification. 1H NMR (CDCl3) δ 7.68 (s, 1H), 7.47 (t, 2H), 7.35 (t, 1H), 7.11 (t, 1H), 4.17 (q, 2H), 4.11 (q, 2H), 1.16 (t, 3H), 1.06 (t, 3H). 13C NMR (CDCl3) δ 193.3, 167.1, 162.9, 161.2, 140.1, 130.7, 130.5, 123.7, 121.4, 114.1, 73.2, 62.0, 56.8, 16.6, 15.6.
4.1.31. (Z)-Ethyl 3-ethoxy-2-(3-(trifluoromethyl)benzoyl)acrylate (5d)
Ethyl (4-trifluoromethylbenzoyl) acetate (1.0 g, 3.84 mmol) and triethylorthoformate (0.91 g, 6.15 mmol) were heated to reflux for 30 min. Acetic anhydride (1.18 g, 11.53 mmol) was then added, and refluxing continued for about 12 h. The reaction mixture was cooled and diluted with EtOAc, and water was added to the solution, which was then stirred for 10 min to decompose excess triethylorthoformate. The aqueous layer was extracted with EtOAc. The organic layers were washed with water and brine, dried over Na2SO4 and concentrated in vacuo to yield 1.08 g (89%) of the title compound, which was used without further purification. 1H NMR (CDCl3) δ 8.06 (d, 1H), 7.97 (d, 1H), 7.87 (s, 1H), 7.74 (d, 2H), 4.16 (q, 2H), 4.11 (q, 2H), 1.27 (t, 6H). 13C NMR (CDCl3) δ 193.2, 164.1, 130.7, 130.3, 127.8, 127.3, 126.8, 114.1, 73.6, 62.1, 16.6, 15.5.
4.1.32. (Z)-Methyl 2-(ethoxymethylene)-5-methoxy-3-oxopentanoate (5e)
Methyl 5-methoxy-3-oxovalerate (2.0 g, 12.48 mmol) and triethylorthoformate (3.0 g, 19.96 mmol) were heated to reflux for 30 min. Acetic anhydride (3.82 g, 37.44 mmol) was then added, and refluxing continued for about 6 h. The reaction mixture was cooled and diluted with EtOAc, and water was added to the solution, which was then stirred for 10 min to decompose excess triethylorthoformate. The aqueous layer was extracted with EtOAc. The organic layers were washed with water and brine, dried over Na2SO4 and concentrated in vacuo to yield 2.27 g (84%) of the title compound, which was used without further purification. 1H NMR (CDCl3) δ 7.90 (s, 1H), 4.25 (q, 2H), 3.66 (t, 2H), 3.35 (s, 3H), 3.23 (t, 2H), 1.31 (t, 3H). 13C NMR (CDCl3) δ 200.5, 185.9, 165.7, 108.1, 67.7, 67.6, 60.3, 51.4, 38.8, 15.2.
4.1.33. Ethyl 3-(p-tolyl)-1H-pyrazole-4-carboxylate (6a)
A solution of hydrazine monohydrate (0.2 ml, 4.2 mmol) in dry ethanol (1 mL) was added dropwise at 0 °C to a solution of (Z)-Ethyl 3-ethoxy-2-(4-methylbenzoyl)acrylate (5a) (1.0 g, 3.81 mmol) in ethanol (5 mL). The reaction mixture was stirred at room temperature for 12 h and the resulting solid was filtered, washed with water and cold ethanol, and dried to afford 0.77 g (88%) of 6a as a white solid, mp 83.5 °C. 1H NMR (CDCl3) δ 8.04 (s, 1H), 7.59 (d, 2H), 7.27 (d, 2H), 4.27 (q, 2H), 2.40 (s, 3H), 1.31 (t, 3H). 13C NMR (CDCl3) δ 164.8, 149.3, 142.8, 140.8, 130.4, 130.3, 127.9, 113.1, 61.6, 22.8, 15.7.
4.1.34. Ethyl 3-isopropyl-1H-pyrazole-4-carboxylate (6b)
A solution of hydrazine monohydrate (0.67 g, 13.40 mmol) in dry ethanol (1 mL) was added dropwise at 0 °C to a solution of (Z)-ethyl 2-(ethoxymethylene)-4-methyl-3-oxopentanoate (5b) (2.49 g, 11.62 mmol) in ethanol (10 mL). The reaction mixture was stirred at room temperature for 12 h and the solvent was removed, solidified by pet. ether to afford 1.28 g (61%) of 6-2 as a white solid, mp 71.5 °C. 1H NMR (CDCl3) δ 11.46 (brs, 1H), 7.96 (s, 1H), 4.32 (q, 2H), 3.69 (m, 1H), 1.36 (s, 6H), 1.33 (t, 3H). 13C NMR (CDCl3) δ 165.1, 142.1, 111.6, 61.3, 27.1, 22.9, 15.8.
4.1.35. Ethyl 3-(3-methoxyphenyl)-1H-pyrazole-4-carboxylate (6c)
A solution of hydrazine monohydrate (0.22 g, 4.35 mmol) in dry ethanol (1 mL) was added dropwise at 0 °C to a solution of (Z)-ethyl 3-ethoxy-2-(3-methoxybenzoyl)acrylate (5c) (1.1 g, 3.95 mmol) in ethanol (5 mL). The reaction mixture was stirred at room temperature for 12 h, and the solvent was removed. The crude was resolved in EtOAc, washed with water, and concentrated to afford 0.97 g (98%) of 6c as a red oil, which was used without further purification. 1H NMR (CDCl3) δ 8.05 (s, 1H), 7.36 (t, 1H), 7.29 (t, 3H), 6.98 (d, 1H), 4.28 (q, 2H), 3.81 (s, 3H), 1.30 (t, 3H). 13C NMR (CDCl3) δ 164.6, 160.7, 141.6, 132.2, 130.7, 122.8, 116.5, 116.2, 112.8, 61.6, 56.8, 15.7.
4.1.36. Ethyl 3-(4-(trifluoromethyl)phenyl)-1H-pyrazole-4-carboxylate (6d)
A solution of hydrazine monohydrate (0.22 g, 4.35 mmol) in dry ethanol (1 mL) was added dropwise at 0 °C to a solution of (Z)-ethyl 3-ethoxy-2-(3-methoxybenzoyl)acrylate (5d) (1.08 g, 3.41 mmol) in ethanol (5 mL). The reaction mixture was stirred at room temperature for 12 h and the solvent was removed. The crude was resolved in EtOAc, washed with water, and concentrated to afford a red oil which was solidified by EtOAc to give 0.79 g (82%) of 6d as a pale red solid, mp 177.2 °C. 1H NMR (MeOD) δ 8.12 (s, 1H), 7.84 (d, 2H), 7.67 (d, 2H), 4.28 (q, 2H), 1.29 (t, 3H). 13C NMR (MeOD) δ 164.7, 136.1, 131.2, 131.1, 130.7, 127.4, 126.7, 126.2, 125.7, 112.6, 61.4, 14.4.
4.1.37. Methyl 3-(2-methoxyethyl)-1H-pyrazole-4-carboxylate (6e)
A solution of hydrazine monohydrate (0.57 g, 11.54 mmol) in dry ethanol (1 mL) was added dropwise at 0 °C to a solution of (Z)-methyl 2-(ethoxymethylene)-5-methoxy-3-oxopentanoate (5e) (2.27 g, 10.49 mmol) in ethanol (7 mL). The reaction mixture was stirred at room temperature for 12 h, and the solvent was removed. The crude was resolved in EtOAc, washed with water, and concentrated to afford a red oil which was solidified by EtOAc to give 1.72 g (89%) of 6e as a yellow oil. 1H NMR (CDCl3) δ 7.90 (s, 1H), 4.24 (q, 2H), 3.76 (s, 1H), 3.65 (t, 2H), 3.33 (s, 3H), 1.28 (t, 3H). 13C NMR (CDCl3) δ 165.2, 148.3, 141.5, 122.4, 72.1, 60.3, 52.5, 27.3.
4.1.38. 1-Isopropyl-3-(p-tolyl)-1H-pyrazole-4-carboxylic acid (7a)
A solution of 6a (0.5 g, 2.17 mmol), 1-Bromo-2-methyl-propane (0.55 g, 3.25 mmol) and cesium carbonate (2.12 g, 6.51 mmol) in acetonitrile (10 mL) was stirred at room temperature for 16 h, and the reaction was stopped. Water (10 mL) was added into the reaction mixture, and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with brine (3 × 10 mL), dried over Na2SO4, filtered and concentrated to give crude. 1H NMR (CDCl3) δ 8.00 (s, 1H), 7.67 (d, 2H), 7.22 (d, 2H), 4.54 (m, 1H), 4.25 (q, 2H), 2.38 (s, 3H), 1.56 (d, 6H), 1.31 (t, 3H). The crude residue in ethanol (12 mL) was treated with 1N aqueous potassium hydroxide (7.5 mL) at 75°C for 2.5 h. The reaction was cooled, acidified with 5N HCl and the ethanol removed in vacuo. The solids were filtered, washed with water, hexanes and dried in vacuo to afford 0.31 g (58%) of 7-1 as a beige powder, mp 129.0 °C. 1H NMR (CDCl3) δ 8.06 (s, 1H), 7.66 (d, 2H), 7.21 (d, 2H), 4.53 (m, 1H), 2.38 (s, 3H), 1.56 (d, 6H). 13C NMR (CDCl3) δ 169.8, 154.6, 139.7, 134.6, 131.0, 130.6, 130.1, 55.9, 24.1, 24.0, 22.9.
4.1.39. 1-Butyl-3-(p-tolyl)-1H-pyrazole-4-carboxylic acid (7b)
A solution of 6a (0.5 g, 2.17 mmol), 1-Bromobutane (0.29 g, 3.25 mmol) and cesium carbonate (2.12 g, 6.51 mmol) in acetonitrile (10 mL) was stirred at room temperature for 16 h, and the reaction was stopped. Water (10 mL) was added into the reaction mixture and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with brine (3 × 10 mL), dried over Na2SO4, filtered and concentrated. The crude residue in ethanol (12 mL) was treated with 1N aqueous potassium hydroxide (7.5 mL) at 75°C for 3.5 h. The reaction was cooled, acidified with 5N HCl and the ethanol removed in vacuo. Solvent was removed and the resulting yellow oil was purified by flash column chromatography (CHCl3/MeOH 9:1) to afford 7-2 as pale-yellow oil (0.5 g, 50% yield). 1H NMR (CDCl3) δ 8.06 (s, 1H), 7.66 (d, 2H), 7.21 (d, 2H), 4.53 (m, 1H), 2.38 (s, 3H), 1.56 (d, 6H). 13C NMR (CDCl3) δ 169.8, 154.6, 139.7, 134.6, 131.0, 130.6, 130.1, 55.9, 24.1, 24.0, 22.9.
4.1.40. 1-(Naphthalen-2-ylmethyl)-3-(p-tolyl)-1H-pyrazole-4-carboxylic acid (7c)
A solution of 6a (0.18 g, 0.78 mmol), 2-(bromomethyl)naphthalene (0.26 g, 1.17 mmol) and cesium carbonate (0.76 g, 2.34 mmol) in acetonitrile (5 ml) was stirred at room temperature for 16 h, and the reaction was stopped. Water (10 mL) was added into the reaction mixture and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with brine (3 × 10 mL), dried over Na2SO4, filtered and concentrated. The crude residue in ethanol (10 mL) was treated with 1N aqueous potassium hydroxide (7.5 mL) at 75°C for 3.5 h. The reaction was cooled, acidified with 5N HCl and the ethanol removed in vacuo to afford 7c as a pale-yellow solid (0.2 g, 77% yield), mp 151.2 °C. 1H NMR (CDCl3) δ 8.06 (s, 1H), 7.75 (m, 3H), 7.71 (d, 2H), 7.53 (d, 2H), 7.48 (m, 1H), 7.20 (m, 3H), 5.45 (s, 2H), 2.40 (s, 3H). 13C NMR (CDCl3) δ 169.3, 155.2, 139.8, 134.7, 131.3, 130.6, 130.5, 130.4, 129.9, 129.7, 129.6, 129.3, 129.2, 129.1, 128.9, 128.0, 59.9, 22.8.
4.1.41. 1-(Cyclopropylmethyl)-3-(p-tolyl)-1H-pyrazole-4-carboxylic acid (7d)
A solution of 6a (0.5 g, 2.17 mmol), bromomethylcyclopropane (0.43 g, 3.25 mmol) and cesium carbonate (2.12 g, 6.51 mmol) in acetonitrile (10 mL) was stirred at room temperature for 16 h. Water (10 mL) was added into the reaction mixture and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with brine (3 × 10 mL), dried over Na2SO4, filtered and concentrated to give the crude. 1H NMR (CDCl3) δ 8.09 (s, 1H), 7.68 (d, 2H), 7.26 (d, 2H), 4.24 (q, 2H), 4.02 (t, 2H), 2.42 (s, 3H), 1.33 (brs, 1H), 1.22 (t, 3H), 0.72 (t, 2H), 0.48 (t, 2H). The crude residue in ethanol (10 mL) was treated with 1N aqueous potassium hydroxide (7.5 mL) at 75°C for 3.5 h. The reaction was cooled, acidified with 5N HCl and the ethanol removed in vacuo. The solids were filtered, washed with water hexanes and dried over in vacuo to afford 7d as a pale-yellow solid (0.3 g, 55% yield), mp 142.6 °C. 1H NMR (CDCl3) δ 8.18 (s, 1H), 7.69 (d, 2H), 7.23 (d, 2H), 4.04 (d, 2H), 2.44 (s, 3H), 1.36 (brs, 1H), 0.74 (t, 2H), 0.44 (t, 2H). 13C NMR (CDCl3) δ 169.7, 154.9, 143.7, 139.7, 136.6, 130.6, 130.5, 130.1, 112.0, 58.7, 22.8, 12.0, 5.5, 5.3.
4.1.42. 1-(3-Methoxybenzyl)-3-(p-tolyl)-1H-pyrazole-4-carboxylic acid (7e)
A solution of 6a (0.1 g, 0.43 mmol), 3-methoxybenzyl bromide (0.13 g, 0.65 mmol) and cesium carbonate (0.42 g, 1.29 mmol) in acetonitrile (5 mL) was stirred at room temperature for 16 h. Water (10 mL) was added to the reaction mixture, and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with brine (3 × 10 mL), dried over Na2SO4, filtered and concentrated to give the crude. 1H NMR (CDCl3) δ 8.05 (s, 1H), 7.90 (d, 2H), 7.24 (m, 3H), 6.89 (d, 2H). 6.84 (s, 1H), 5.29 (s, 2H), 4.21 (q, 2H), 3.75 (s, 3H), 2.41 (s, 3H), 1.21 (t, 3H). The crude residue in ethanol (5 mL) was treated with 1N aqueous potassium hydroxide (3.5 mL) at 75°C for 3.5 h. The reaction was cooled, acidified with 5N HCl and the solvent removed in vacuo. The solids were filtered, washed with hexanes and dried over in vacuo to afford 7e as a white solid (0.1 g, 77% yield), mp 169.6 °C. 1H NMR (CDCl3) δ 7.95 (s, 1H), 7.68 (d, 2H), 7.31 (t, 1H), 7.22 (t, 2H), 6.90 (d, 2H), 6.84 (s, 1H), 5.29 (b, 2H), 3.73 (s, 3H), 2.41 (s, 3H). 13C NMR (CDCl3) δ 168.9, 155.2, 139.8, 137.3, 131.5, 131.2, 130.6, 130.5, 130.1, 121.8, 115.4, 115.3, 57.8, 56.7, 22.7.
4.1.43. 3-(p-Tolyl)-1-(3-(trifluoromethyl)benzyl)-1H-pyrazole-4-carboxylic acid (7f)
A solution of 6a (0.1 g, 0.43 mmol), 3-(trifluoromethyl)benzyl bromide (0.15 g, 0.65 mmol) and cesium carbonate (0.42 g, 1.29 mmol) in acetonitrile (5 mL) was stirred at room temperature for 6 h, and the reaction was stopped. Water (10 mL) was added to the reaction mixture, and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with brine (3 × 10 mL), dried over Na2SO4, filtered and concentrated to give the crude. 1H NMR (CDCl3) δ 7.97 (s, 1H), 7.68 (d, 2H), 7.58 (s, 1H), 7.51 (m, 3H). 7.23 (d, 2H), 5.39 (s, 2H), 4.26 (q, 2H), 2.41 (s, 3H), 1.26 (t, 3H). The crude residue in ethanol (5 mL) was treated with 1N aqueous potassium hydroxide (3.5 mL) at 75°C for 5.5 h. The reaction was cooled, acidified with 5N HCl and the ethanol removed in vacuo. The solids were filtered, washed with water hexanes and dried over in vacuo to afford 7f as a white solid (0.11 g, 77% yield), mp 149.9 °C. 1H NMR (CDCl3) δ 8.01 (s, 1H), 7.67 (d, 2H), 7.52 (s, 1H), 7.49 (d, 2H), 7.21 (m, 3H), 5.39 (s, 2H), 2.41 (s, 3H). 13C NMR (CDCl3) δ 169.2, 156.4, 143.9, 140.1, 137.65 132.7, 131.1, 130.7, 130.6, 130.1, 126.8, 126.2, 119.5, 118.5, 57.2, 22.7.
4.1.44. 1-(4-Hydroxybutyl)-3-(p-tolyl)-1H-pyrazole-4-carboxylic acid (7g)
A solution of 6a (0.1 g, 0.43 mmol), 4-bromo-1-butanol (0.10 g, 0.65 mmol) and cesium carbonate (0.42 g, 1.29 mmol) in acetonitrile (5 mL) was stirred at room temperature for 6 h, and the reaction was stopped. Water (10 mL) was added to the reaction mixture, and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with brine (3 × 10 mL), dried over Na2SO4, filtered and concentrated to give the crude. 1H NMR (CDCl3) δ 7.98 (s, 1H), 7.65 (d, 2H), 7.20 (d, 2H), 4.24 (q, 2H). 4.09 (t, 2H), 3.45 (t, 2H), 2.41 (s, 3H), 1.67 (t, 2H), 1.57 (t, 2H), 1.42 (t, 3H). The crude residue in ethanol (5 mL) was treated with 1N aqueous potassium hydroxide (3.5 mL) at 75°C for 5.5 h. The reaction was cooled; the ethanol was removed in vacuo, acidified with 5N HCl, and dried in vacuo to afford 7g as an oil (0.09 g, 82% yield). 1H NMR (CDCl3) δ 7.99 (s, 1H), 7.65 (d, 2H), 7.19 (d, 2H), 4.14 (q, 2H), 3.62 (t, 2H), 2.40 (s, 3H), 1.96 (t, 2H), 1.55 (t, 2H). 13C NMR (CDCl3) δ 168.8, 154.9, 139.7, 137.3, 131.1, 130.6, 130.1, 130.7, 112.0, 63.1, 53.7, 30.5, 27.9, 22.8.
4.1.45. 3-Isopropyl-1-(3-methoxybenzyl)-1H-pyrazole-4-carboxylic acid (7h)
A solution of 6b (0.5 g, 2.74 mmol), 3-methoxybenzyl bromide (0.83 g, 4.11 mmol) and cesium carbonate (2.68 g, 8.22 mmol) in acetonitrile (10 mL) was stirred at room temperature for 16 h and the reaction was stopped. Water (10 mL) was added into the reaction mixture and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with brine (3 × 10 mL), dried over Na2SO4, filtered and concentrated to give crude. 1H NMR (CDCl3) δ 7.73 (s, 1H), 7.29 (d, 1H), 6.87 (t, 1H), 6.83 (d, 1H), 6.77 (s, 1H), 5.22 (s, 2H), 4.26 (q, 2H), 3.82 (s, 3H), 3.54 (m, 1H), 1.35 (d, 6H), 1.26 (t, 3H). The crude residue in ethanol (12 mL) was treated with 1N aqueous potassium hydroxide (7.5 mL) at 75°C for 4.5 h. The reaction was cooled; the ethanol was removed in vacuo, acidified with 5N HCl, and dried in vacuo to afford 7h as an white solid (0.59 g, 78% yield), mp 111.9 °C. 1H NMR (CDCl3) δ 7.79 (s, 1H), 7.29 (t, 1H), 6.88 (d, 1H), 6.84 (d, 1H), 6.78 (s, 1H), 5.23 (s, 2H), 3.79 (s, 3H), 3.55 (m, 1H), 1.32 (d, 6H). 13C NMR (CDCl3) δ 169.6, 163.0, 161.5, 138.0, 136.4, 131.4, 121.7, 115.3, 115.1, 111.8, 57.5, 56.7, 28.0, 23.3.
4.1.46. 3-Isopropyl-1-(3-methoxybenzyl)-1H-pyrazole-4-carboxylic acid (7i)
A solution of 6c (0.3 g, 1.22 mmol), 2-iododpropane (0.31 g, 1.83 mmol) and cesium carbonate (1.19 g, 3.66 mmol) in acetonitrile (5 mL) was stirred at room temperature for 16 h, and the reaction was stopped. Water (10 mL) was added to the reaction mixture, and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with brine (3 × 10 mL), dried over Na2SO4, filtered and concentrated to give crude. 1H NMR (CDCl3) δ 8.01 (s, 1H), 7.39 (d, 1H), 7.32 (t, 2H), 6.92 (d, 1H), 4.26 (q, 2H), 4.14 (m, 1H), 3.85 (s, 3H), 1.43 (d, 6H), 1.17 (t, 3H). 13C NMR (CDCl3) δ 164.6, 161.6, 142.5, 133.5, 130.8, 123.5, 117.0, 116.1, 61.4, 56.7, 55.8, 51.9, 24.2, 15.7. The crude residue in ethanol (12 mL) was treated with 1N aqueous potassium hydroxide (7.5 mL) at 75°C for 12 h. The reaction was cooled; the ethanol was removed in vacuo, acidified with 5N HCl, and dried in vacuo to afford 7i as a yellow oil (0.25 g, 78% yield). 1H NMR (CDCl3) δ 8.08 (s, 1H), 7.39 (d, 2H), 7.32 (t, 1H), 6.95 (d, 1H), 4.51 (m, 1H), 3.85 (s, 3H), 1.58 (d, 6H). 13C NMR (CDCl3) δ 169.6, 160.6, 154.3, 143.5, 134.9, 134.7, 130.9, 123.3, 116.9, 115.9, 56.7, 55.9, 24.1, 24.0.
4.1.47. 1-(3-Methoxybenzyl)-3-(3-methoxyphenyl)-1H-pyrazole-4-carboxylic acid (7j)
A solution of 6c (0.3 g, 1.22 mmol), benzyl-3-methoxybenzene (0.37 g, 1.83 mmol) and cesium carbonate (1.19 g, 3.66 mmol) in acetonitrile (5 mL) was stirred at room temperature for 6 h, and the reaction was stopped. Water (10 mL) was added to the reaction mixture and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with brine (3 × 10 mL), dried over Na2SO4, filtered and concentrated to give crude. 1H NMR (CDCl3) δ 7.92 (s, 1H), 7.39 (s, 1H), 7.32 (t, 3H), 6.94 (d, 3H), 6.84 (s, 1H), 5.31 (s, 2H), 4.24 (q, 2H), 3.85 (s, 3H), 3.76 (s, 3H), 1.86 (t, 3H). The crude residue in ethanol (12 mL) was treated with 1N aqueous potassium hydroxide (7.5 mL) at 75°C for 12 h. The reaction was cooled; the ethanol was removed in vacuo, acidified with 5N HCl, and dried in vacuo to afford 7j as a yellow oil (0.27 g, 66% yield). 1H NMR (CDCl3) δ 7.96 (s, 1H), 7.38 (d, 2H), 7.32 (t, 3H), 6.92 (d, 2H), 6.84 (s, 1H), 5.29 (s, 2H), 3.83 (s, 3H), 3.73 (s, 3H). 13C NMR (CDCl3) δ 168.7, 160.6, 161.1, 143.5, 137.7, 137.4, 134.6, 131.5, 130.4, 123.2, 121.8, 116.2, 115.9, 115.4, 115.3, 57.9, 56.7.
4.1.48. 1-Isopropyl-3-(4-(trifluoromethyl)phenyl)-1H-pyrazole-4-carboxylic acid (7k)
A solution of 6d (0.3 g, 1.06 mmol), 2-iodopropane (0.36 g, 1.83 mmol) and cesium carbonate (1.19 g, 3.66 mmol) in acetonitrile (5 mL) was stirred at room temperature for 6 h, and the reaction was stopped. Water (10 mL) was added to the reaction mixture, and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with brine (3 × 10 mL), dried over Na2SO4, filtered and concentrated to give crude. 1H NMR (CDCl3) δ 8.00 (s, 1H), 7.92 (s, 1H), 7.93 (d, 2H), 7.64 (d, 2H), 4.25 (q, 2H), 4.10 (m, 1H), 1.61 (d, 6H), 1.28 (t, 3H). The crude residue in ethanol (10 mL) was treated with 2N aqueous potassium hydroxide (5.5 mL) at 75°C for 12 h. The reaction was cooled; the ethanol was removed in vacuo. The residue was taken up with EtOAc and water and acidified with 5N HCl to pH 3. Organic phases were dried over Na2SO4, filtered and concentrated in vacuo to afford 7k as a yellow solid (0.21 g, 67% yield), mp 161.1 °C. 1H NMR (CDCl3) δ 10.17 (brs, 1H), 8.10 (s, 1H), 7.93 (d, 2H), 7.62 (d, 2H), 4.51 (brs, 1H), 1.52 (s, 6H). 13C NMR (CDCl3) δ 168.1, 151.5, 143.9, 142.1, 135.9, 133.5, 132.7, 130.3, 129.6, 125.3, 54.7, 22.4, 22.3.
4.1.49. 1-(3-Methoxybenzyl)-3-(4-(trifluoromethyl)phenyl)-1H-pyrazole-4-carboxylic acid (7l)
A solution of 6d (0.2 g, 0.7 mmol), 3-methoxy benzyl bromide (0.21 g, 1.05 mmol) and cesium carbonate (0.68 g, 2.1 mmol) in acetonitrile (5 mL) was stirred at room temperature for 12 h. Water (10 mL) was added to the reaction mixture, and the aqueous layer was extracted with ethyl acetate (3×15 mL). The combined organic layers were washed with brine (3 × 10 mL), dried over Na2SO4, filtered and concentrated to give crude. 1H NMR (CDCl3) δ 7.96 (t, 2H), 7.66 (d, 2H), 7.29 (d, 2H), 6.88 (d, 2H), 6.83 (s, 1H), 5.28 (s, 2H), 4.23 (q, 2H), 3.79 (s, 3H), 1.26 (t, 3H). The crude residue in ethanol (5 mL) was treated with 2N aqueous potassium hydroxide (4 mL) at 75°C for 12 h. The reaction was cooled; the ethanol was removed in vacuo. The residue was taken up with EtOAc/water and acidified with 5N HCl to pH 3. Organic phases were dried over Na2SO4, filtered and concentrated in vacuo to afford 7a as a yellow solid (0.21 g, 80% yield), mp 141.7 °C. 1H NMR (CDCl3) δ 7.92 (s, 1H), 7.87 (d, 2H), 7.23 (t, 1H), 6.84 (d, 2H), 6.77 (s, 1H), 5.24 (s, 2H), 3.74 (s, 3H). 13C NMR (CDCl3) δ 166.1, 160.8, 159.6, 142.4, 139.2, 135.9, 135.6, 130.0, 129.5, 129.1, 125.4, 119.8, 113.6, 113.0, 112.7, 64.8, 55.0.
4.1.50. 3-Isopropyl-1-(3-methoxybenzyl)-N-(4-methyl-3-(morpholinosulfonyl)phenyl)-1H-pyrazole-4-carboxamide (4m)
To a solution of 7h (0.12 g, 0.44 mmol) in CH2Cl2 (5 mL) was added EDCI (0.1 g, 0.53 mmol) and DMAP (0.07 g, 0.53 mmol). To the reaction was added 4-methyl-3-(morpholinosulfonyl)aniline (0.11 g, 0.44 mmol). The reaction mixture was stirred at room temperature for 6 h and then treated with 10% HCl. The organic layer was collected, and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4, evaporated under a vacuum. The resulting yellow oil was purified by flash column chromatography (H/EtOAc 1:1) to afford 4m as a white solid (0.18 g, 83% yield), mp 179.7 °C. 1H NMR (CDCl3) δ 8.10 (s, 1H), 7.79 (dd, 3H), 7.31 (m, 2H), 6.83 (t, 2H), 6.82 (s, 1H), 5.28 (s, 2H), 3.81 (s, 3H), 3.66 (t, 4H), 3.59 (m, 1H), 3.11 (t, 4H), 2.60 (s, 3H), 1.39 (d, 6H). 13C NMR (CDCl3) δ 163.2, 161.2, 160.0, 138.8, 138.5, 136.3, 134.5, 135.1, 131.8, 131.4, 126.1, 122.6, 122.7, 121.7, 115.2, 115.1, 67.6, 57.4, 56.7, 46.8, 28.4, 23.6, 21.5. Anal. Calcd for C26H32N4O5S: C, 60.92; H, 6.29; N, 10.93. Found: C, 60.45; H, 5.97; N, 10.51.
4.1.51. 1-Isopropyl-3-(3-methoxyphenyl)-N-(4-methyl-3-(morpholinosulfonyl)phenyl)-1H-pyrazole-4-carboxamide (4n)
To a solution of 7i (0.25 g, 0.96 mmol) in CH2Cl2 (5 mL) was added EDCI (0.22 g, 1.15 mmol) and DMAP (1.15 g, 1.15 mmol). To the reaction was added 4-methyl-3-(morpholinosulfonyl)aniline (0.25 g, 0.96 mmol). The reaction mixture was stirred at room temperature for 12 h and then treated with 10% HCl. The organic layer was collected, and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4, evaporated under a vacuum. The resulting yellow oil was purified by flash column chromatography (H/EtOAc 1:1) to afford 4n as a white solid (0.26 g, 55% yield), mp 158.0 °C. 1H NMR (CDCl3) δ 8.15 (s, 1H), 7.72 (s, 1H), 7.53 (s, 1H), 7.47 (d, 2H), 7.24 (d, 2H), 7.19 (s, 1H), 7.05 (m, 1H), 4.57 (m, 1H), 3.86 (s, 3H), 3.74 (t, 4H), 3.17 (t, 4H), 2.57 (s, 3H), 1.61 (s, 6H). 13C NMR (CDCl3) δ 163.2, 161.7, 150.6, 142.2, 137.7, 136.9, 134.8, 134.5, 132.9, 132.6, 131.7, 125.6, 123.6, 123.1, 122.0, 117.6, 67.7, 56.8, 56.0, 46.9, 24.2, 21.7. Anal. Calcd for C25H30N4O5S · ¼ H2O: C, 59.53; H, 5.92; N, 11.10. Found: C, 59.76; H, 5.67; N, 11.10.
4.1.52. 1-(3-Methoxybenzyl)-3-(3-methoxyphenyl)-N-(4-methyl-3-(morpholinosulfonyl)phenyl)-1H-pyrazole-4-carboxamide (4o)
To a solution of 7j (0.25 g, 0.73 mmol) in CH2Cl2 (5 mL) was added EDCI (0.17 g, 0.88 mmol) and DMAP (0.11 g, 0.88 mmol). To the reaction was added 4-methyl-3-(morpholinosulfonyl)aniline (0.19 g, 0.73 mmol). The reaction mixture was stirred at room temperature for 12 h and then treated with 10% HCl. The organic layer was collected, and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4, evaporated under a vacuum. The resulting yellow oil was solidified by MeOH to afford 4o as a white solid (0.31 g, 74% yield), mp 190.1 °C. 1H NMR (CDCl3) δ 8.04 (s, 1H), 7.72 (s, 1H), 7.54 (s, 1H), 7.48 (t, 2H), 7.34 (s, 1H), 7.28 (m, 3H), 7.20 (d, 1H), 6.94 (t, 1H), 6.88 (s, 1H), 3.82 (s, 3H), 3.80 (s, 3H), 3.74 (t, 4H), 3.17 (t, 4H), 2.57 (s, 3H). 13C NMR (CDCl3) δ 163.2, 161.6, 160.0, 151.4, 137.7, 137.4, 135.7, 134.9, 134.5, 131.7, 131.6, 125.0, 123.1, 122.1, 122.0, 116.9, 116.2, 115.5, 115.4, 67.8, 58.1, 56.9, 56.7, 46.9, 21.7. Anal. Calcd for C30H32N4O5S · ½ H2O: C, 61.51; H, 5.50; N, 9.56. Found: C, 61.50; H, 5.28; N, 9.39.
4.1.53. 1-Isopropyl-N-(4-methyl-3-(morpholinosulfonyl)phenyl)-3-(4-(trifluoromethyl)phenyl)-1H-pyrazole-4-carboxamide (4p)
To a solution of 7k (0.2 g, 0.67 mmol) in CH2Cl2 (5 mL) was added EDCI (0.15 g, 0.81 mmol) and DMAP (0.08 g, 0.81 mmol). To the reaction was added 4-methyl-3-(morpholinosulfonyl)aniline (0.17 g, 0.67 mmol). The reaction mixture was stirred at room temperature for 12 h, quenched with water and then treated with 10% HCl. The organic layer was collected, and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4, evaporated under a vacuum. The resulting yellow oil was purified by flash column chromatography (H/EtOAc 1:1) to afford 4p as a yellow solid (0.18 g, 51% yield), mp 192.2 °C. 1H NMR (CDCl3) δ 8.12 (s, 1H), 8.09 (s, 1H), 7.87 (d, 2H), 7.81 (d, 2H), 7.65 (d, 2H), 7.24 (d, 1H), 4.53 (m, 1H), 3.64 (t, 4H), 3.07 (t, 4H), 2.54 (s, 3H), 1.53 (s, 6H). 13C NMR (CDCl3) δ 161.5, 148.8, 138.6, 136.2, 134.9, 133.7, 133.3, 130.4, 129.3, 125.7, 119.4, 66.2, 66.1, 45.3, 45.2, 22.5, 20.1, 20.0. Anal. Calcd for C25H27F3N4O4S · ½ H2O: C, 55.03; H, 4.98; N, 10.44. Found: C, 55.23; H, 5.03; N, 10.26.
4.1.54. 1-(3-Methoxybenzyl)-N-(4-methyl-3-(morpholinosulfonyl)phenyl)-3-(4-(trifluoromethyl)phenyl)-1H-pyrazole-4-carboxamide (4q)
To a solution of 7l (0.06 g, 0.16 mmol) in CH2Cl2 (2 mL) was added EDCI (0.04 g, 0.019 mmol) and DMAP (0.02 g, 0.019 mmol). To the reaction was added 4-methyl-3-(morpholinosulfonyl)aniline (0.04 g, 0.16 mmol). The reaction mixture was stirred at room temperature for 12 h, quenched with water and then treated with 10% HCl. The organic layer was collected and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over Na2SO4, evaporated under a vacuum. The resulting yellow oil was purified by flash column chromatography (H/EtOAc 1:1) to afford 4q as a yellow solid (0.022 g, 61% yield), mp 208.2 °C. 1H NMR (CDCl3) δ 7.98 (s, 1H), 7.89 (d, 2H), 7.81 (s, 1H), 7.74 (d, 2H), 7.62 (d, 1H), 7.47 (s, 1H), 7.27 (t, 1H), 7.26 (t, 1H), 6.94 9m, 3H), 5.34 (s, 2H), 3.82 (s, 3H), 3.72 (t, 4H), 3.15 (t, 4H), 2.58 (s, 3H). 13C NMR (CDCl3) δ 161.5, 148.8, 138.6, 136.2, 134.9, 133.7, 133.3, 130.4, 129.3, 125.7, 119.4, 66.2, 66.1, 45.3, 45.2, 22.5, 20.1, 20.0. Anal. Calcd for C30H29F3N4O5S · ½ H2O: C, 57.77; H, 4.68; N, 8.98. Found: C, 58.08; H, 4.64; N, 8.64.
4.1.55. 1-Isopropyl-N-methyl-N-(4-methyl-3-(morpholinosulfonyl)phenyl)-3-(p-tolyl)-1H-pyrazole-4-carboxamide (8a)
A mixture of 4f (0.1 g, 0.21 mmol), NaH (60% dispersion in oil) (0.018 g, 0.46 mmol) and anhydrous DMF (10 mL) was stirred at room temperature for 30 min. The reaction mixture was cooled to 0 °C, and iodomethane (0.1 mL, 1.47 mmol) was added to it. The resulting mixture was stirred at room temperature under an atmosphere of N2 and monitored by TLC. The reaction was stopped, water (30 mL) was added to the reaction mixture, and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were washed with water and brine (3 × 10 mL), dried over Na2SO4, filtered and concentrated. The resulting yellow oil was purified by flash column chromatography (H/EtOAc 1:1) to afford 8a as a clear oil (0.07 g, 70% yield). 1H NMR (CDCl3) δ 7.34 (s, 1H), 7.15 (d, 2H), 7.09 (t, 2H), 6.94 (d, 2H), 6.87 (d, 1H), 4.26 (m, 1H), 3.67 (t, 4H), 3.27 (s, 3H), 3.05 (t, 4H), 2.55 (s, 3H), 2.41 (s, 3H), 1.31 (d, 6H). 13C NMR (CDCl3) δ 166.5, 144.0, 140.8, 136.8, 134.4, 131.6, 130.8, 130.7, 130.4, 129.6, 129.3, 128.5, 127.4, 67.6, 51.5, 46.6, 39.0, 22.8, 22.7, 21.6. HRMS (ESI); Calcd for C26H32N4O4S (M+1): 497.2217. Found: 497.2215.
4.1.56. 3-Isopropyl-1-(3-methoxybenzyl)-N-methyl-N-(4-methyl-3-(morpholinosulfonyl)phenyl)-1H-pyrazole-4-carboxamide (8b)
A mixture of 4m (0.3 g, 0.58 mmol), NaH (60% dispersion in oil) (0.046 g, 1.27 mmol) and anhydrous DMF (15 mL) was stirred at room temperature for 30 min. The reaction mixture was cooled to 0 °C, and iodomethane (0.25 mL, 4.09 mmol) was added to it. The resulting mixture was stirred at room temperature under an atmosphere of N2 and monitored by TLC. The reaction was stopped, water (50 mL) was added to the reaction mixture and the aqueous layer was extracted with ethyl acetate (3 × 25 mL). The combined organic layers were washed with water and brine (3 × 10 mL), dried over Na2SO4, filtered and concentrated. The resulting yellow oil was purified by flash column chromatography (H/EtOAc 1:1) to afford 8b as a clear oil (0.27 g, 87% yield). 1H NMR (CDCl3) δ 7.45 (s, 1H), 7.18 (dd, 3H), 6.78 (d, 1H), 6.52 (d, 2H), 6.50 (d, 1H), 4.95 (s, 2H), 3.71 (s, 3H), 3.63 (t, 4H), 3.34 (s, 3H), 3.31 (m, 1H), 2.82 (t, 4H), 2.53 (s, 3H), 1.23 (d, 6H). 13C NMR (CDCl3) δ 166.5, 144.0, 140.8, 136.8, 134.4, 131.6, 130.8, 130.7, 130.4, 129.6, 129.3, 128.5, 127.4, 67.6, 51.5, 46.6, 39.0, 22.8, 22.7, 21.6. HRMS (ESI); Calcd for C27H34N4O5S (M+1): 527.2323. Found: 527.2321.
4.1.57. 1-(3-Methoxybenzyl)-3-(3-methoxyphenyl)-N-methyl-N-(4-methyl-3-(morpholinosulfonyl)phenyl)-1H-pyrazole-4-carboxamide (8c)
A mixture of 4o (0.1 g, 0.17 mmol), NaH (60% dispersion in oil) (0.02 g, 0.51 mmol) and anhydrous DMF (10 mL) was stirred at room temperature for 30 min. The reaction mixture was cooled to 0 °C, and iodomethane (0.075 mL, 1.21 mmol) was added to it. The resulting mixture was stirred at room temperature under an atmosphere of N2 and monitored by TLC. The reaction was stopped, water (50 mL) was added into the reaction mixture and the aqueous layer was extracted with ethyl acetate (3 × 25 mL). The combined organic layers were washed with water and brine (3 × 10 mL), dried over Na2SO4, filtered and concentrated. The resulting yellow oil was purified by flash column chromatography (H/EtOAc 1:1) to afford 8c as a clear oil (0.08 g, 87% yield). 1H NMR (CDCl3) δ 7.45 (s, 1H), 7.21 (t, 2H), 7.14 (t, 1H), 6.85 (d, 2H), 6.80 (t, 3H), 6.72 (d, 2H), 6.70 (s, 1H), 5.12 (s, 2H), 3.72 (s, 6H), 3.58 (t, 4H), 3.26 (s, 3H), 2.84 (t, 4H), 2.41 (s, 3H). 13C NMR (CDCl3) δ 164.3, 159.0, 158.6, 148.3, 140.8, 135.5, 134.4, 133.9, 132.6, 131.6, 131.3, 129.0, 128.3, 127.0, 119.3, 118.8, 113.3, 112.8, 111.3, 65.2, 54.3, 54.2, 44.0, 19.0. HRMS (ESI); Calcd for C31H34N4O6S (M+1): 591.2272. Found: 591.2270.
4.1.58. N-((1-Isopropyl-3-(p-tolyl)-1H-pyrazol-4-yl)methyl)-4-methyl-3-(morpholinosulfonyl)aniline (9a)
To a solution of 4f (0.1 g, 0.21 mmol) in anhydrous THF (5 mL), 1 N borane-THF complex (7 mL) was added at 0 °C under N2. The reaction mixture was heated to reflux for 3 h. After it was cooled to 0 °C, 1N methanolic HCl (3 mL) was added dropwise to quench excess borane reagent and MeOH (5 mL) was added. The reaction mixture was heated to reflux for 2 h. The solvent was evaporated at reduced pressure. The oily residue was dissolved in EtOAc and neutralized with 2N NaOH. The resulting mixture was extracted with EtOAc (3 × 15 mL). The combined extracts were washed with 0.5 N KOH, dried over Na2SO4, and evaporated in vacuo. The resulting yellow oil was purified by flash column chromatography (H/EtOAc 1:1) to afford 9a as colorless oil (0.08 g, 81% yield). 1H NMR (CDCl3) δ 7.55 (d, 2H), 7.47 (s, 1H), 7.20 (d, 2H), 7.14 (s, 1H), 7.10 (d, 1H), 6.72 (d, 1H), 4.52 (m, 1H), 4.27 (s, 2H), 3.71 (t, 4H), 3.12 (t, 4H), 2.51 (s, 3H), 2.36 (s, 3H), 1.53 (d, 6H), 13C NMR (CDCl3) δ 150.5, 147.6, 138.8, 136.7, 135.1, 130.7, 128.8, 128.5, 127.6, 118.7, 115.7, 69.0, 46.7, 33.1, 24.1, 24.0. HRMS (ESI); Calcd for C25H32N4O3S (M+1): 469.6116. Found: 469.2715.
4.1.59. N-((3-Isopropyl-1-(3-methoxybenzyl)-1H-pyrazol-4-yl)methyl)-4-methyl-3-(morpholinosulfonyl)aniline (9b)
To a solution of 4m (0.1 g, 0.19 mmol) in anhydrous THF (5 mL), 1 N borane-THF complex (7 mL) was added at 0 °C under N2. The reaction mixture was heated to reflux for 3 h. After it was cooled to 0 °C, 1N methanolic HCl (3 ml) was added dropwise to quench excess borane reagent and additional MeOH was added. The reaction mixture was heated to reflux for 3 h. The solvent was evaporated at reduced pressure. The oily residue was dissolved in EtOAc and neutralized with 2N NaOH. The resulting mixture was extracted with EtOAc (3 × 15 mL). The combined extracts were washed with 0.5 N KOH, dried over Na2SO4, and evaporated in vacuo. The resulting yellow oil was purified by flash column chromatography (H/EtOAc 1:1) to afford 9b as colorless oil (0.07 g, 77% yield). 1H NMR (CDCl3) δ 7.22 (d, 2H), 7.21 (s, 1H), 7.12 (s, 1H), 7.10 (d, 1H), 6.83 (d, 1H), 6.77 (d, 1H), 6.72 (t, 2H), 5.18 (s, 2H), 4.11 (t, 2H), 3.76 (s, 3H), 3.69 (t, 4H), 3.11 (t, 4H), 3.05 (m, 1H), 2.48 (s, 3H), 1.26 (d, 6H). 13C NMR (CDCl3) δ 161.3, 157.7, 147.6, 139.6, 136.4, 135.1, 131.2, 130.5, 127.1, 121.3, 118.5, 116.6, 115.6, 114.8, 114.7, 67.7, 57.1, 56.6, 46.7, 39.8, 26.5, 24.0, 23.6, 21.1. HRMS (ESI); Calcd for C26H34N4O4S (M+1): 499.6376. Found: 499.3676.
4.1.60. N-((1-(3-Methoxybenzyl)-3-(3-methoxyphenyl)-1H-pyrazol-4-yl)methyl)-4-methyl-3-(morpholinosulfonyl)aniline (9c)
To a solution of 4o (0.1 g, 0.17 mmol) in anhydrous THF (5 mL), 1 N borane-THF complex (8 mL) was added at 0 °C under N2. The reaction mixture was heated to reflux for 3 h. After it was cooled to 0 °C, 1N methanolic HCl (3 mL) was added dropwise to quench excess borane reagent and additional MeOH was added. The reaction mixture was heated to reflux for 2 h. The solvent was evaporated at reduced pressure. The oily residue was dissolved in EtOAc and neutralized with 2N NaOH. The resulting mixture was extracted with EtOAc (3 × 15 mL). The combined extracts were washed with 0.5 N KOH, dried over Na2SO4, and evaporated in vacuo. The resulting yellow oil was purified by flash column chromatography (H/EtOAc 1:1) to afford 9-c as colorless oil (0.065 g, 72% yield). 1H NMR (CDCl3) δ 7.38 (s, 1H), 7.32 (m, 5H), 7.18 (d, 1H), 7.11 (s, 1H), 6.89 (t, 2H), 6.81 (d, 2H), 6.69 (t, 1H), 5.28 (s, 2H), 4.26 (s, 2H), 3.81 (s, 3H), 3.76 (s, 3H), 3.71 (t, 4H), 3.11 (t, 4H), 2.49 (s, 3H). 13C NMR (CDCl3) δ 161.3, 161.2, 151.1, 147.3, 139.1, 136.4, 135.1, 131.7, 131.3, 127.4, 121.5, 118.6, 117.7, 115.7, 115.3, 115.1, 116.2, 114.8, 114.1, 67.7, 57.5, 56.6, 46.7, 40.5, 21.2. HRMS (ESI); Calcd for C30H34N4O5S (M+Na): 565.6798. Found: 585.2798.
4.2. Biology
4.2.1. Materials
DMSO and charcoal/dextran-treated FBS were obtained from Fisher Scientific (Pittsburgh, PA). GW4064 was purchased from R&D Systems, Inc. (Minneapolis, MN); LCA and rifampicin were purchased from Sigma-Aldrich (St. Louis, MO); TO901317 was obtained from Cayman Chemical Company (Ann Arbor, MI); Z26476908 (E16) was purchased from Enamine LLC (Monmouth Junction, NJ). Black polypropylene 384-well plates were purchased from Matrical Bioscience (Spokane, WA). Black 384-well tissue culture-treated clear bottom plates were obtained from Corning Incorporated (Corning, NY). White tissue culture-treated 384-well plates were purchased from PerkinElmer (Waltham, MA). GST-hFXR-LBD, Tb-anti-GST antibody, coregulator buffer G, 1 M DTT, GeneBLAzer FXR-UAS-bla HEK 293T cells, DMEM, phenol red-free DMEM, dialyzed FBS, non-essential amino acids, sodium pyruvate, HEPES, hygromycin B, zeocin, penicillin/ streptomycin, G418 and CCF4-AM substrate were purchased from Invitrogen (Carlsbad, CA).
All chemicals were initially solubilized in DMSO as 10 mM stocks and then diluted in corresponding assay buffer or medium to indicated concentrations. All experiments were repeated in triplicate.
4.2.2. FXR TR-FRET binding assay
The FXR TR-FRET binding assay was performed as previously described23 with modifications. Briefly, in a black polypropylene 384-well plate, DMSO, 10 µM GW4064, and titrations of GW4064, lithocholic acid or the synthesized chemicals were mixed with 10 nM GST-hFXR-LBD, 1.5 nM Tb-anti-GST antibody, and 10 nM DY246 in a 20 µL/well of coregulator buffer G supplemented with 10 mM DTT. The final DMSO concentration was 0.4% for all wells. The plate was spun down after a brief shake and incubated at room temperature for 20 min. The TR-FRET emission signals at 520 nm and 490 nm for each well were determined using a PHERAstar plate reader (BMG LABTECH, Cary, NC) and an excitation wavelength of 337 nm, 100 µs delay time, and 200 µs integration time. The 520 nm/490 nm ratio from each well was then calculated. The DMSO group and 10 µM GW4064 group served as negative (0% inhibition) and positive (100% inhibition) controls, respectively. The % inhibition of tested chemicals at given concentration was calculated based on negative and positive controls using equation 1.
| Equation 1 |
For those chemicals with a maximal % inhibition > 50% and activities that behaved in a dose-dependent manner, the inhibitory activities at different concentrations were fit into a one-site competitive binding equation with GraphPad PRISM 6.01 (GraphPad Software, Inc., La Jolla, CA) to derive IC50 values.
4.2.3. FXR cell-based transactivation assay
FXR transactivation assays were performed using GeneBLAzer FXR-UAS-bla HEK 293T cells (Invitrogen). Cells were maintained according to the manufacturer’s instructions. Briefly, cells were grown in DMEM supplemented with 10% dialyzed FBS, 0.1 mM non-essential amino acids, 25 mM HEPES, 100 µg/mL of hygromycin B, 100 µg/ml of zeocin, 100 units/mL penicillin and 100 µg/mL streptomycin in regular tissue culture flasks. Upon transactivation assay, DMSO, 400 nM GW4064, 10 µM GW4064, and titrations of GW4064, LCA or chemicals without 400 nM GW4064 (for FXR agonistic activity determination) or with 400 nM GW4064 (for FXR antagonistic activity determination) were mixed with cells (20,000 cells/well in a 30 µL) in assay medium (phenol red-free DMEM supplemented with 2% charcoal/dextran-treated FBS, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 100 units/mL penicillin and 100 µg/mL streptomycin) in black 384-well tissue culture-treated clear bottom plates (Corning Incorporated, Corning, NY). As a background control, the wells in column 24 of each plate were filled with 30 µL assay medium along with 0.5% DMSO. The final DMSO concentration was 0.5% in each assay well. Plates were incubated (16 h, cell culture incubator at 37°C), after which 6 µl/well of loading solution with CCF4-AM substrate was added. Plates were then incubated in the dark for 90 min at room temperature before measuring the fluorescent emissions at 460 nm and 535 nm (using excitation at 400 nm) with an Envision plate reader with bottom read capacity. After background signal subtraction for individual emission channels, emission signals at 460 and 535 nm from each well were used to determine the ratio of 460 nm/535 nm. For agonist assays, the DMSO group and 10 µM GW4064 group served as negative (0% activation) and positive (100% activation) controls, respectively. The % activation was calculated using equation 2.
| Equation 2 |
For antagonistic assays in which 400 nM GW4064 was included in the assay conditions, the DMSO group and 400 nM GW4064 group served as positive (100% inhibition) and negative (0% inhibition) controls, respectively. The % inhibition was calculated using equation 3.
| Equation 3 |
For those chemicals with a maximal % inhibition > 50% and activities that behaved in a dose-dependent manner, the inhibitory activities at different concentrations were fit into a sigmoidal dose-response equation with GraphPad PRISM 6.01 to derive IC50 values.
4.2.4. Cell-based cytotoxicity assay
Cytotoxicity assays were performed side-by-side with the FXR agonistic assay using GeneBLAzer FXR-UAS-bla HEK 293T cells and the CellTiter-Glo® Luminescent Cell Viability Assay reagent (Promega). Briefly, cells were maintained in DMEM supplemented with 10% dialyzed FBS, 0.1 mM non-essential amino acids, 25 mM HEPES, 100 µg/mL of hygromycin B, 100 µg/mL of zeocin, 100 units/mL penicillin and 100 µg/mL streptomycin in regular tissue culture flasks. For the cytotoxicity assay, DMSO, titrations of GW4064, LCA or of chemicals were mixed with cells (20,000 cells/well in a 30 µL) in assay medium (phenol red-free DMEM supplemented with 2% charcoal/dextran-treated FBS, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 100 units/mL penicillin and 100 µg/mL streptomycin) in black 384-well tissue culture-treated clear bottom plates. As a positive control, the wells in column 24 of each plate were filled with 30 µL assay medium along with 0.5% DMSO. The final DMSO concentration was 0.5% in each assay well. After a 16 h incubation in a cell culture incubator at 37°C, the cell plates were cooled at room temperature for 20 min and then CellTiter-Glo® reagent 20 µL/well was added followed by an incubation for 20 min in the dark to allow the development of optimal luminescence signal. The luminescence signal from each well was collected with an Envision plate reader equipped with Ultra-sensitive Luminescence detector. The DMSO group and no cell group served as negative (0% activation) and positive (100% activation) controls, respectively. The % inhibition was calculated using equation 4 below.
| Equation 4 |
ACKNOWLEDGMENTS
We thank Drs. Arthur D. Riggs and Wendong Huang for support and encouragement. We also thank Min Lin for technical assistance and Dr. Keely Walker for editing the manuscript. This work was supported by the American Lebanese Syrian Associated Charities (ALSAC), St. Jude Children’s Research Hospital (St. Jude), National Institute of General Medical Sciences [Grant GM086415] (to TC), and National Cancer Institute [Grant P30-CA21765].
ABBREVIATIONS USED
- HTS
High-Throughput Screen
- FXR
farnesoid X receptor
- NRs
nuclear receptors
- Cyp7A1
cytochrome 7A1
- BAs
bile acids
- BSEP
bile salt export pump
- SAR
structure-activity relationship
- LBD
ligand-binding domain
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Nuclear Receptor Nomenclature Committee. A unified nomenclature system for the nuclear receptor superfamily. Cell. 1999;97:161–163. doi: 10.1016/s0092-8674(00)80726-6. [DOI] [PubMed] [Google Scholar]
- 2.Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T, Noonan DJ, Burka LT, McMorris T, Lamph WW, Evans RM, Weinberger C. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995;81:687–693. doi: 10.1016/0092-8674(95)90530-8. [DOI] [PubMed] [Google Scholar]
- 3.Wang H, Chen J, Hollister K, Sower LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell. 1999;3:543–553. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- 4.Fiorucci S, Mencarelli A, Distrutti E, Palladino G, Cipriani S. Targeting farnesoid-X-receptor: from medicinal chemistry to disease treatment. Curr. Med. Chem. 2010;17:139–159. doi: 10.2174/092986710790112666. [DOI] [PubMed] [Google Scholar]
- 5.Fiorucci S, Cipriani S, Baldelli F, Mencarelli A. Bile acidactivated receptors in the treatment of dyslipidemia and related disorders. Prog. Lipid Res. 2010;49:171–185. doi: 10.1016/j.plipres.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Chen WD, Wang YD, Zhang L, Shiah S, Wang M, Yang F, Yu D, Forman BM, Huang W. Farnesoid X receptor alleviates age-related proliferation defects in regenerating mouse livers by activating forkhead box m1b transcription. Hepatology. 2010;51(3):953–962. doi: 10.1002/hep.23390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fiorucci S, Baldelli F. Farnesoid X receptor agonists in biliary tract disease. Curr. Opin. Gastroenterol. 2009;25:252–259. doi: 10.1097/MOG.0b013e328324f87e. [DOI] [PubMed] [Google Scholar]
- 8.Maloney PR, Parks DJ, Haffner CD, Fivush AM, Chandra G, Plunket KD, Greech KL, Moore PR, Wilson JG, Lewis MC, Jones SA, Willson TM. Identification of a Chemical Tool for the Orphan Nuclear Receptor FXR. J. Med. Chem. 2000;43:2971–2974. doi: 10.1021/jm0002127. [DOI] [PubMed] [Google Scholar]
- 9.Donna Yu, Mattern Daniell L, Forman Barry M. An improved synthesis of 6α-ethyl chenodeoxycholic acid (6ECDCA), a potent and selective agonist for the farnesoid X receptor (FXR) Steroids. 2012;77:1335–1338. doi: 10.1016/j.steroids.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 10.Fiorucci S, Zampella A, Distrutti E. Development of FXR, PXR and CAR agonists and antagonists for treatment of liver disorders. Curr Top Med Chem. 2012;12:605–624. doi: 10.2174/156802612799436678. [DOI] [PubMed] [Google Scholar]
- 11.Renga B, Migliorati M, Mencarelli A, Cipriani S, D’Amore C, Distrutti E, Fiorucci S. Farnesoid X receptor suppresses constitutive androstane receptor activity at the multidrug resistance protein-4 promoter. Biochim. Biophys. Acta. 2011;1809:157–165. doi: 10.1016/j.bbagrm.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 12.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 13.Urizar NL, Liverman AB, Dodds DT, Silva FV, Ordentlich P, Yan YZ, Gonzalez FJ, Heyman RA, Mangelsdorf DJ, Moore DD. A natural product that lowers cholesterol as an antagonist ligand for FXR. Science. 2002;296:1703–1706. doi: 10.1126/science.1072891. [DOI] [PubMed] [Google Scholar]
- 14.Urizar NL, Moore DD. Guggulipid: a natural cholesterollowering agent. Annu. Rev. Nutr. 2003;23:303–313. doi: 10.1146/annurev.nutr.23.011702.073102. [DOI] [PubMed] [Google Scholar]
- 15.Pellicciari R, Costantino G, Fiorucci S. Farnesoid X Receptor: From Structure to Potential Clinical Applications. J. Med. Chem. 2005;17:1–21. doi: 10.1021/jm0582221. [DOI] [PubMed] [Google Scholar]
- 16.Sepe V, Bifulco G, Renga B, D’Amore C, Fiorucci S, Zampella A. Discovery of Sulfated Sterols from Marine Invertebrates as a New Class of Marine Natural Antagonists of Farnesoid-X-Receptor. J. Med. Chem. 2011;54:1314–1320. doi: 10.1021/jm101336m. [DOI] [PubMed] [Google Scholar]
- 17.Renga1 B, Mencarelli A, D’Amore C, Cipriani S, D’Auria MV, Sepe V, Chini MG, Monti MC, Bifulco G, Zampella A, Fiorucci S. Discovery that theonellasterol a marine sponge sterol is a highly selective FXR antagonist that protects against liver injury in cholestasis. PLoS ONE. 2012;7:e30443. doi: 10.1371/journal.pone.0030443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nam SJ, Ko H, Shin M, Ham J, Chin J, Kim Y, Kim H, Shin K, Choi H, Kang H. Farnesoid X-activated receptor antagonists from a marine sponge Spongia sp. Bioorg. Med. Chem. Lett. 2006;16:5398–5402. doi: 10.1016/j.bmcl.2006.07.079. [DOI] [PubMed] [Google Scholar]
- 19.Di Leva FS, Festa C, D'Amore C, De Marino S, Renga B, D'Auria MV, Novellino E, Limongelli V, Zampella A, Fiorucci S. Binding Mechanism of the Farnesoid X Receptor Marine Antagonist Suvanine Reveals a Strategy To Forestall Drug Modulation on Nuclear Receptors. Design, Synthesis, and Biological Evaluation of Novel Ligands. J. Med. Chem. 2013;56:4701–4717. doi: 10.1021/jm400419e. [DOI] [PubMed] [Google Scholar]
- 20.Dussault I, Beard R, Lin M, Hollister K, Chen J, Xiao JH, Chandraratna R, Forman BM. Identification of gene selective modulators of the bile acid receptor FXR. J. Biol. Chem. 2003;278:7027–7033. doi: 10.1074/jbc.M209863200. [DOI] [PubMed] [Google Scholar]
- 21.Huang H, Yu Y, Gao Z, Zhang Y, Li C, Xu X, Jin H, Yan W, Ma R, Zhu J, Shen X, Jiang H, Chen L, Li J. Discovery and Optimization of 1,3,4-Trisubstituted-pyrazolone Derivatives as Novel, Potent, and Nonsteroidal Farnesoid X Receptor (FXR) Selective Antagonists. J. Med. Chem. 2012;55:7037–7053. doi: 10.1021/jm3002718. [DOI] [PubMed] [Google Scholar]
- 22.Sayin SI, Wahlstroem A, Felin J, Jaentti S, Marschall H-U, Bamberg K, Angelin B, Hyoetylaeinen T, Oresic M, Baeckhed F. Gut Microbiota Regulates Bile Acid Metabolism by Reducing the Levels of Tauro-beta-muricholic Acid, a Naturally Occurring FXR Antagonist. Cell Metabolism. 2013;17:225–235. doi: 10.1016/j.cmet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 23.Yu DD, Lin W, Chen T, Forman BM. Development of Time Resolved Fluorescence Resonance Energy Transfer-based Assay for FXR Antagonist Discovery. Bioorganic & Medicinal Chemistry. 2013;21:4266–4278. doi: 10.1016/j.bmc.2013.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(a) Pendri A, Dodd DS, Chen J, Cvijic ME, Kang L, Baska RA, Carlson KE, Burford NT, Sun C, Ewing WR, Gerritz SW. Solid phase synthesis of 1,5-diarylpyrazole-4-carboxamides: Discovery of antagonists of the CB-1 receptor. Comb. Sci. 2012;14:197–201. doi: 10.1021/co200147y. [DOI] [PubMed] [Google Scholar]; (b) WO/2007/052843. TAKEDA PHARMACEUTICAL COMPANY LIMITED, HETEROCYCLIC AMIDE COMPOUND AND USE THEREOF. 2007
- 25.Lamberth C. Pyrazole chemistry in crop protection. Heterocycles. 2007;71:1467. [Google Scholar]
- 26.Akwabi-Ameyaw A, Caravella JA, Chen L, Creech KL, Deaton DN, Madauss KP, Marr HB, Miller AB, Navas F, III, Parks DJ, Spearing PK, Todd D, Williams SP, Wisely GB. Conformationally constrained farnesoid X receptor (FXR) agonists: alternative replacements of the stilbene. Bioorg. Med. Chem. Lett. 2011;21:6154–6160. doi: 10.1016/j.bmcl.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 27.Hashimoto Y. Retinobenzoic acids and nuclear retinoic acid receptors. Cell Struct. Funct. 1991;16:113–123. doi: 10.1247/csf.16.113. [DOI] [PubMed] [Google Scholar]
- 28.Soisson SM, Parthasarathy G, Adams AD, Sahoo S, Sitlani A, Sparrow C, Cui J, Becker JW. Identification of a potent synthetic FXR agonist with an unexpected mode of binding and activation. Proc Natl Acad Sci U S A. 2008;105:5337–5342. doi: 10.1073/pnas.0710981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Babaoglu K, Shoichet BK. Deconstructing fragment-based inhibitor discovery. Nat. Chem. Biol. 2006;2:720–723. doi: 10.1038/nchembio831. [DOI] [PubMed] [Google Scholar]
- 30.Zuercher WJ, Buckholz RG, Campobasso N, Collins JL, Galardi CM, Gampe RT, Hyatt SM, Merrihew SL, Moore JT, Oplinger JA, Reid PR, Spearing PK, Stanley TB, Stewart EL, Willson TM. Discovery of Tertiary Sulfonamides as Potent Liver X Receptor Antagonists. J. Med. Chem. 2010;53:3412–3416. doi: 10.1021/jm901797p. [DOI] [PubMed] [Google Scholar]
- 31.Bebernitz GR, Argentieri G, Battle B, Brennan C, Balkan B, Burkey BF, Eckhardt M, Gao J, Kapa P, Strohschein RJ, Schuster HF, Wilson M, Xu DD. The Effect of 1,3-Diaryl-[1H]-pyrazole-4-acetamides on Glucose Utilization in ob/ob Mice. J. Med. Chem. 2001;44:2601–2611. doi: 10.1021/jm010032c. [DOI] [PubMed] [Google Scholar]
- 32.Baraldi PG, Tabrizi MA, Preti D, Bovero A, Fruttarolo F, Romagnoli R, Zaid NA, Moorman AR, Varani K, Borea PA. New 2-Arylpyrazolo[4,3-c]quinoline Derivatives as Potent and Selective Human A3 Adenosine Receptor Antagonists. J. Med. Chem. 2005;48:5001–5008. doi: 10.1021/jm050125k. [DOI] [PubMed] [Google Scholar]
- 33.Lepore SD, He Y. Use of sonication for the coupling of sterically hindered substrates in the phenolic Mitsunobu reaction. J. Org. Chem. 2003;68:8261–8263. doi: 10.1021/jo0345751. [DOI] [PubMed] [Google Scholar]
- 34.Petzinger E, Wickboldt A, Pagels P, Starke D, Kramer W. Hepatobiliary Transport of Bile Acid Amino Acid, Bile Acid Peptide, and Bile Acid Oligonucleotide Conjugates in Rats. HEPATOLOGY. 1999;30:1257–1268. doi: 10.1002/hep.510300529. [DOI] [PubMed] [Google Scholar]
- 35.Ozers MS, Marks BD, Gowda K, Kupcho KR, Ervin KM, De Rosier T, Qadir N, Eliason HC, Riddle SM, Shekhani MS. The Androgen Receptor T877A Mutant Recruits LXXLL and FXXLF Peptides Differently than Wild-Type Androgen Receptor in a Time-Resolved Fluorescence Resonance Energy Transfer Assay. Biochemistry. 2007;46:683–695. doi: 10.1021/bi061321b. [DOI] [PubMed] [Google Scholar]
- 36.Li G, Lin W, Araya JJ, Chen T, Timmermann BN, Guo GL. A tea catechin, epigallocatechin-3-gallate, is a unique modulator of the farnesoid X receptor. Toxicol Appl Pharmacol. 2012;258:268–74. doi: 10.1016/j.taap.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Gupta R, Huang L, Lown JW. CC-1065 functional analogs possessing different electron-withdrawing substituents and leaving groups: synthesis, kinetics, and sequence specificity of reaction with DNA and biological evaluation. J. Med. Chem. 1993;36:4172–4182. doi: 10.1021/jm00078a005. [DOI] [PubMed] [Google Scholar]