

Abstract
Functional magnetic resonance imaging (fMRI) provides a unique view of the working human mind. The blood-oxygen-level-dependent (BOLD) signal, detected in fMRI, reflects changes in deoxyhemoglobin driven by localized changes in brain blood flow and blood oxygenation, which are coupled to underlying neuronal activity by a process termed neurovascular coupling. Over the past 10 years, a range of cellular mechanisms, including astrocytes, pericytes, and interneurons, have been proposed to play a role in functional neurovascular coupling. However, the field remains conflicted over the relative importance of each process, while key spatiotemporal features of BOLD response remain unexplained. Here, we review current candidate neurovascular coupling mechanisms and propose that previously overlooked involvement of the vascular endothelium may provide a more complete picture of how blood flow is controlled in the brain. We also explore the possibility and consequences of conditions in which neurovascular coupling may be altered, including during postnatal development, pathological states, and aging, noting relevance to both stimulus-evoked and resting-state fMRI studies.
Keywords: neurovascular coupling, fMRI, astrocytes, pericytes, vascular endothelium
INTRODUCTION
Noninvasive assessment of human brain function is a major challenge. Techniques such as electroencephalography (EEG) and magnetoencephalography (MEG) provide direct measurement of neuronal activity but are faced with sensitivity, localization, and resolution issues and are rarely used for functional brain imaging research. All other current methods of human functional brain imaging rely on proxy measures of neuronal activity that are related to local blood flow, oxygenation, or metabolism (Raichle 1998). The most common of these functional brain imaging techniques is functional magnetic resonance imaging (fMRI).
WHAT IS THE fMRI BOLD SIGNAL?
fMRI relies upon the measurement of T2* relaxation, which is sensitive primarily to local concentrations of paramagnetic deoxyhemoglobin (HbR) (Ogawa et al. 1990). The so-called fMRI blood-oxygen-level-dependent (BOLD) signal increases with decreasing HbR and can be analyzed to produce localized maps of functional activity in the human brain. However, despite widespread use for almost 20 years, the fMRI BOLD signal is still poorly understood (Attwell et al. 2010, Girouard & Iadecola 2006, Logothetis 2010, Sirotin & Das 2009).
Interpretation of the fMRI BOLD signal is intrinsically linked to understanding the underlying physiological and metabolic processes in the brain that modulate blood flow. A prevailing misconception is that BOLD provides a direct measurement of neuronal oxygen consumption. However, this is generally not the case; classic positive BOLD signals, seen in response to functional stimuli, represent a decrease in HbR and thus an overoxygenation of the responding region (Attwell & Iadecola 2002). These positive BOLD responses correspond to a local, actively actuated, increase in blood flow and volume, which brings blood in sufficient excess to increase local oxygenation levels (Raichle 1998). This response typically begins within ~500 ms and peaks 3–5 s after stimulus onset (Figure 1), even for short stimuli lasting less than 1 s (Hirano et al. 2011, Martindale et al. 2003, Yeşilyurt et al. 2008), with more complex dynamics for prolonged stimuli (Figures 2 and 4) (Martindale et al. 2005). The active process linking local neuronal activity to an orchestrated increase in local blood flow is termed neurovascular coupling. The spatiotemporal properties of the BOLD response to stimulus are therefore strongly dependent upon neurovascular coupling and the dynamic properties of the vasculature that alters blood flow through its physical dilation and constriction.
Figure 1.
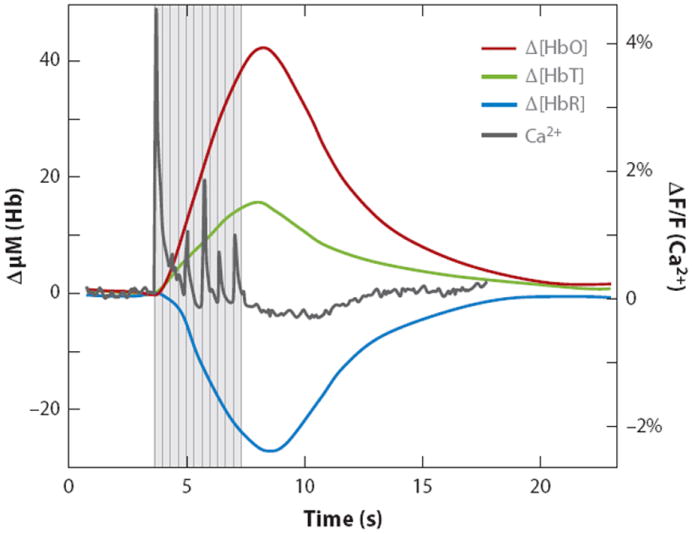
A typical stimulus-evoked response in the rat somatosensory cortex. Stimulus was 4 s of ~1 mA, 3 Hz forepaw stimulation. Data were acquired using multispectral optical intrinsic signal imaging of the exposed cortex, averaged over the responding region. Dark gray trace shows calcium response to the same stimulation, measured using bulk cortical injection of calcium sensitive fluorophore Oregon green 488 BAPTA-1 AM (Bouchard et al. 2009). Figure reproduced from Hillman (2007). Notably, there is a distinct increase in total hemoglobin (HbT) corresponding to vessel dilation and an increase in the number of red blood cells per unit volume of cortex, consistent with an increase in blood flow. Oxyhemoglobin (HbO) increases while deoxyhemoglobin (HbR) decreases, indicating a net overoxygenation of the region. The fMRI BOLD signal is sensitive to changes in HbR, where stimulus-evoked ‘positive BOLD’ corresponds to the decrease in HbR shown here. The response begins within ~500 ms of stimulus onset and peaks at 3–5 s before slowly returning to baseline. Note that the first large calcium response (corresponding to neuronal activity) precedes marked hemodynamic changes.
Figure 2.
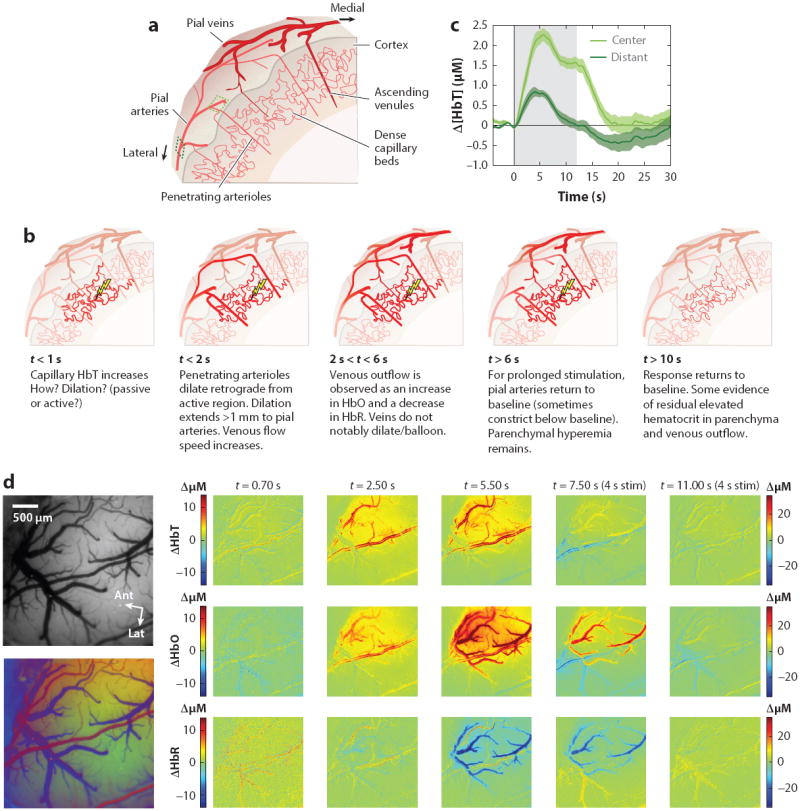
Vascular evolution of normal stimulus-evoked functional hyperemia. (a) Schematic cut through of the mammalian cortex. Major cortical blood vessels are located on the pial surface with penetrating arterioles diving perpendicularly into the cortex and branching into dense capillary beds within the cortical layers. Blood drains from these capillaries via perpendicularly oriented ascending venules, which join a network of large draining veins on the pial surface. (b) Schematic sequence of the vascular dynamic response to functional stimulation (see text for citations). (c) Dynamics of the Δ [HbT] response to a 12-s duration, 3 Hz, ~1 mA electrical hindpaw stimulation recorded using optical imaging of the rat somatosensory cortex. ‘Center’ and ‘Distant’ vessels sampled are indicated in panel a. Time courses reveal that the response is more sustained within the central region, while more distant arteries return to baseline earlier, after a peak at 3–5 seconds. Δ [HbT] is independent of oxygenation dynamics and here represents only hyperemia due to arterial/arteriolar dilation and increased concentrations of red blood cells in the capillary beds. (d) A sequence of optical intrinsic signal imaging (OISI) data acquired on the rat somatosensory cortex in response to 4-s, 3 Hz hindpaw stimulation. Left: images showing the field of view under green (530 nm) illumination. Below: a composite based on oxygenation-dependent baseline reflectance highlighting arteries (red), veins (blue), and parenchyma (green). Time sequence: [HbT] changes show an initial increase in parenchymal signal by 0.7 s after stimulus onset (color scale for 0.7 s maps shown at left; all other time points use color scale at right). Increased contrast of pial arteries corresponds to dilation (confirmed by full width half maximum calculation). Increased contrast of the intervening parenchymal space corresponds to an increase in the number of red blood cells per unit volume within the capillary beds (as well as diving and ascending arterioles and venules). [HbR] (deoxyhemoglobin) changes show a distinctly different pattern and can be seen to localize to the shape of the draining veins, with decreases delayed relative to initial changes in [HbT]. Data reproduced from Chen et al. (2011, 2014) and Bouchard et al. (2009).
Figure 4.

Proposed model of nonlinear neurovascular coupling incorporating fast and slow propagated vasodilation. Neuronal activity at the capillary level could either directly or indirectly cause an increase in endothelial intracellular calcium. An initial large-amplitude increase in endothelial calcium could initiate endothelial hyperpolarization, which would be rapidly propagated with minimal attenuation to drive relaxation of perivascular SMCs all the way up to the pial arteries (Wölfle et al. 2011). The same initial increase in endothelial calcium could also drive a slower propagating wave of increased calcium within the endothelium (Tallini et al. 2007), bringing NO and prostanoid-dependent vasodilation over a shorter distance. Combined, these two effects would generate spatiotemporal nonlinearities consistent with the properties of functional hyperemia. A lower threshold in endothelial calcium (Marrelli 2001) for slow propagation might explain continued parenchymal hyperemia, but only transient (initial) long-range dilation as shown in Figure 2.
For classical interpretation of fMRI BOLD signals (Boynton et al. 1996), it is assumed that neurovascular coupling is so robust that any increase in neuronal activity generates a proportional increase in local blood flow, irrespective of brain region, brain development, and pathological state (Logothetis 2010). We return to the possible implications of these conditions in later sections. However, we begin by reviewing what is currently known about the cause, manifestations, and mechanisms of neurovascular coupling that drive stimulus-evoked functional hyperemia leading to positive BOLD in the normal brain.
WHY DOES BLOOD FLOW INCREASE?
The BOLD response is clearly an essential component of normal brain function (Girouard & Iadecola 2006, Mogi & Horiuchi 2011, Schroeter et al. 2007), yet its relationship to neuronal metabolism is less clear. Questioning whether neurovascular coupling simply matches supply to demand, several studies have maintained animals in hyperoxic or hyperglycemic states, with the surprising result that all continued to exhibit functional hyperemia in response to stimulation despite plentiful availability of oxygen and/or glucose (Lindauer et al. 2010, Wolf et al. 1997). Hypoglycemia in humans produced similar results (Powers et al. 1996). These studies demonstrate that blood flow increases are not triggered simply by local sensing of depleted nutrients.
The fact that blood flow increases are high enough to generate local hyperoxygenation, far exceeding oxygen consumption, also suggests an indirect relation between oxygen supply and demand. While one recent study suggested that high oxygen gradients are required to supply all active cells (Devor et al. 2011), others have inferred that the role of the hemodynamic response is to provide higher levels of glucose rather than oxygen (Fox & Raichle 1986, Fox et al. 1988, Heeger & Ress 2002, Paulson et al. 2009). Roles for functional hyperemia in waste removal and heat regulation have also been proposed (Yablonskiy et al. 2000).
The relative delay in the peak of increased blood flow (Figure 1) further confirms that neurons do not rely upon functional hyperemia to meet their initial needs for increased oxygen and glucose, since neuronal firing may have ended prior to measurable changes in blood flow. One explanation proposed for this discrepancy is that neurons, or associated support cells, may maintain sufficient stores of nutrients such as glycogen to support initial neuronal responses (Brown & Ransom 2007, Heeger & Ress 2002), which may be necessary if the speed of the vascular response is physically limited. A later blood flow peak may serve to replenish supplies or, in the case of prolonged stimulation, could be required to sustain neuronal responses once initial stores have been depleted. One model that fits with this view is the astrocyte-neuron lactate shuttle (Pellerin et al. 2007, Pellerin & Magistretti 1994), which posits that astrocytes undergo anaerobic glycolysis in response to elevated glutamate levels, preserving local oxygen supplies for neuronal use, while supplying lactate as an energy substrate to neurons (Schurr 2005). Astrocytes, located between neurons and blood vessels, are well positioned to serve this role, although the lactate shuttle model itself remains controversial (Hertz 2004, Kasischke et al. 2004).
So while the underlying purpose of functional hyperemia remains unresolved, the observations above point toward functional hyperemia being driven by an actively triggered process, initiated soon after stimulus onset, but which is not, at least initially, causally dependent on local metabolic needs (Attwell & Iadecola 2002).
SPATIOTEMPORAL PROPERTIES OF THE HEMODYNAMIC RESPONSE
The sometimes counterintuitive properties of the BOLD response described above provide an important framework for determining the mechanisms initiating and sustaining functional hyperemia. Adding to this picture, recent in vivo optical imaging and two-photon microscopy studies (see sidebar) have sought to further define the precise vascular and spatiotemporal dynamics of the cortical hemodynamic response to stimulation (Blinder et al. 2013, Chen et al. 2011, Drew et al. 2011, Hillman et al. 2007, Sirotin et al. 2009, Stefanovic et al. 2007, Tian et al. 2010). The properties of functional hyperemia that have emerged are illustrated in Figure 2.
Vascular Features of Functional Hyperemia
Capillary hyperemia
Capillaries are increasingly thought to physically expand during functional hyperemia (Stefanovic et al. 2007, Villringer et al. 1994). Whether active or passive, this increase in capillary diameter could explain observed increases in parenchymal [HbT] (Chen et al. 2011, Culver et al. 2005, Hillman et al. 2007, Sirotin et al. 2009), as well as the deeper location of cerebral blood volume (CBV) changes compared with BOLD in MION-MRI (Zhao et al. 2006). Localized parenchymal HbT increases occur very early, prior to dilation of pial arteries, and may be the underlying cause of observations initially interpreted as the initial dip (see sidebar, The Initial Dip) (Chen et al. 2011, Sirotin et al. 2009).
Arteriolar/arterial dilation
Penetrating arterioles dilate during functional hyperemia (Tian et al. 2010), although their relative timing in relation to capillary dilations has yet to be shown. Specific branches of pial arteries also dilate rapidly, extending up to >1 mm away from the center of the responding region (Li et al. 2003, Ngai & Winn 2002) with recent evidence supporting high-speed retrograde propagation of vasodilation (Chen et al. 2011, Iadecola et al. 1997, Chen et al. 2014).
Venous changes
Increases in arterial, arteriolar, and possibly capillary diameters decrease resistance and increase blood flow. Veins are less likely to exhibit diameter increases, but instead increase their speed of blood flow (Bouchard et al. 2009, Drew et al. 2011, Hillman et al. 2007). Venous oxygenation levels increase after a short delay, as increased blood flow increases the net oxygenation level of blood leaving the capillary beds. The onset delay of venous oxygenation increases is usually attributed to capillary transit time but may also have contributions from oxygen consumption dynamics (Hillman et al. 2007).
Short- and long-duration stimuli
Short stimuli generate responses with a similar temporal shape irrespective of stimulus duration, and with nonlinear scaling of amplitude (up to stimuli 3–4 s long) (Hirano et al. 2011, Martindale et al. 2003, Yesilyurt et al. 2008). Longer-duration stimuli tend to show a peak-then-plateau pattern (Figures 2c and 4a) (Dunn et al. 2005). More distant pial arteries tend to return toward baseline after the initial peak, whereas the plateau phase is more localized to the central parenchyma (Berwick et al. 2008) and can be more variable in amplitude than the initial peak (Drew et al. 2011, Martindale et al. 2005). Often in fMRI, the plateau is higher than the initial peak (Mandeville et al. 1999).
Cessation of stimulus
As blood flow decreases, there is some evidence for residual high HbT in the capillary beds, which combined with frequently observed post-stimulus arterial vasocon-striction (Devor et al. 2008) could play a role in the poststimulus undershoot observed in fMRI (Buxton 2012, Hillman et al. 2007).
BOLD Signal Representations of Vascular Changes
In relation to fMRI BOLD, it is important to consider the manifestation of the dynamics described above in terms of changes in [HbR]. Although the dilation of arteries and arterioles can be significant (5–25%) (Tian et al. 2010), their small volume and high oxygenation means that arteries themselves contribute relatively little to the fMRI BOLD signal (Hillman et al. 2007).
Veins, however, exhibit large increases in [HbR] that contribute significantly to the fMRI BOLD signal. However, these increases can be expected to be both delayed and superficially weighted with respect to the active capillary beds and may even be shifted medially as a result of the drainage patterns of the venous system (Turner 2002). These features are visible in the optical imaging sequence shown in Figure 2d.
Linearity versus Nonlinearity of the BOLD Response
Initial approaches to modeling and interpreting the fMRI BOLD signal assumed linear relations in which a simple hemodynamic response function (HRF) could be convolved with some driving function representing neuronal activity (Boynton et al. 1996, Buxton et al. 1998, Heeger & Ress 2002). The linearity of the BOLD response was subsequently questioned by numerous reports (Devor et al. 2003; Friston et al. 2000; Hewson-Stoate et al. 2005; Hirano et al. 2011; Martin et al. 2006, 2013; Martindale et al. 2005; Sheth et al. 2004; Yesilyurt et al. 2008), which identified nonlinear scaling in the amplitude and duration of the hemodynamic response to stimuli of different frequencies, amplitudes, and durations. A 5-ms visual stimulus, for example, yields a BOLD response almost identical to that from a 250-ms stimulus, both peaking at 3–5 s after stimulus onset. A linear relation would predict a response up to 50 times larger in the latter case (Yesilyurt et al. 2008). The peak-plateau properties of the response for longer stimulus durations add further complexity to HRF-based modeling and quantification (Kennerley et al. 2012, Martin et al. 2013, Martindale et al. 2005). The vascular properties of the hemodynamic response (Figure 2) indicate a more complex spatiotemporal neurovascular relationship than originally assumed (Buxton 2012, Hwan Kim et al. 2013).
In a seminal study that acquired neuronal and fMRI BOLD data simultaneously in primates, linear modeling required site specific HRFs and found fit discrepancies for long-duration stimuli (Logothetis et al. 2001). This study highlighted the importance of considering which measure of neuronal activity should or could be expected to correlate with the BOLD signal and found that local field potentials (LFPs) better predicted BOLD than postsynaptic multiunit spiking activity (MUA) (Cardoso et al. 2012, Logothetis 2010, Sirotin & Das 2009). Such questions will remain unanswered until a comprehensive picture of the cellular and vascular mechanisms of neurovascular coupling is identified.
COUPLING MECHANISMS: HOW DOES BLOOD FLOW CHANGE?
Early investigators proposed that local metabolic factors modulate local blood flow in the brain (Friedland & Iadecola 1991, Roy & Sherrington 1890). The possibility of direct action of neuronally derived substances such as glutamate and nitric oxide (NO) on the vasculature persisted (Attwell & Iadecola 2002) until the past 10 years, when a number of seminal studies introduced additional possible cellular mediators of neurovascular coupling, including astrocytes (Takano et al. 2006), interneurons (Cauli et al. 2004), and pericytes (Peppiatt et al. 2006). Each of these cellular candidates is described below (pathways illustrated in Figure 3).
Figure 3.

Candidate neurovascular coupling pathways [modified from Félétou & Vanhoutte (2004) and Attwell et al. (2010)]. Astrocytes can sense glutamate via metabotropic glutamate receptors (mGluR) and increase their intracellular calcium (Ca2+), which can generate arachidonic acid (AA) from phospholipase A2 (PLA2) which is converted by COX1 (or 3) to prostaglandins (PG) and by P450 epoxygenase to epoxyeicosatrienoic acid (EETs). Both PGs and EETs can relax smooth muscle cells (SMCs) through conversion of adenosine triphosphate (ATP) to cyclic adenosine monophosphate (cAMP). Endothelial cells can increase their intracellular calcium through transient receptor potential (TRP) cation channels, and in response to receptor (R) binding, through IP3-mediated release of calcium from intracellular stores [endoplasmic reticulum (ER)]. Endothelial receptor targets include acetylcholine (ACh), bradykinin (BK), adenosine diphosphate (ADP), ATP, uridine triphosphate (UTP), and adenosine. Receptor binding can activate phospholipase C (PLC) (or PLA2), which via diacyl-glycerol (DAG) can also produce EETs and AA derivatives including prostacyclin (PGI2), both of which can drive SMC relaxation via cAMP, while increased intracellular calcium can drive the production of endothelial nitric oxide (NO), which can affect SMC relaxation through conversion of guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP). Intracellular calcium increases also lead to endothelial hyperpolarization through opening of calcium-dependent potassium channels (KCa). Endothelial hyperpolarization could be coupled to adjacent SMCs through myoendothelial gap junctions (MEGJs) or some other endothelium-derived hyperpolarizing factor (EDHF) such as K+ efflux through endothelial SKCa and IKCa channels by activating KIR and/or the Na+/K+ ATPase. SMC hyperpolarization causes relaxation through inactivation of voltage-dependent calcium channels (Cav). Endothelial hyperpolarization can spread rapidly to adjacent endothelial cells, likely via gap junctions. Pericytes possess many SMC-like properties and could relax in response to NO and PGI2 from astrocytes, neurons, or endothelial cells or in response to neuropeptides such as vasointenstinal peptides (VIPs). Pericytes or astrocytes could also be involved in signaling to endothelial cells. Question marks represent many other potential signaling pathways yet to be identified. Additional abbreviation: NMB, nucleus basalis of Meynert.
Astrocytes
Astrocyte end-feet ensheath diving arterioles, capillaries, and ascending venules throughout the cortex, and almost all astrocytes, have a process in contact with a blood vessel (McCaslin et al. 2011). Astrocyte involvement in neurovascular coupling has become widely accepted following a study that performed in vivo uncaging of calcium in astrocytic end-feet resulting in dilation of adjacent penetrating arterioles. The response to uncaging was blocked by SC-560 (COX1), methyl arachidonyl fluorophosphonate (MAFP) (PLA2), and indomethacin (COX1 and -2) but was unaffected by NS-398 (COX2), L-NAME (NO), MS-PPOH and miconazole (EETS), and caffeine (adenosine). Researchers proposed that a buildup of glutamate (implying local neuronal activity) is sensed by metabotropic glutamate receptors (mGluR5s) on astrocytes, leading to increased intracellular calcium and generation of arachidonic acid derivatives such as prostaglandins capable of causing vasodilation through direct action on perivascular smooth muscle.
However, recent studies have begun to question the role of astrocytes in neurovascular coupling. Although increases in astrocytic intracellular calcium have been observed to correlate to stimulation in many cases (Schummers et al. 2008, Wang et al. 2006, Winship et al. 2007), a recent comprehensive in vivo two-photon study suggested that measurable increases in astrocytic calcium occur after the onset of arteriolar dilation (Nizar et al. 2013). The same study also showed that mice lacking astrocytic inositol trisphosphate (IP3) type-2 receptors (necessary for generating intracellular calcium increases, which in turn would trigger the release of vasoactive arachidonic acid derivatives) still exhibited normal stimulus-evoked functional hyperemia (confirmed by Takata et al. 2013). A further recent study found that astrocytes do not express mGluR5s in the adult brain, the presumed pathway for glutamate-mediated astrocyte activation (Sun et al. 2013). Finally, astrocytes do not directly contact pial arteries above the cortical surface, making astrocytes unlikely mediators of pial artery dilations (Chen et al. 2011, McCaslin et al. 2011). While an additional recent study has countered some of these concerns with new observations of higher speed changes in astrocytic calcium (Lind et al. 2013), the role of astrocytes as the primary mediators of functional hyperemia in the cortex has become less certain.
Pericytes
Pericytes are small cells that wrap around vessels, particularly capillaries, and are involved in vasculogenesis and the blood–brain barrier (Armulik et al. 2005, Winkler et al. 2011). In 2006, Peppiatt et al. showed that pericytes in acute in vitro retina and cerebellar slices evoked capillary vasocon-striction in response to electrical stimulation, GABA, and ATP in the retina, and noradrenaline in the cerebellum, and that this preconstriction could be reversed by glutamate. Another in vitro whole-retina study observed capillary dilation in response to bradykinin and cholinergic agonists, as well as histamine (Schönfelder et al. 1998). Cultured pericytes relax in response to prostacyclin PGI2, NO (endothelial or neuronal), vasoactive intestinal peptide (VIP), and low pH, whereas constriction (wrinkling) has been observed in response to endothelial-derived ET-1, thromboxane A2, and angiotensin II and the catecholamines serotonin, histamine, and noradrenaline (Hamilton et al. 2010). The presence of contractile proteins in pericytes suggests that active (rather than passive) capillary dilation could play a role in functional hyperemia.
However, discriminating active from passive effects in vivo can be very challenging, and the interplay between capillary, arteriolar, and arterial diameters, and their relative influence on vascular resistance and flow is not yet well understood. One recent in vivo study examining pericyte function failed to observe capillary dilations in response to bicuculline despite upstream dilation (Fernández-Klett et al. 2010). Further work is needed to define both the behavior of capillaries during stimulus-evoked functional hyperemia and the potential role of pericytes in mediating capillary dilation.
Neuronal Networks and Interneurons
Afferents from the basal forebrain are known to modulate regional blood flow via acetylcholine (ACh) release (Arnerić et al. 1988, Chédotal et al. 1994, Hotta et al. 2011, Kocharyan et al. 2007, Takata et al. 2013, Vaucher & Hamel 1995). Researchers have also demonstrated the release of vasoactive substances by cortical interneurons including VIP and NO (dilation) and neuropeptide Y (NPY) and somatostatin (SOM) (constriction) (Cauli et al. 2004). It has been proposed that such local and distributed neuronal networks play a role in neurovascular coupling, potentially providing an alternate route to local coupling within the cortex (Piché et al. 2010, Sato & Sato 1995). More conservative models suggest that interneurons may fine-tune local hemodynamics, with astrocytes or pericytes perhaps acting as intermediaries (Attwell & Iadecola 2002, Cauli & Hamel 2010). However, a recent study returned to the possible importance of deep brain regions, showing that norepinephrine release from locus coeruleus afferents could generate large-scale cerebral vasoconstrictions, demonstrating that this effect may play a role in controlling and constraining functional hyperemia (Bekar et al. 2012, Kozberg et al. 2013).
Propagated Vasodilation
Overall, because no single coupling mechanism has been demonstrated incontrovertibly, the prevailing view is that some combination of the mechanisms above must work together to generate the blood flow response (Attwell et al. 2010). However, one component of neurovascular coupling that has been overlooked in recent years is the vasculature itself, and the potential importance of propagated vasodilation in the generation of functional hyperemia.
Retrograde propagation of vasodilation was first explored in the brain in 1997 (Iadecola et al. 1997) but was only recently demonstrated to occur in the cortex during functional hyperemia (Chen et al. 2011, 2014; Tian et al. 2010). Early studies explored propagation of vasodilation in vessels within the hamster cheek pouch but observed speeds that were too slow to account for the properties of the brain’s hemodynamic response (Duling & Berne 1970). However, recent work has identified a more rapidly propagated vasodilation mechanism, mediated via endothelial hyperpolarization (Bagher & Segal 2011, Figueroa & Duling 2009). This hyperpolarization can propagate electrically within the vascular endothelium itself, traveling distances exceeding 1 mm with limited attenuation (Wölfle et al.2011) and causing self-dilation via myoendothelial coupling to encircling smooth muscle cells (SMCs) via myoendothelial gap junctions or some endothelium-derived hyperpolarizing factor (EDHF). This EDHF-type dilation has been shown to be independent of NO and COX pathways, persisting in the presence of indomethacin and L-NAME. A second form of endothelially propagated vasodilation has also been characterized, which is linked to a slowly moving endothelial calcium wave (Tallini et al. 2007) that can propagate up to 500 microns and cause dilation of SMCs via endothelial release of NO and prostanoids such as prostacyclin (whose production in endothelial cells depends primarily on COX1 but can also involve COX2) (de Wit & Griffith 2010).
Both forms of endothelium-dependent vasodilation can be initiated by IP3-mediated increases in intracellular endothelial calcium and can be generated experimentally by iontophoresis of ACh into the vascular endothelium. Short applications (<0.2–1 s) result in vasodilation closely resembling the BOLD HRF (Wölfle et al. 2011). Other identified initiators of EDHF-type propagated vasodilation include adenosine, ATP, UTP, K+ ions, and bradykinin (Marrelli et al. 2003, Rosenblum 1986, Winter & Dora 2007, You et al. 1997). Studies in isolated rat middle cerebral arteries have demonstrated that EDHF-type propagated vasodilation requires an endothelial intracellular calcium increase exceeding 340 nM, whereas slower NO/prostanoid-dependent vasodilation requires a lower threshold of 220 nM (Marrelli 2001). These endothelial and SMC pathways are incorporated into Figure 3.
An Integrated Model?
Despite having been extensively observed and characterized in peripheral vessels, endothelially mediated vasodilation in the brain has not been considered in recent models of functional neurovascular coupling. Incorporating propagated vasodilation presents a potentially more complete picture of how functional hyperemia may be generated (Figure 4). EDHF-type propagated vasodilation provides an elegant mechanism to explain the rapid dilation of distant pial arteries. The possibility that endothelial signaling is initiated at the capillary level, close to active neurons, and travels retrograde along an integrative vascular route would explain the selective recruitment of specific arterial branches and the generation of an optimally localized increase in blood flow (Erinjeri & Woolsey 2002, Sarelius & Pohl 2010). Most neurovascular coupling research in recent years has focused on extravascular interactions with vascular SMCs (Cauli & Hamel 2010, Takano et al. 2006) and mechanisms by which vasoactive prostanoids, EETs, and NO could be generated by perivascular cells such as neurons and astrocytes. Recognizing that these same substances can be released by the vascular endothelium itself removes the requirement for extravascular signaling at the level of diving arterioles. Initiation of functional hyperemia could thus occur at capillary endothelial cells, perhaps via pericytes, astrocytes, or interneurons as intermediaries, or potentially via direct ACh or other neurogenic signaling to endothelial receptors (Arnerić et al. 1988).
We further hypothesize that fast and slow components of propagated vasodilation (Tallini et al. 2007) could provide a physical basis for the nonlinearities of the BOLD response. As shown in Figure 4, rapid and transient EDHF-dependent dilation of distant pial arteries combined with a slower, more spatially restricted NO/prostanoid-dependent dilation sustaining capillary hyperemia could elegantly explain the spatiotemporal patterns illustrated in Figure 2. Differing intracellular calcium thresholds (Marrelli 2001) may trigger fast EDHF-type dilation in response to large, initial neuronal responses, whereas slower NO and prostanoid-dependent dilation may be sustained by lower levels of ongoing activity. The possibly differing latencies and regenerative properties of these fast and slow responses could explain the influence of stimulus frequency, amplitude, and anesthesia on the resultant hemodynamic response. Such two phase models have previously been proposed, based empirically on the spatiotemporal dynamics of the hemodynamic response (Cauli & Hamel 2010, Martindale et al. 2005, Tomita 2007); however, until now these models have not had a defined cellular and mechanistic basis. The involvement of at least two distinct endothelial mechanisms (EDHF, NO and prostanoid-dependent) may explain pharmacology results that have, almost without exception, failed to completely eliminate functional hyperemia through blockade of a single pathway (Lecrux et al. 2011, Takano et al. 2006). These same studies may need to be reinterpreted if the sensitivity of the vascular endothelium to the same pharmacological manipulations was not considered. Although further work is needed to develop this integrative model, one recent study has already confirmed a role for endothelial propagation of vasodilation in the brain during functional hyperemia (Chen et al. 2014). Endothelial involvement in neurovascular coupling would be highly consistent with the established importance of endothelium dependent vasodilation in the rest of the body (Andresen et al. 2006, Marrelli 2001).
A more detailed understanding of neurovascular coupling mechanisms could directly address the question of whether fMRI BOLD responses are truly neurogenic, and explain many other effects that continue to confound fMRI interpretation such as “negative BOLD” (see sidebar, Positive versus Negative BOLD, and Figure 5). Understanding the mechanistic basis of spatiotemporal non-linearities in the BOLD response could permit the design of fMRI stimulation paradigms that might better probe the timing and spatial extent of neuronal activity.
Figure 5.
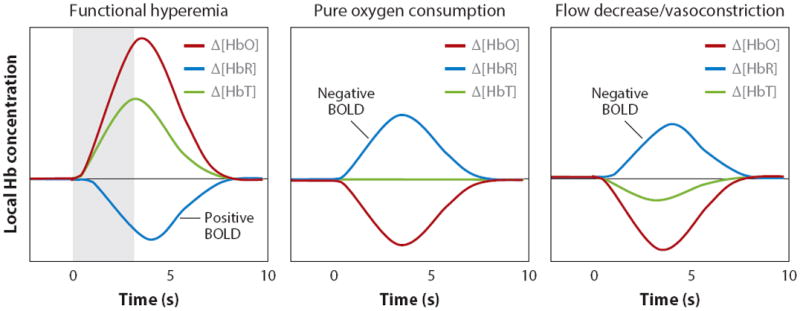
Conditions for positive and negative BOLD. Left: Normal ‘positive BOLD’ in which functional hyperemia increases [HbT] and [HbO]. A decrease in [HbR] occurs because of the wash-in of oxygenated blood. Middle: Possible response when oxygen consumption occurs in the absence of functional hyperemia. This sign of increased metabolic activity would be measured as ‘negative BOLD’. Right: Arteriolar vasoconstriction would decrease [HbT] and [HbO], assuming high arterial oxygen saturation. The consequent decrease in flow would cause deoxygenation, even if oxygen consumption does not change. If there were an associated decrease in the volume of the capillary beds (where oxygen saturation is <98%), the HbR decrease caused by this volume decrease would compete with increasing HbR owing to increasing relative oxygen extraction.
IS NEUROVASCULAR COUPLING ALWAYS NORMAL?
A range of recent reports have explored conditions under which the stimulus-evoked fMRI BOLD response is altered or abnormal, including during postnatal development (Kozberg et al. 2013), in diseases such as Alzheimer’s and stroke (Girouard & Iadecola 2006, Hamilton et al. 2010), diabetes (Mogi & Horiuchi 2011), aging, and drug use (D’Esposito et al. 2003, Schroeter et al. 2007). While most of these studies inferred that altered BOLD responses were a reflection of altered underlying neuronal activity, it is becoming increasingly recognized that in some situations, neurovascular coupling itself could be altered.
The emerging technique of resting state functional connectivity mapping (RS-FCM) has provided additional interesting findings in relation to the diseased brain. RS-FCM records a subject’s brain in the absence of specific stimuli. Resulting BOLD data show seemingly random fluctuations in signal throughout the brain; however, analysis of this data can reveal distinct spatial patterns corresponding to regions where signals are temporally correlated (Fox et al. 2005). These regions are inferred to be functionally connected. Similar functional connectivity networks have been found across subjects; however, abnormal networks have also been identified in a wide range of neurological and psychological disorders including Alzheimer’s, schizophrenia, depression, autism, attention deficit hyperactivity disorder, glioma, and multiple sclerosis (Greicius et al. 2004, Rocca et al. 2010, Zhang & Raichle 2010). It is routinely assumed that RS-FCM is detecting alterations in the neuronal connectivity or processing of the brain in these conditions. However, since fMRI measures are purely hemodynamic in origin, it is important to consider that alterations in apparent functional connectivity networks could, in some cases, be the result of altered neurovascular coupling. Although this possibility retains the potential utility of RS-FCM for clinical diagnosis, the possibility that neurovascular impairment could be a symptom or cause of a pathological state exhibiting abnormal functional connectivity could provide new insights into the condition and could help to elucidate new therapies (Nicolakakis & Hamel 2011).
We should not overlook that intact neurovascular coupling is essential for normal brain function. Neurovascular deficits could feasibly manifest as attentional or psychological in the short term and neurodegenerative in the long term (Roy & Sherrington 1890, Schroeter et al. 2007). Improving our understanding of neurovascular coupling in the resting, developing, and diseased brain is a critical next step. Development of fMRI-based neurovascular assessment tools and neurovascular therapies could be exciting new clinical frontiers.
THE ROLE OF OPTICAL IMAGING IN UNDERSTANDING BOLD.
Oxy- and deoxyhemoglobin (HbO and HbR) exhibit strong, oxygen-dependent optical absorption in the visible to near-infrared (NIR) wavelength range, making it possible to use light to image changes in both [HbO] and [HbR] (Hillman 2007). NIR spectroscopy (NIRS) can acquire signals noninvasively from the human cortex, providing [HbR] measurements equivalent to BOLD fMRI signals (Eggebrecht et al. 2012), in addition to HbO and total hemoglobin (HbT = HbO + HbR). Optical intrinsic signal imaging (OISI) uses visible light to image the exposed cortex, allowing both the vascular dynamics of superficial vessels and local changes in HbO, HbR, and HbT to be recorded in vivo across many species, often in conjunction with electrophysiology (Boorman et al. 2010; Devor et al. 2007, Rayshubskiy et al. 2013, Sirotin et al. 2009, Vanzetta et al. 2005). Laser Doppler and laser speckle flow imaging can measure blood flow dynamics (Dunn et al. 2005, Nielsen & Lauritzen 2001), while fluorescent reporters of neuronal activity including calcium-sensitive dyes (Bouchard et al. 2009), GCaMP (Akerboom et al. 2012), metabolic cofactor flavin adenine dinucleotide (Shibuki et al. 2003), and phosphorescent reporters of intra and extravascular PO2 (Parpaleix et al. 2013, Sakadzic et al. 2010, Vanzetta & Grinvald 1999) can also be imaged dynamically in vivo. In vivo two-photon microscopy permits functional assessment of individual cells and vessels (Nizar et al. 2013). Optical imaging data (as shown in Figures 1 and 2) have played a vital role in shaping our understanding of the origins and mechanisms of the fMRI BOLD signal (Berwick et al. 2008, Chen et al. 2011, Devor et al. 2011, Dunn et al. 2003, Lindauer et al. 2001, Mayhew et al. 1998, Sheth et al. 2004).
THE INITIAL DIP.
Early optical imaging studies reported a transient increase in cortical HbR shortly after stimulus onset (corresponding to an “initial dip” in the fMRI BOLD signal) (Malonek & Grinvald 1996). This “dip” was found to localize to a more discrete region of the cortex than changes at later time points and was interpreted as a direct marker of neuronal oxygen consumption. Optical intrinsic signal imaging studies using 610–630-nm light (assumed to be wholly sensitive to HbR) routinely observed this dip (Chen et al. 2005, Das & Gilbert 1997). As fMRI became more widely used, however, relatively few researchers reported an initial dip in the BOLD signal (Hu & Yacoub 2012, Raichle 1998). Optical imaging studies employing additional wavelengths similarly found less evidence for an initial dip (Chen et al. 2011, Lindauer et al. 2001), instead observing localized, early increases in [HbT] rather than increases in [HbR] (Chen et al. 2011). A recent study in awake behaving primates demonstrated that early HbT increases (assessed at isosbestic wavelength 530 nm) could in fact account for initial absorption increases at 610–630 nm, suggesting contamination and misinterpretation of earlier optical initial dip intrinsic signal data (Sirotin et al. 2009). While the mechanistic basis of rapid, localized [HbT] increases is still being explored, it is likely a result of active neurovascular coupling. Therefore, although an initial dip may still occur in the brain, for example if functional hyperemia is delayed, optical intrinsic signal imaging maps produced using only 610–630 nm are unlikely to represent early oxygen consumption. Studies that have directly measured brief, local decreases in PO2 prior to increases in blood flow (Ances et al. 2001, Li et al. 2010, Parpaleix et al. 2013) are consistent with this conclusion because local oxygen depletion need not have measurable effects on the oxygenation level of red blood cells within adjacent capillaries. Few recent fMRI and optical imaging studies have noted marked initial dips in their data (Chen et al. 2011, Devor et al. 2008, Harris et al. 2010, Hirano et al. 2011, Martin et al. 2013).
POSITIVE VERSUS NEGATIVE BOLD: STIMULUS-EVOKED VERSUS RESTING STATE.
If oxygen consumption were to increase in the absence of functional hyperemia (e.g., if neurovascular coupling were impaired or neuronal activity failed to reach some threshold level), the resulting increase in [HbR] would produce a negative BOLD signal (Figure 5b). Such a region would be classically interpreted as exhibiting a decrease in neuronal activity or even local inhibition (Devor et al. 2007). Vasoconstriction could equally result in an increase in [HbR] and thus negative BOLD (Figure 5c), although here, the resultant changes in [HbO] and [HbR] will depend on the properties of the vascular compartment that changed volume (see Figure 5 caption). It is important to consider that the evidence presented above suggests that vasodilation (leading to positive BOLD) is a triggered response to a specific stimulation event. This could mean that a reduction in neuronal activity would not necessarily be expected to evoke a coupled vasoconstriction by analogous mechanisms. While our above discussions of mechanisms focused on mediators of vasodilation more than constriction, a primary candidate to explain observed vasoconstrictions might be the effect of a global influence such as the norepinephrine network, uniformly driving vasoconstriction in the absence of counteracting vasodilation (Bekar et al. 2012, Kozberg et al. 2013). A reactive and possibly threshold dependent model of functional blood flow regulation raises questions regarding whether resting-state fMRI, in which only baseline fluctuations in the BOLD signal are measured, can be assumed to represent the properties of underlying neuronal activity in the same way as stimulus-evoked responses.
SUMMARY POINTS.
Stimulus-evoked positive BOLD is an actively driven process governed by neurovascular coupling mechanisms and the properties of the brain’s vasculature.
Although a range of candidate mechanisms for neurovascular coupling have been proposed, a consensus has yet to be reached in the field.
Incorporation of propagated vasodilation and endothelial signaling into existing models may provide new explanations for the sensitivities and properties of the fMRI BOLD response.
Resting-state functional connectivity mapping has brought fMRI to clinical populations where neurovascular coupling may be impaired. Abnormal coupling may be the cause of alterations in functional connectivity networks in some pathologies.
The continuing utility and significance of both stimulus evoked and resting-state fMRI BOLD are inextricably tied to improving our understanding of the cellular and vascular bases of neurovascular coupling in health and disease.
Acknowledgments
The author thanks Mariel Kozberg, Mohammed Shaik, Brenda Chen, Aleksandr Rayshubskiy, Costantino Iadecola, David Attwell, Karl Kasischke, Ingrid Sarelius, and Caryl Hill for their comments and contributions. The author acknowledges support from NINDS 1R01NS063226, 1R01NS076628, and R21NS053684, the Human Frontier Science Program (HFSP), NSF (CAREER 0954796 and student fellowships), and NCATS UL1 RR024156.
Footnotes
DISCLOSURE STATEMENT
The author is not aware of any affliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- Akerboom J, Chen T-W, Wardill TJ, Tian L, Marvin JS, et al. Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci. 2012;32:13819–40. doi: 10.1523/JNEUROSCI.2601-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ances BM, Buerk DG, Greenberg JH, Detre JA. Temporal dynamics of the partial pressure of brain tissue oxygen during functional forepaw stimulation in rats. Neurosci Lett. 2001;306:106–10. doi: 10.1016/s0304-3940(01)01868-7. [DOI] [PubMed] [Google Scholar]
- Andresen J, Shafi NI, Bryan RM., Jr Endothelial influences on cerebrovascular tone. J Appl Physiol. 2006;100:318–27. doi: 10.1152/japplphysiol.00937.2005. [DOI] [PubMed] [Google Scholar]
- Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–23. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- Arnerić SP, Honig MA, Milner TA, Greco S, Iadecola C, Reis DJ. Neuronal and endothelial sites of acetylcholine synthesis and release associated with microvessels in rat cerebral cortex: ultrastructural and neurochemical studies. Brain Res. 1988;454:11–30. doi: 10.1016/0006-8993(88)90799-8. [DOI] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, MacVicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–43. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Iadecola C. The neural basis of functional brain imaging signals. Trends Neurosci. 2002;25:621–25. doi: 10.1016/s0166-2236(02)02264-6. [DOI] [PubMed] [Google Scholar]
- Bagher P, Segal SS. Regulation of blood flow in the microcirculation: role of conducted vasodilation. Acta Physiol. 2011;202:271–84. doi: 10.1111/j.1748-1716.2010.02244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekar LK, Wei HS, Nedergaard M. The locus coeruleus-norepinephrine network optimizes coupling of cerebral blood volume with oxygen demand. J Cereb Blood Flow Metab. 2012;32:2135–45. doi: 10.1038/jcbfm.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berwick J, Johnston D, Jones M, Martindale J, Martin C, et al. Fine detail of neurovascular coupling revealed by spatiotemporal analysis of the hemodynamic response to single whisker stimulation in rat barrel cortex. J Neurophysiol. 2008;99:787–98. doi: 10.1152/jn.00658.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinder P, Tsai PS, Kaufhold JP, Knutsen PM, Suhl H, Kleinfeld D. The cortical angiome: an interconnected vascular network with noncolumnar patterns of blood flow. Nat Neurosci. 2013;16:889–97. doi: 10.1038/nn.3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boorman L, Kennerley AJ, Johnston D, Jones M, Zheng Y, et al. Negative blood oxygen level dependence in the rat: a model for investigating the role of suppression in neurovascular coupling. J Neurosci. 2010;30:4285–94. doi: 10.1523/JNEUROSCI.6063-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard MB, Chen BR, Burgess SA, Hillman EMC. Ultra-fast multispectral optical imaging of cortical oxygenation, blood flow, and intracellular calcium dynamics. Opt Express. 2009;17:15670–78. doi: 10.1364/OE.17.015670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boynton GM, Engel SA, Glover GH, Heeger DJ. Linear systems analysis of functional magnetic resonance imaging in human V1. J Neurosci. 1996;16:4207–21. doi: 10.1523/JNEUROSCI.16-13-04207.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, Ransom BR. Astrocyte glycogen and brain energy metabolism. Glia. 2007;55:1263–71. doi: 10.1002/glia.20557. [DOI] [PubMed] [Google Scholar]
- Buxton RB. Dynamic models of BOLD contrast. NeuroImage. 2012;62:953–61. doi: 10.1016/j.neuroimage.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxton RB, Wong EC, Frank LR. Dynamics of blood flow and oxygenation changes during brain activation: the balloon model. Magn Reson Med. 1998;39:855–64. doi: 10.1002/mrm.1910390602. [DOI] [PubMed] [Google Scholar]
- Cardoso MMB, Sirotin YB, Lima B, Glushenkova E, Das A. The neuroimaging signal is a linear sum of neurally distinct stimulus- and task-related components. Nat Neurosci. 2012;15:1298–306. doi: 10.1038/nn.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauli B, Hamel E. Revisiting the role of neurons in neurovascular coupling. Front Neuroenerg. 2010;2:9. doi: 10.3389/fnene.2010.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauli B, Tong X-K, Rancillac A, Serluca N, Lambolez B, et al. Cortical GABA interneurons in neurovascular coupling: relays for subcortical vasoactive pathways. J Neurosci. 2004;24:8940–49. doi: 10.1523/JNEUROSCI.3065-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chédotal A, Umbriaco D, Descarries L, Hartman BK, Hamel E. Light and electron microscopic immunocytochemical analysis of the neurovascular relationships of choline acetyltransferase and vasoactive intestinal polypeptide nerve terminals in the rat cerebral cortex. J Comp Neurol. 1994;343:57–71. doi: 10.1002/cne.903430105. [DOI] [PubMed] [Google Scholar]
- Chen BR, Bouchard MB, McCaslin AFH, Burgess SA, Hillman EMC. High-speed vascular dynamics of the hemodynamic response. NeuroImage. 2011;54:1021–30. doi: 10.1016/j.neuroimage.2010.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BR, Kozberg MG, Bouchard MB, Shaik MA, Hillman EMC. A critical role for the vascular endothelium in functional neurovascular coupling in the brain. J Am Heart Assoc. 2014 doi: 10.1161/JAHA.114.000787. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LM, Friedman RM, Roe AW. Optical imaging of SI topography in anesthetized and awake squirrel monkeys. J Neurosci. 2005;25:7648–59. doi: 10.1523/JNEUROSCI.1990-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culver JP, Siegel AM, Franceschini MA, Mandeville JB, Boas DA. Evidence that cerebral blood volume can provide brain activation maps with better spatial resolution than deoxygenated hemoglobin. NeuroImage. 2005;27:947–59. doi: 10.1016/j.neuroimage.2005.05.052. [DOI] [PubMed] [Google Scholar]
- D’Esposito M, Deouell LY, Gazzaley A. Alterations in the BOLD fMRI signal with ageing and disease: a challenge for neuroimaging. Nat Rev Neurosci. 2003;4:863–72. doi: 10.1038/nrn1246. [DOI] [PubMed] [Google Scholar]
- Das A, Gilbert CD. Distortions of visuotopic map match orientation singularities in primary visual cortex. Nature. 1997;387:594–98. doi: 10.1038/42461. [DOI] [PubMed] [Google Scholar]
- de Wit C, Griffith T. Connexins and gap junctions in the EDHF phenomenon and conducted vasomotor responses. Pflügers Arch - Eur J Physiol. 2010;459:897–914. doi: 10.1007/s00424-010-0830-4. [DOI] [PubMed] [Google Scholar]
- Devor A, Dunn AK, Andermann ML, Ulbert I, Boas DA, Dale AM. Coupling of total hemoglobin concentration, oxygenation, and neural activity in rat somatosensory cortex. Neuron. 2003;39:353–59. doi: 10.1016/s0896-6273(03)00403-3. [DOI] [PubMed] [Google Scholar]
- Devor A, Hillman EMC, Tian P, Waeber C, Teng IC, et al. Stimulus-induced changes in blood flow and 2-deoxyglucose uptake dissociate in ipsilateral somatosensory cortex. J Neurosci. 2008;28:14347–57. doi: 10.1523/JNEUROSCI.4307-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor A, Sakadžić S, Saisan PA, Yaseen MA, Roussakis E, et al. “Overshoot” of O2 is required to maintain baseline tissue oxygenation at locations distal to blood vessels. J Neurosci. 2011;31:13676–81. doi: 10.1523/JNEUROSCI.1968-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor A, Tian P, Nishimura N, Teng IC, Hillman EMC, et al. Suppressed neuronal activity and concurrent arteriolar vasoconstriction may explain negative blood oxygenation level dependent signal. J Neurosci. 2007;27:4452–59. doi: 10.1523/JNEUROSCI.0134-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew PJ, Shih AY, Kleinfeld D. Fluctuating and sensory-induced vasodynamics in rodent cortex extend arteriole capacity. Proc Natl Acad Sci USA. 2011;108:8473–78. doi: 10.1073/pnas.1100428108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duling BR, Berne RM. Propagated vasodilation in the microcirculation of the hamster cheek pouch. Circ Res. 1970;26:163–70. doi: 10.1161/01.res.26.2.163. [DOI] [PubMed] [Google Scholar]
- Dunn AK, Devor A, Bolay H, Andermann M, Moskowitz M, et al. Simultaneous imaging of total cerebral hemoglobin concentration, oxygenation, and blood flow during functional activation. Opt Lett. 2003;28:28–30. doi: 10.1364/ol.28.000028. [DOI] [PubMed] [Google Scholar]
- Dunn AK, Devor A, Dale AM, Boas DA. Spatial extent of oxygen metabolism and hemodynamic changes during functional activation of the rat somatosensory cortex. NeuroImage. 2005;27:279–90. doi: 10.1016/j.neuroimage.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Eggebrecht AT, White BR, Ferradal SL, Chen C, Zhan Y, et al. A quantitative spatial comparison of high-density diffuse optical tomography and fMRI cortical mapping. NeuroImage. 2012;61:1120–28. doi: 10.1016/j.neuroimage.2012.01.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erinjeri JP, Woolsey TA. Spatial integration of vascular changes with neural activity in mouse cortex. J Cereb Blood Flow Metab. 2002;22:353–60. doi: 10.1097/00004647-200203000-00013. [DOI] [PubMed] [Google Scholar]
- Félétou M, Vanhoutte PM. EDHF: new therapeutic targets? Pharmacol Res. 2004;49:565–80. doi: 10.1016/j.phrs.2003.10.017. [DOI] [PubMed] [Google Scholar]
- Fernández-Klett F, Offenhauser N, Dirnagl U, Priller J, Lindauer U. Pericytes in capillaries are contractile in vivo, but arterioles mediate functional hyperemia in the mouse brain. Proc Natl Acad Sci USA. 2010;107:22290–95. doi: 10.1073/pnas.1011321108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa XF, Duling BR. Gap junctions in the control of vascular function. Antioxid Redox Signal. 2009;11:251–66. doi: 10.1089/ars.2008.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MD, Snyder AZ, Vincent JL, Corbetta M, Van Essen DC, Raichle ME. The human brain is intrinsically organized into dynamic, anticorrelated functional networks. Proc Natl Acad Sci USA. 2005;102(27):9673–78. doi: 10.1073/pnas.0504136102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox PT, Raichle ME. Focal physiological uncoupling of cerebral blood flow and oxidative metabolism during somatosensory stimulation in human subjects. Proc Natl Acad Sci USA. 1986;83:1140–44. doi: 10.1073/pnas.83.4.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox PT, Raichle ME, Mintun MA, Dence C. Nonoxidative glucose consumption during focal physiologic neural activity. Science. 1988;241:462–64. doi: 10.1126/science.3260686. [DOI] [PubMed] [Google Scholar]
- Friedland RP, Iadecola C. Roy and Sherrington (1890): a centennial reexamination of “On the Regulation of the Blood-Supply of the Brain”. Neurology. 1991;41:10–14. doi: 10.1212/wnl.41.1.10. [DOI] [PubMed] [Google Scholar]
- Friston KJ, Mechelli A, Turner R, Price CJ. Nonlinear responses in fMRI: the balloon model, Volterra kernels, and other hemodynamics. NeuroImage. 2000;12:466–77. doi: 10.1006/nimg.2000.0630. [DOI] [PubMed] [Google Scholar]
- Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol. 2006;100:328–35. doi: 10.1152/japplphysiol.00966.2005. [DOI] [PubMed] [Google Scholar]
- Greicius MD, Srivastava G, Reiss AL, Menon V. Default mode network activity distinguishes Alzheimer’s disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci USA. 2004;101:4637–42. doi: 10.1073/pnas.0308627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton NB, Attwell D, Hall CN. Pericyte-mediated regulation of capillary diameter: a component of neurovascular coupling in health and disease. Front Neuroenerg. 2010;2:5. doi: 10.3389/fnene.2010.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris S, Jones M, Zheng Y, Berwick J. Does neural input or processing play a greater role in the magnitude of neuroimaging signals? Front Neuroenerg. 2010;2:15. doi: 10.3389/fnene.2010.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeger DJ, Ress D. What does fMRI tell us about neuronal activity? Nat Rev Neurosci. 2002;3:142–51. doi: 10.1038/nrn730. [DOI] [PubMed] [Google Scholar]
- Hertz L. The astrocyte-neuron lactate shuttle: a challenge of a challenge. J Cereb Blood Flow Metab. 2004;24:1241–48. doi: 10.1097/00004647-200411000-00008. [DOI] [PubMed] [Google Scholar]
- Hewson-Stoate N, Jones M, Martindale J, Berwick J, Mayhew J. Further nonlinearities in neurovascular coupling in rodent barrel cortex. NeuroImage. 2005;24:565–74. doi: 10.1016/j.neuroimage.2004.08.040. [DOI] [PubMed] [Google Scholar]
- Hillman EMC. Optical brain imaging in-vivo: techniques and applications from animal to man. J Biomed Opt. 2007;12:051402. doi: 10.1117/1.2789693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman EMC, Devor A, Bouchard M, Dunn AK, Krauss GW, et al. Depth-resolved optical imaging and microscopy of vascular compartment dynamics during somatosensory stimulation. NeuroImage. 2007;35:89–104. doi: 10.1016/j.neuroimage.2006.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano Y, Stefanovic B, Silva AC. Spatiotemporal evolution of the functional magnetic resonance imaging response to ultrashort stimuli. J Neurosci. 2011;31:1440–47. doi: 10.1523/JNEUROSCI.3986-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotta H, Uchida S, Kagitani F, Maruyama N. Control of cerebral cortical blood flow by stimulation of basal forebrain cholinergic areas in mice. J Physiol Sci. 2011;61:201–9. doi: 10.1007/s12576-011-0139-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Yacoub E. The story of the initial dip in fMRI. NeuroImage. 2012;62:1103–8. doi: 10.1016/j.neuroimage.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwan Kim J, Khan R, Thompson JK, Ress D. Model of the transient neurovascular response based on prompt arterial dilation. J Cereb Blood Flow Metab. 2013;33:1429–39. doi: 10.1038/jcbfm.2013.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Yang G, Ebner TJ, Chen G. Local and propagated vascular responses evoked by focal synaptic activity in cerebellar cortex. J Neurophysiol. 1997;78:651–59. doi: 10.1152/jn.1997.78.2.651. [DOI] [PubMed] [Google Scholar]
- Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science. 2004;305:99–103. doi: 10.1126/science.1096485. [DOI] [PubMed] [Google Scholar]
- Kennerley AJ, Harris S, Bruyns-Haylett M, Boorman L, Zheng Y, et al. Early and late stimulus-evoked cortical hemodynamic responses provide insight into the neurogenic nature of neurovascular coupling. J Cereb Blood Flow Metab. 2012;32:468–80. doi: 10.1038/jcbfm.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocharyan A, Fernandes P, Tong X-K, Vaucher E, Hamel E. Specific subtypes of cortical GABA interneurons contribute to the neurovascular coupling response to basal forebrain stimulation. J Cereb Blood Flow Metab. 2007;28:221–31. doi: 10.1038/sj.jcbfm.9600558. [DOI] [PubMed] [Google Scholar]
- Kozberg MG, Chen BR, DeLeo SE, Bouchard MB, Hillman EMC. Resolving the transition from negative to positive blood oxygen level-dependent responses in the developing brain. Proc Natl Acad Sci USA. 2013;110:4380–85. doi: 10.1073/pnas.1212785110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecrux C, Toussay X, Kocharyan A, Fernandes P, Neupane S, et al. Pyramidal neurons are “neurogenic hubs” in the neurovascular coupling response to whisker stimulation. J Neurosci. 2011;31:9836–47. doi: 10.1523/JNEUROSCI.4943-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Freeman RD. Neurometabolic coupling in the lateral geniculate nucleus changes with extended age. J Neurophysiol. 2010;104(1):414–25. doi: 10.1152/jn.00270.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Luo Q, Luo W, Chen S, Cheng H, Zeng S. Spatiotemporal characteristics of cerebral blood volume changes in rat somatosensory cortex evoked by sciatic nerve stimulation and obtained by optical imaging. J Biomed Opt. 2003;8:629–35. doi: 10.1117/1.1609199. [DOI] [PubMed] [Google Scholar]
- Lind BL, Brazhe AR, Jessen SB, Tan FCC, Lauritzen MJ. Rapid stimulus-evoked astrocyte Ca2+ elevations and hemodynamic responses in mouse somatosensory cortex in vivo. Proc Natl Acad Sci USA. 2013;110(48):E4678–87. doi: 10.1073/pnas.1310065110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindauer U, Leithner C, Kaasch H, Rohrer B, Foddis M, et al. Neurovascular coupling in rat brain operates independent of hemoglobin deoxygenation. J Cereb Blood Flow Metab. 2010;30:757–68. doi: 10.1038/jcbfm.2009.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindauer U, Royl G, Leithner C, Kühl M, Gold L, et al. No evidence for early decrease in blood oxygenation in rat whisker cortex in response to functional activation. NeuroImage. 2001;13:988–1001. doi: 10.1006/nimg.2000.0709. [DOI] [PubMed] [Google Scholar]
- Logothetis NK. Neurovascular uncoupling: much ado about nothing. Front Neuroenerg. 2010;2:2. doi: 10.3389/fnene.2010.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetis NK, Pauls J, Augath M, Trinath T, Oeltermann A. Neurophysiological investigation of the basis of the fMRI signal. Nature. 2001;412:150–57. doi: 10.1038/35084005. [DOI] [PubMed] [Google Scholar]
- Malonek D, Grinvald A. Interactions between electrical activity and cortical microcirculation revealed by imaging spectroscopy: implications for functional brain mapping. Science. 1996;272:551–54. doi: 10.1126/science.272.5261.551. [DOI] [PubMed] [Google Scholar]
- Mandeville JB, Marota JJA, Ayata C, Zaharchuk G, Moskowitz MA, et al. Evidence of a cerebrovascular postarteriole Windkessel with delayed compliance. J Cereb Blood Flow Metab. 1999;19:679–89. doi: 10.1097/00004647-199906000-00012. [DOI] [PubMed] [Google Scholar]
- Marrelli SP. Mechanisms of endothelial P2Y1- and P2Y2-mediated vasodilatation involve differential [Ca2+]i responses. Am J Physiol - Heart Circ Physiol. 2001;281:H1759–66. doi: 10.1152/ajpheart.2001.281.4.H1759. [DOI] [PubMed] [Google Scholar]
- Marrelli SP, Eckmann MS, Hunte MS. Role of endothelial intermediate conductance KCa channels in cerebral EDHF-mediated dilations. Am J Physiol - Heart Circ Physiol. 2003;285:H1590–99. doi: 10.1152/ajpheart.00376.2003. [DOI] [PubMed] [Google Scholar]
- Martin C, Martindale J, Berwick J, Mayhew J. Investigating neural-hemodynamic coupling and the hemodynamic response function in the awake rat. NeuroImage. 2006;32:33–48. doi: 10.1016/j.neuroimage.2006.02.021. [DOI] [PubMed] [Google Scholar]
- Martin C, Zheng Y, Sibson NR, Mayhew JEW, Berwick J. Complex spatiotemporal haemodynamic response following sensory stimulation in the awake rat. NeuroImage. 2013;66:1–8. doi: 10.1016/j.neuroimage.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martindale J, Berwick J, Martin C, Kong Y, Zheng Y, Mayhew J. Long duration stimuli and nonlinearities in the neural-haemodynamic coupling. J Cereb Blood Flow Metab. 2005;25:651–61. doi: 10.1038/sj.jcbfm.9600060. [DOI] [PubMed] [Google Scholar]
- Martindale J, Mayhew J, Berwick J, Jones M, Martin C, et al. The hemodynamic impulse response to a single neural event. J Cereb Blood Flow Metab. 2003;23:546–55. doi: 10.1097/01.WCB.0000058871.46954.2B. [DOI] [PubMed] [Google Scholar]
- Mayhew J, Hu D, Zheng Y, Askew S, Hou Y, et al. An evaluation of linear model analysis techniques for processing images of microcirculation activity. NeuroImage. 1998;7:49–71. doi: 10.1006/nimg.1997.0311. [DOI] [PubMed] [Google Scholar]
- McCaslin AFH, Chen BR, Radosevich AJ, Cauli B, Hillman EMC. In-vivo 3D morphology of astrocyte-vasculature interactions in the somatosensory cortex: implications for neurovascular coupling. J Cereb Blood Flow Metab. 2011;31:795–806. doi: 10.1038/jcbfm.2010.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogi M, Horiuchi M. Neurovascular coupling in cognitive impairment associated with diabetes mellitus. Circ J. 2011;75:1042–48. doi: 10.1253/circj.cj-11-0121. [DOI] [PubMed] [Google Scholar]
- Ngai AC, Winn HR. Pial arteriole dilation during somatosensory stimulation is not mediated by an increase in CSF metabolites. Am J Physiol Heart Circ Physiol. 2002;282:H902–7. doi: 10.1152/ajpheart.00128.2001. [DOI] [PubMed] [Google Scholar]
- Nicolakakis N, Hamel E. Neurovascular function in Alzheimer’s disease patients and experimental models. J Cereb Blood Flow Metab. 2011;31:1354–70. doi: 10.1038/jcbfm.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen AN, Lauritzen M. Coupling and uncoupling of activity-dependent increases of neuronal activity and blood flow in rat somatosensory cortex. J Physiol. 2001;533:773–85. doi: 10.1111/j.1469-7793.2001.00773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizar K, Uhlirova H, Tian P, Saisan PA, Cheng Q, et al. In vivo stimulus-induced vasodilation occurs without IP3 receptor activation and may precede astrocytic calcium increase. J Neurosci. 2013;33:8411–22. doi: 10.1523/JNEUROSCI.3285-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa S, Lee TM, Kay AR, Tank DW. Brain magnetic resonance imaging with contrast dependent on blood oxygenation. Proc Natl Acad Sci USA. 1990;87:9868–72. doi: 10.1073/pnas.87.24.9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpaleix A, Houssen YG, Charpak S. Imaging local neuronal activity by monitoring PO2 transients in capillaries. Nat Med. 2013;19:241–46. doi: 10.1038/nm.3059. [DOI] [PubMed] [Google Scholar]
- Paulson OB, Hasselbalch SG, Rostrup E, Knudsen GM, Pelligrino D. Cerebral blood flow response to functional activation. J Cereb Blood Flow Metab. 2009;30:2–14. doi: 10.1038/jcbfm.2009.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, et al. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia. 2007;55:1251–56. doi: 10.1002/glia.20528. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA. 1994;91:10625–29. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppiatt CM, Howarth C, Mobbs P, Attwell D. Bidirectional control of CNS capillary diameter by pericytes. Nature. 2006;443:700–4. doi: 10.1038/nature05193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piché M, Uchida S, Hara S, Aikawa Y, Hotta H. Modulation of somatosensory-evoked cortical blood flow changes by GABAergic inhibition of the nucleus basalis of Meynert in urethane-anaesthetized rats. J Physiol. 2010;588:2163–71. doi: 10.1113/jphysiol.2010.187633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers WJ, Hirsch IB, Cryer PE. Effect of stepped hypoglycemia on regional cerebral blood flow response to physiological brain activation. Am J Physiol - Heart Circ Physiol. 1996;270:H554–59. doi: 10.1152/ajpheart.1996.270.2.H554. [DOI] [PubMed] [Google Scholar]
- Raichle ME. Behind the scenes of functional brain imaging: a historical and physiological perspective. Proc Natl Acad Sci USA. 1998;95:765–72. doi: 10.1073/pnas.95.3.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayshubskiy A, Wojtasiewicz TJ, Mikell CB, Bouchard MB, Timerman D, et al. Direct, intraoperative observation of ~0.1 Hz hemodynamic oscillations in awake human cortex: implications for fMRI. NeuroImage. 2013;87:323–31. doi: 10.1016/j.neuroimage.2013.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocca MA, Valsasina P, Absinta M, Riccitelli G, Rodegher ME, et al. Default-mode network dysfunction and cognitive impairment in progressive MS. Neurology. 2010;74:1252–59. doi: 10.1212/WNL.0b013e3181d9ed91. [DOI] [PubMed] [Google Scholar]
- Rosenblum WI. Endothelial dependent relaxation demonstrated in vivo in cerebral arterioles. Stroke. 1986;17:494–97. doi: 10.1161/01.str.17.3.494. [DOI] [PubMed] [Google Scholar]
- Roy CS, Sherrington CS. On the regulation of the blood supply of the brain. J Physiol. 1890;11:85–108. doi: 10.1113/jphysiol.1890.sp000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakadzić S, Roussakis E, Yaseen MA, Mandeville ET, Srinivasan VJ, et al. Two-photon high-resolution measurement of partial pressure of oxygen in cerebral vasculature and tissue. Nat Meth. 2010;7:755–59. doi: 10.1038/nmeth.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarelius I, Pohl U. Control of muscle blood flow during exercise: local factors and integrative mechanisms. Acta Physiol. 2010;199:349–65. doi: 10.1111/j.1748-1716.2010.02129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A, Sato Y. Cholinergic neural regulation of regional cerebral blood flow. Alzheimer Dis Assoc Disord. 1995;9:28–38. doi: 10.1097/00002093-199505000-00007. [DOI] [PubMed] [Google Scholar]
- Schönfelder U, Hofer A, Paul M, Funk RHW. In situ observation of living pericytes in rat retinal capillaries. Microvasc Res. 1998;56:22–29. doi: 10.1006/mvre.1998.2086. [DOI] [PubMed] [Google Scholar]
- Schroeter ML, Cutini S, Wahl MM, Scheid R, Yves von Cramon D. Neurovascular coupling is impaired in cerebral microangiopathy—an event-related Stroop study. NeuroImage. 2007;34:26–34. doi: 10.1016/j.neuroimage.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Schummers J, Yu H, Sur M. Tuned responses of astrocytes and their influence on hemodynamic signals in the visual cortex. Science. 2008;320:1638–43. doi: 10.1126/science.1156120. [DOI] [PubMed] [Google Scholar]
- Schurr A. Lactate: the ultimate cerebral oxidative energy substrate? J Cereb Blood Flow Metab. 2006;26:142–52. doi: 10.1038/sj.jcbfm.9600174. [DOI] [PubMed] [Google Scholar]
- Sheth SA, Nemoto M, Guiou M, Walker M, Pouratian N, Toga AW. Linear and nonlinear relationships between neuronal activity, oxygen metabolism, and hemodynamic responses. Neuron. 2004;42:347–55. doi: 10.1016/s0896-6273(04)00221-1. [DOI] [PubMed] [Google Scholar]
- Shibuki K, Hishida R, Murakami H, Kudoh M, Kawaguchi T, et al. Dynamic imaging of somatosensory cortical activity in the rat visualized by flavoprotein autofluorescence. J Physiol. 2003;549:919–27. doi: 10.1113/jphysiol.2003.040709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirotin YB, Das A. Anticipatory haemodynamic signals in sensory cortex not predicted by local neuronal activity. Nature. 2009;457:475–79. doi: 10.1038/nature07664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirotin YB, Hillman EMC, Bordier C, Das A. Spatiotemporal precision and hemodynamic mechanism of optical point spreads in alert primates. Proc Natl Acad Sci USA. 2009;106:18390–95. doi: 10.1073/pnas.0905509106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanovic B, Hutchinson E, Yakovleva V, Schram V, Russell JT, et al. Functional reactivity of cerebral capillaries. J Cereb Blood Flow Metab. 2007;28:961–72. doi: 10.1038/sj.jcbfm.9600590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, McConnell E, Pare J-F, Xu Q, Chen M, et al. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science. 2013;339:197–200. doi: 10.1126/science.1226740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Tian G-F, Peng W, Lou N, Libionka W, et al. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–67. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- Takata N, Nagai T, Ozawa K, Oe Y, Mikoshiba K, Hirase H. Cerebral blood flow modulation by basal forebrain or whisker stimulation can occur independently of large cytosolic Ca2+ signaling in astrocytes. PLoS One. 2013;8:e66525. doi: 10.1371/journal.pone.0066525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallini YN, Brekke JF, Shui B, Doran R, Hwang S-M, et al. Propagated endothelial Ca2+ waves and arteriolar dilation in vivo: measurements in Cx40BAC–GCaMP2 transgenic mice. Circ Res. 2007;101:1300–9. doi: 10.1161/CIRCRESAHA.107.149484. [DOI] [PubMed] [Google Scholar]
- Tian P, Teng IC, May LD, Kurz R, Lu K, et al. Cortical depth-specific microvascular dilation underlies laminar differences in blood oxygenation level-dependent functional MRI signal. Proc Natl Acad Sci USA. 2010;107:15246–51. doi: 10.1073/pnas.1006735107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita M. Blood flow control in the brain: possible biphasic mechanism of functional hyperemia. Asian Biomed. 2007;1(1):17–32. [Google Scholar]
- Turner R. How much cortex can a vein drain? Downstream dilution of activation-related cerebral blood oxygenation changes. NeuroImage. 2002;16:1062–67. doi: 10.1006/nimg.2002.1082. [DOI] [PubMed] [Google Scholar]
- Vanzetta I, Grinvald A. Increased cortical oxidative metabolism due to sensory stimulation: implications for functional brain imaging. Science. 1999;286:1555–58. doi: 10.1126/science.286.5444.1555. [DOI] [PubMed] [Google Scholar]
- Vanzetta I, Hildesheim R, Grinvald A. Compartment-resolved imaging of activity-dependent dynamics of cortical blood volume and oximetry. J Neurosci. 2005;25:2233–44. doi: 10.1523/JNEUROSCI.3032-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaucher E, Hamel E. Cholinergic basal forebrain neurons project to cortical microvessels in the rat: electron microscopic study with anterogradely transported Phaseolus vulgaris leucoagglutinin and choline acetyltransferase immunocytochemistry. J Neurosci. 1995;15:7427–41. doi: 10.1523/JNEUROSCI.15-11-07427.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villringer A, Them A, Lindauer U, Einhäupl K, Dirnagl U. Capillary perfusion of the rat brain cortex. An in vivo confocal microscopy study. Circ Res. 1994;75:55–62. doi: 10.1161/01.res.75.1.55. [DOI] [PubMed] [Google Scholar]
- Wang X, Lou N, Xu Q, Tian G, Peng WG, et al. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat Neurosci. 2006;9:816–23. doi: 10.1038/nn1703. [DOI] [PubMed] [Google Scholar]
- Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14:1398–405. doi: 10.1038/nn.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winship IR, Plaa N, Murphy TH. Rapid astrocyte calcium signals correlate with neuronal activity and onset of the hemodynamic response in vivo. J Neurosci. 2007;27:6268–72. doi: 10.1523/JNEUROSCI.4801-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter P, Dora KA. Spreading dilatation to luminal perfusion of ATP and UTP in rat isolated small mesenteric arteries. J Physiol. 2007;582:335–47. doi: 10.1113/jphysiol.2007.135202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf T, Lindauer U, Villringer A, Dirnagl U. Excessive oxygen or glucose supply does not alter the blood flow response to somatosensory stimulation or spreading depression in rats. Brain Res. 1997;761:290–99. doi: 10.1016/s0006-8993(97)00354-5. [DOI] [PubMed] [Google Scholar]
- Wölfle SE, Chaston DJ, Goto K, Sandow SL, Edwards FR, Hill CE. Non-linear relationship between hyperpolarisation and relaxation enables long distance propagation of vasodilatation. J Physiol. 2011;589:2607–23. doi: 10.1113/jphysiol.2010.202580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yablonskiy DA, Ackerman JJH, Raichle ME. Coupling between changes in human brain temperature and oxidative metabolism during prolonged visual stimulation. Proc Natl Acad Sci USA. 2000;97:7603–8. doi: 10.1073/pnas.97.13.7603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeşilyurt B, Uğurbil K, Uludağ K. Dynamics and nonlinearities of the BOLD response at very short stimulus durations. Magn Reson Imaging. 2008;26:853–62. doi: 10.1016/j.mri.2008.01.008. [DOI] [PubMed] [Google Scholar]
- You J, Johnson TD, Childres WF, Bryan RM., Jr Endothelial-mediated dilations of rat middle cerebral arteries by ATP and ADP. Am J Physiol - Heart Circ Physiol. 1997;273:H1472–77. doi: 10.1152/ajpheart.1997.273.3.H1472. [DOI] [PubMed] [Google Scholar]
- Zhang D, Raichle ME. Disease and the brain’s dark energy. Nat Rev Neurol. 2010;6:15–28. doi: 10.1038/nrneurol.2009.198. [DOI] [PubMed] [Google Scholar]
- Zhao F, Wang P, Hendrich K, Ugurbil K, Kim S-G. Cortical layer-dependent BOLD and CBV responses measured by spin-echo and gradient-echo fMRI: insights into hemodynamic regulation. NeuroImage. 2006;30:1149–60. doi: 10.1016/j.neuroimage.2005.11.013. [DOI] [PubMed] [Google Scholar]