

Abstract
To assess the consequences of locus ceruleus (LC) degeneration and subsequent noradrenaline (NA) deficiency in early Alzheimer's disease (AD), mice overexpressing mutant amyloid precursor protein and presenilin-1 (APP/PS1) were crossed with Ear2(−/−) mice that have a severe loss of LC neurons projecting to the hippocampus and neocortex. Testing spatial memory and hippocampal long-term potentiation revealed an impairment in APP/PS1 Ear2(−/−) mice, whereas APP/PS1 or Ear2(−/−) mice showed only minor changes. These deficits were associated with distinct synaptic changes including reduced expression of the NMDA 2A subunit and increased levels of NMDA receptor 2B in APP/PS1 Ear2(−/−) mice. Acute pharmacological replacement of NA by l-threo-DOPS partially restored phosphorylation of β-CaMKII and spatial memory performance in APP/PS1 Ear2(−/−) mice. These changes were not accompanied by altered APP processing or amyloid β peptide (Aβ) deposition. Thus, early LC degeneration and subsequent NA reduction may contribute to cognitive deficits via CaMKII and NMDA receptor dysfunction independent of Aβ and suggests that NA supplementation could be beneficial in treating AD.
Keywords: Alzheimer, locus ceruleus, memory, neurodegeneration, noradrenaline
Introduction
Alzheimer's disease (AD) is the most common cause of dementia characterized by the deposition of the amyloid β peptide (Aβ) in senile plaques and progressing memory loss (Querfurth and LaFerla, 2010). The degeneration of the locus ceruleus (LC), located within the brain stem, is an early event in AD pathogenesis (Forno, 1966). Loss of LC neurons has even been observed in patients suffering from mild cognitive impairment that are at high risk for developing AD over the next decade (Grudzien et al., 2007). The LC consists of approximately 1700 neurons and is divided in a dorsal and a ventral part (Swanson, 1976). The dorsal part projects mostly into the neocortex, where it serves as the sole source of noradrenaline (NA). In addition to direct synaptic transmission, NA influences distant neurons, glial cells, and blood vessels when released from extrasynaptic varicosities (Marien et al., 2004).
Functionally, the LC is involved in the modulation of general arousal, selective attention, learning and memory, stress reactivity, behavioral adaptation, sleep architecture, and inflammation (Sara, 2009). During normal aging, LC neurons and cerebral NA concentrations decline by 25–50%. Some memory tasks may be rescued by activation of the noradrenergic system (Marien et al., 2004). LC neuron loss correlates well with Aβ plaque load and the severity of dementia and is more abundant and corresponds better to AD progression than the degeneration of the nucleus basalis of Meynert (Zarow et al., 2003). There is evidence that LC changes may be an initial event in AD pathogenesis, since LC cell loss does not result from nonspecific retrograde degenerative processes (Marien et al., 2004).
One animal model, to investigate the impact of the LC on AD-like neuropathology, is the application of the neurotoxin N-[2-chloroethyl]-N-ethyl-2-bromobenzylamine (DSP-4) causing the specific destruction of LC neurons (Heneka et al., 2006; Kalinin et al., 2007; Hurko et al., 2010; Jardanhazi-Kurutz et al., 2010). Since LC neurons produce, besides NA, a variety of other neuromodulators, DSP-4 lesions may not isolate the contribution of NA loss itself. Recently, we described a model where NA production was completely abolished by the deletion of the enzyme dopamine β-hydroxylase catalyzing the conversion of dopamine to NA, revealing cognitive deficits and changes in learning-associated proteins (Hammerschmidt et al., 2013).
Nevertheless, the consequences of early LC degeneration on AD remain unclear (Weinshenker, 2008). Since mouse models of AD show only very limited deficits regarding the LC, and do not appear until long after profound forebrain neuropathology is evident (O'Neil et al., 2007), we aimed to assess consequences of early LC loss.
We therefore crossed amyloid precursor protein and presenilin-1 (APP/PS1) mice with mice lacking the orphan nuclear receptor Ear2 (also known as Nr2f6), which is necessary for the development of LC neurons in mice. Loss of Ear2 primarily affects its dorsal division, which projects to the neocortex and hippocampus (Warnecke et al., 2005). Wild-type (wt), APP/PS1, Ear2(−/−), and APP/PS1 Ear2(−/−) mice were tested for spatial memory, synaptic plasticity, APP processing, inflammatory reaction, and synaptic protein abundance (Fig. 1A).
Figure 1.
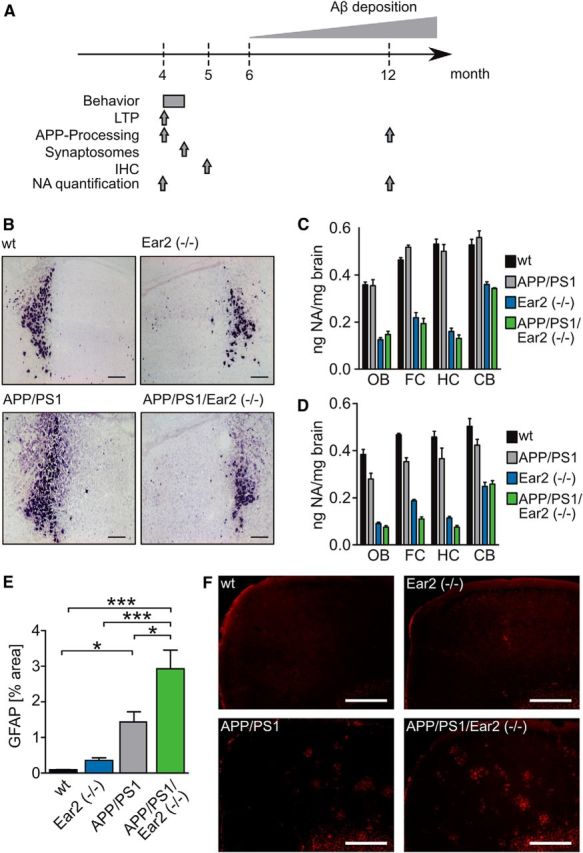
LC degeneration and noradrenergic deficit in LC projection areas. A, Time scheme of the different experiment and analysis conducted. IHC, Immunocytochemistry. B, TH in situ hybridization of wt, APP/PS1, Ear2(−/−), and APP/PS1 Ear2(−/−) mice. Loss of Ear2 causes a strong reduction of TH-positive neurons in the dorsal part of the LC (scale bars, 200 μm). C, Determination of NA level in the olfactory bulb (OB), frontal cortex (FC), hippocampus (HC), and cerebellum (CB) by HPLC in 4-month-old wt, APP/PS1, Ear2(−/−), and APP/PS1 Ear2(−/−) mice (mean of n = 4–5 ± SEM; two-way ANOVA, Bonferroni's post-test; see Table 1). D, Same as B but in 12-month-old mice (mean of n = 4–5 ± SEM; two-way ANOVA, Bonferroni's post-test; see Table 1). E, Quantification of the GFAP-positive area in the frontal cortex of wt, Ear2(−/−), APP/PS1, and APP/PS1 Ear2(−/−) mice at 5 months of age (mean of n = 4–5 mice ± SEM; 4 sections per mouse; one-way ANOVA followed by Tukey's post hoc test, *p < 0.05, ***p < 0.001). F, Representative images of GFAP-stained wt, Ear2(−/−), APP/PS1, and APP/PS1 Ear2(−/−) mice analyzed in E (scale bars, 500 μm).
Materials and Methods
Animals.
Hemizygous APP/PS1 mice expressing a chimeric mouse/human APP with the Swedish mutation and a human PS1 Δexon 9 mutation (Jankowsky et al., 2001) were studied. Ear2(−/−) mice were generated as described previously (Warnecke et al., 2005). All animals were maintained on a C57BL/6 background. Mice were housed in groups under standard conditions at a temperature of 22°C and a 12 h light/dark cycle with ad libitum access to standard food (Altromin) and water. Gender-balanced groups were used for the in situ hybridization, the NA quantification, and the behavioral experiments. For all other experiments, either female or male mice were used. For dissection, mice were killed by deep anesthesia using isoflurane and perfused with 20 ml of PBS through the heart using a peristaltic pump. The skull was removed, and the brain was split into hemispheres. The cerebellum was removed, and the remaining forebrain was immediately frozen on dry ice and stored at −80°C. The other hemisphere was fixed in 4% paraformaldehyde. Animal care and handling were performed according to the Declaration of Helsinki and approved by local ethical committees.
Quantification of NA concentration by HPLC.
Specific brain regions were dissected, weighed, and homogenized in 100, 200, or 300 μl of sample buffer (0.1 m perchloric acid, 0.01% EDTA, and 0.08 ng/μl 3,4-dihydroxybenzylamine, used as an internal standard). Twenty microliters of the supernatant was injected onto a C-18 reverse-phase column (Spherisorb ODS-I, 5 μm, 250 × 4.6 mm; Techlab). The isocratic mobile phase consisted of 0.15 m chloroacetic acid, 0.2 mm EDTA, 0.86 mm sodium octyl sulfate, 6% acetonitrile, and 2.5% tetrahydrofuran, pH 3, at a flow rate of 1.2 ml/min. NA was detected by an electrochemical detector (Coulochem II, model 5200A; ESA).
In situ hybridization for tyrosine hydroxylase.
The procedure for in situ hybridization has been described previously (Yaylaoglu et al., 2005). The following tyrosine hydroxylase (TH)-specific (NM_009377) PCR primers were used: forward 5′-GATTGCAGAGATTGCCTTCC- 3′ and reverse 5′-CCTGTGGGTGGTACCCTATG-3′. The 2100 Bioanalyzer (Agilent) was used for quantification.
Drugs and treatments.
For the l-threo-3,4-dihydroxyphenylserine (DOPS) rescue experiments, male APP/PS1 Ear2(−/−) mice were given injections each day of the Morris water maze (MWM) and were tested 5 h after injection, when brain NA levels peak (Thomas et al., 1998). The injection contained DOPS (500 mg/kg, s.c.), benserizide (125 mg/kg, s.c.), and ascorbic acid (2 mg/ml). Control mice were treated with the corresponding volume of vehicle solution. Because DOPS is converted to NA by the enzyme aromatic acid decarboxylase (AADC) and benserazide is an AADC inhibitor that cannot cross the blood–brain barrier, DOPS plus benserazide treatment selectively restores NA in the brain (Thomas et al., 1998; Murchison et al., 2004).
Behavioral analysis.
Mice were singly housed in the testing room under a reversed light/dark cycle for 2 weeks before the start of behavioral experiments. Approximately equal numbers of male and female mice at the age of 4 months were used, before plaque deposition. Behavior was scored by an observer blind to the genotype and treatment groups. For the open-field (OF) exploration, mice were placed in the center of the dimly lit (20–30 lux) open field arena (61 × 61 × 61 cm). Movements of the animals were tracked by an automatic monitoring system (Ethovision 3; Noldus) for 5 min. The area was virtually divided into a center (40 cm edge lengths), a corridor (7.5 cm along the walls), and four corner squares (with 10 cm edge lengths). The experiment was repeated on 3 consecutive days. The MWM test was conducted in a circular tank (diameter, 1 m) filled with opacified water at 24°C. The water basin was dimly lit (20–30 lux) and surrounded by a white curtain. The maze was virtually divided into four quadrants. In one of the quadrants, a hidden platform (15 × 15 cm) was present 1.5 cm below the water surface. Mice were trained to find the platform by means of three intermaze cues placed asymmetrically at the walls of the tank. They were let into the water in a quasi-random fashion to prevent strategy learning. Mice were allowed to search for the platform for 40 s. Mice that did not reach the platform in the allotted time were placed manually on it and stayed there for 15 s. Mice received four training trials per day for 8 consecutive days. Movements of the mice were recorded by a tracking system (Ethovision 3; Noldus). On the ninth day, the mice were tested for latency to find the cued/visual platform.
Long-term potentiation in vitro.
Electrophysiological recordings were performed on brain slices from 4-month-old male mice. Brains were sliced sagitally in 400 μm sections using a Vibratome (Integraslice 7550 PSDS; Camden Instruments). Hippocampal slices were isolated and maintained at 29°C in an oxygen-enriched atmosphere in artificial CSF (aCSF; containing 124 mm NaCl, 4 mm KCl, 1.24 mm NaH2PO4, 1.3 mm MgSO4, 26 mm NaHCO3, 10 mm d-glucose, and 1 mm CaCl2) for 30 min, followed by incubation in aCSF at elevated CaCl2 (2 mm) for an additional 30 min. Extracellular field EPSPs (fEPSPs) were recorded in the pyramidal cell layer of the cornu ammonis 1 (CA1) region using glass microelectrodes (resistance, 0.7–2 MΩ) filled with aCSF. The stimulating electrode was placed in the CA2 region within the Schaffer collaterals. Stimuli were applied at a constant current at a duration of 0.1 ms. The initial slope of evoked fEPSPs was measured as an index of synaptic strength. Basal synaptic transmission was assessed by plotting the stimulus intensity (in microamperes) against the peak slope of the evoked fEPSP to generate input–output relationships. Paired-pulse facilitation (PPF) was performed by a two-stimuli protocol using stimuli intervals of 30, 50, 75, and 100 ms at 30% maximal stimulation intensity. For long-term potentiation (LTP) experiments, a 20 min baseline was recorded with an interpulse interval of 1 min at a stimulus intensity that evoked a response of ∼30% of maximum fEPSP. The LTP was induced by a theta burst consisting of four trains of 10 pulses at 100 Hz separated by 200 ms.
Extraction of brain lysates.
Extraction of forebrains from male mice was performed as described previously (Kummer et al., 2011). Briefly, snap-frozen forebrain hemispheres were homogenized in PBS, and protein was extracted in RIPA (25 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.5% sodium desoxycholate, 1% NP-40, and 0.1% SDS). After centrifugation at 100,000 × g for 30 min at 4°C, the resulting supernatant (RIPA-soluble fraction) was saved, and the pellet was sonified in 25 mm Tris-HCl, pH 7.5, and 2% SDS (RIPA-insoluble fraction).
Synaptosomal preparation.
Forebrains of male mice were homogenized in 9 volumes of 0.32 m sucrose buffer (50 mm Tris acetate, 1 mm EDTA, 5 mm pyrophosphate, 5 mm NaF, 2 mm Na2VO4, 1 mm PMSF, and protease inhibitor mixture, pH 7.4) using a Teflon-glass homogenizer (0.15 mm clearance) at 700 rpm, and homogenates were centrifuged at 800 × g for 5 min. The supernatant was layered on a discontinuous sucrose density gradient consisting of 1.4 and 1.0 m sucrose and centrifuged at 54,000 × g for 90 min, and the synaptosomal fraction was recovered from the interface of the 1.4 m/1.0 m sucrose layers, diluted with H2O, and centrifuged at 17,000 × g for 15 min.
Western blot analysis of brain extracts and synaptosomal fractions.
Protein concentrations were determined using the BCA Protein Assay kit (Pierce). Protein samples were separated by 4–12% NuPAGE (Invitrogen) using MES or 3-(N-morpholino) propanesulfonic acid buffer and transferred to nitrocellulose membranes. For detection of Aβ, blots were boiled for 5 min in water. Aβ was detected using antibody 6E10 (catalog #SIG-39300-500 RRID:AB_10175291; Covance), APP and C-terminal fragment (CTF) using antibody C-terminal 140 (kind gift from Dr. J. Walter, University of Bonn, Bonn, Germany), BDNF using antiserum sc-546 (catalog #sc-546 RRID:AB_630940; Santa Cruz Biotechnology), PS1 using antibody PS1-NT (catalog #529591 RRID:AB_2172922; EMD Biosciences) or polyclonal antibody 3109 (kind gift from Dr. J. Walter; Prager et al., 2007). Tubulin (catalog #e7 RRID:AB_528499; Developmental Studies Hybridoma Bank) was used as a loading control. In synaptosomal fractions, protein levels of CaMKII (catalog #sc-9035 RRID:AB_634551; Santa Cruz Biotechnology), CaMKII T286 phosphorylation (catalog #PPS002 RRID:AB_2290672; R&D Systems), NR2A (catalog #454580-10UG RRID:AB_10683116; Merck), and NR2B (catalog #454582-10UG RRID:AB_10690211; Merck) were assessed. RabGDP dissociation inhibitor (GDI; catalog #130 011 RRID:AB_1966443; Synaptic Systems) was used as an internal control. Primary antibody incubation was followed by incubation with appropriate horseradish peroxidase-conjugated or infrared dye-coupled secondary antibodies. Immunoreactivity was detected by enhanced chemiluminescence reaction (Millipore), and luminescence intensities were analyzed using the Chemidoc XRS documentation system (Bio-Rad). In case of infrared dye-coupled secondary antibodies, signals were detected using the Odyssey Clx imaging system (Li-COR).
Sandwich ELISA for Aβ.
32 Quantitative determination of Aβ1–40 and Aβ1–42 from brain extracts was performed using the human Amyloid Aβ1–40 and Aβ1–42 ELISA kit (Millipore) according to the manufacturer's instructions. Samples were cleared by centrifugation at 100,000 × g for 20 min and diluted to meet the concentration range of the standard curve.
Histochemistry.
Serial sagittal cryosections (40 μm) of male mice were immunostained free-floating using antiserum against GFAP (catalog #Z0334 RRID:AB_10013382, 1:800; Dako). For that, sections were washed in PBS, permeabilized using PBST (PBST and 0.1% Tx-100), and blocked for 1 h in 20% goat serum in PBST. Primary antibodies were added in 10% goat serum on PBST for 18 h at 4°C while gently rocking. Secondary antibodies were added to 10% goat serum in PBST for 1.5 h at room temperature. Sections were mounted with ImmuMount (Thermo Fisher Scientific) on superfrost slides. Sections were analyzed using a BX61 microscope equipped with a disk-spinning unit (Olympus). Quantitative assessment of the stained areas was done using the Fiji-ImageJ 1.48p software bundle (ImageJ, RRID:nif-0000-30467).
Statistical analysis.
Behavioral data of the MWM test were analyzed by two-way ANOVA with repeated measurements (independent between subjects variables: APP/PS1 transgene, Ear2(−/−) and DOPS treatment; dependent between subjects variables: latency or distance; independent within subject variable: day). If a significant effect of the independent variables was found in the ANOVA, the Student's t test for intergroup comparison was performed subsequently. Statistical evaluation was done with SPSS 17 (RRID:rid_000042; IBM) and Prism 5.03 (RRID:rid_000081; GraphPad).
Results
NA reduction in LC projection areas by Ear2(−/−)
Ear2(−/−) caused a 70% loss of LC neurons (Warnecke et al., 2005), predominantly in the dorsal division (Fig. 1B; Warnecke et al., 2005), resulting in a 60–75% decrease of NA levels in projection areas like the hippocampus, frontal cortex, cerebellum, and olfactory bulb in 4- and 12-month-old Ear2(−/−) and APP/PS1 Ear2(−/−) mice (Fig. 1C,D; Table 1). We also observed a minor reduction of NA in the 12-month-old APP/PS1 mice compared with wild-type mice, demonstrating that the APP/PS1 transgenes already have a negative impact on NA production in the LC (Fig. 1D). Since NA has been described to suppress inflammation, we looked for indications of astrogliosis, an early symptom for neuroinflammation. Detection of GFAP by immunohistochemistry at 5 months of age revealed that APP/PS1 and APP/PS1 Ear2(−/−) mice have increased GFAP-positive areas over wt and Ear2(−/−) mice (Fig. 1E,F). In addition, APP/PS1 Ear2(−/−) mice show an even higher percentage of GFAP-positive areas compared with APP/PS1 mice, suggesting that partial loss of the LC and decreased NA concentration favor the occurrence of astrogliosis.
Table 1.
Statistical analysis of noradrenaline concentrations in 4- and 12-month-old mice (see Fig. 1C,D)
| Comparison | Brain area | 4 month p valuea | 12 month p valuea |
|---|---|---|---|
| wt vs APP/PS1 | OB | n.s. | <0.01 |
| FC | n.s. | <0.01 | |
| HC | n.s. | <0.05 | |
| CB | n.s. | <0.05 | |
| wt vs Ear2(−/−) | OB | <0.001 | <0.001 |
| FC | <0.001 | <0.001 | |
| HC | <0.001 | <0.001 | |
| CB | <0.001 | <0.001 | |
| wt vs APP/PS1/Ear2(−/−) | OB | <0.001 | <0.001 |
| FC | <0.001 | <0.001 | |
| HC | <0.001 | <0.001 | |
| CB | <0.001 | <0.001 | |
| APP/PS1 vs Ear2(−/−) | OB | <0.001 | <0.001 |
| FC | <0.001 | <0.001 | |
| HC | <0.001 | <0.001 | |
| CB | <0.001 | <0.001 | |
| APP/PS1 vs APP/PS1/Ear2(−/−) | OB | <0.001 | <0.001 |
| FC | <0.001 | <0.001 | |
| HC | <0.001 | <0.001 | |
| CB | <0.001 | <0.001 | |
| Ear2(−/−) vs APP/PS1/Ear2(−/−) | OB | n.s. | n.s. |
| FC | n.s. | n.s. | |
| HC | n.s. | n.s. | |
| CB | n.s. | n.s. |
aTwo-way ANOVA followed by Bonferroni's post hoc test.
OB, Olfactory bulb; FC, frontal cortex; HC, hippocampus; CB, cerebellum.
Ear2(−/−) increases cognitive deficits in APP/PS1 mice
Testing the exploration behavior in the OF test, Ear2(−/−) did not affect horizontal activity (Fig. 2A). The presence of the APP/PS1 transgene induced a slight hyperactivity (Fig. 2A). No differences in center time were observed between the genotypes, suggesting that Ear(−/−) deficiency does not influence anxiety-like behavior in this setup.
Figure 2.

Spatial memory is affected by APP/PS1 transgene and Ear2(−/−) mice. A) Open-field behavior. wt, APP/PS1, Ear2(−/−), and APP/PS1 Ear2(−/−) mice were in tested for time spent in the center, distance traveled, time spent in corridor, and time spent in the corners (mean of n = 10 ± SEM; one-way ANOVA, Tukey's post hoc test). B, C, Latency (B) and distance (C) to reach the platform in the Morris water maze test. The bar graphs represent the area under the curve for latency and distance to the platform for each genotype [mean of n = 10 ± SEM; Student's t test, *p < 0.05 wt vs APP/PS1, #p < 0.05 wt vs Ear2(−/−), ′′p > 0.01, ′′′p > 0.001 wt vs APP/PS1 Ear2(−/−)]. D, Result from the visual platform trial conducted at day 9. Shown is the mean latency to reach the cued platform (mean of n = 10 ± SEM; one-way ANOVA, Tukey's post hoc test, *p < 0.05).
In the MWM test (Fig. 2B,C), all mice learned to find a hidden platform by spatial navigation. Two-way ANOVA with repeated measurements revealed differences for distance for the factors Ear2(−/−) and APP/PS1 and for latency for the factor APP/PS1 (Table 2). However, Ear2(−/−) and APP/PS1 Ear2(−/−) mice had increased escape latencies (Fig. 2B) and longer distances to platform (Fig. 2C) compared with wild-type controls. There was no difference between APP/PS1 and APP/PS1 Ear2(−/−). Testing the ability to find the visual platform on day 9 revealed a difference between wt and APP/PS1 Ear(−/−) mice, but not between the APP/PS1 transgenic or Ear2(−/−) groups (Fig. 2 D).
Table 2.
Statistical analysis of the Morris water maze
| Factor | F value (time) | p value (time) | F value (distance) | p value (distance) |
|---|---|---|---|---|
| Ear2(−/−) | F(1,54) = 5.67 | #p < 0.05 | F(1,53) = 4.32 | #p < 0.05 |
| APP/PS1 | F(1,54) = 2.99 | p > 0.05 | F(1,53) = 10.42 | **p < 0.01 |
Data are intersubject effects of two-way ANOVA with repeated measurements.
Loss of Ear2 impairs long-term but not short-term synaptic plasticity
Since synaptic plasticity is considered one of the major cellular mechanisms that underlies spatial memory formation, we recorded hippocampal LTP of CA1 neurons along the Schaffer collateral in vitro. fEPSPs were recorded in the pyramidal cell layer. Peak slopes of fEPSP recordings, averaged before and after LTP induction, were recorded (Fig. 3A). Early and late stages of LTP were assessed at 10 min (T1) and 60 min (T2) after induction, respectively. APP/PS1 and Ear2(−/−) mice showed a modest but significant reduction in the early (T1) and late (T2) components of LTP, compared with wt mice, but the ability to form LTP was strongly decreased in the APP/PS1 and APP/PS1 Ear2(−/−) mice (Fig. 3B,C).
Figure 3.

Impaired long-term and unaltered short-term plasticity in hippocampal CA1 in APP/PS1 transgenic and Ear2(−/−) mice. A, Representative fEPSP responses obtained before and after an LTP-inducing stimulus. Bottom plots depict the fEPSP slope as a percentage of baseline (bl) as the mean of 5 min. B, C, Quantification of the magnitude of LTP. The fEPSP slope as a percentage of the baseline is depicted at time point 1 (T1), 10 min after decay of post-tetanic potentiation, and time point 2 (T2), 60 min after LTP induction [mean of n = 5 ± SEM; Student's t test; wt vs APP/PS1: *p < 0.05 for T1, ***p < 0.001 for T2; wt vs Ear2(−/−): #p < 0.05 for T1 and T2; Ear2(−/−) vs APP/PS1 Ear2(−/−): **p < 0.01 for T1 and T2; APP/PS1 vs APP/PS1 Ear2(−/−): #p < 0.05 for T1 and T2; wt vs APP/PS1 Ear2(−/−): ′′′p < 0.001 for T1 and T2]. D, Ratio of paired fEPSP at interpulse intervals of 30, 50, 75, and 100 ms (mean of n = 5 ± SEM; Student's t test).
Next, we assessed short-term synaptic plasticity by PPF in the CA1 region using varying interstimuli intervals. Changes in PPF during synaptic potentiation can indicate a change in the expression of synaptic proteins at the presynaptic site. PPF was similar in all genotypes tested (Fig. 3D). Furthermore, PPF was the same before and after LTP induction, suggesting that suppression of LTP was caused by changes at the postsynaptic site.
Ear2 modulates plasticity-related proteins at the synapse
To reveal why Ear2 deficiency causes reduced spatial memory, synaptic fractions from all mice groups were analyzed for changes in learning-associated proteins (Fig. 4). We observed that phosphorylation levels at threonine 268 of α- and β-Ca2+/calmodulin-dependent protein kinase (CaMKII) were lower in APP/PS1 Ear2(−/−) independent on the overall expression of α- or β-CaMKII (Fig. 4); even so, this effect did not reach statistical significance. In addition, we found that Ear2 deficiency caused decreased expression of the NMDAR subunit NR2A in APP/PS1 Ear2(−/−) mice, whereas the NR2B subunit was upregulated (Fig. 4A,D,E).
Figure 4.
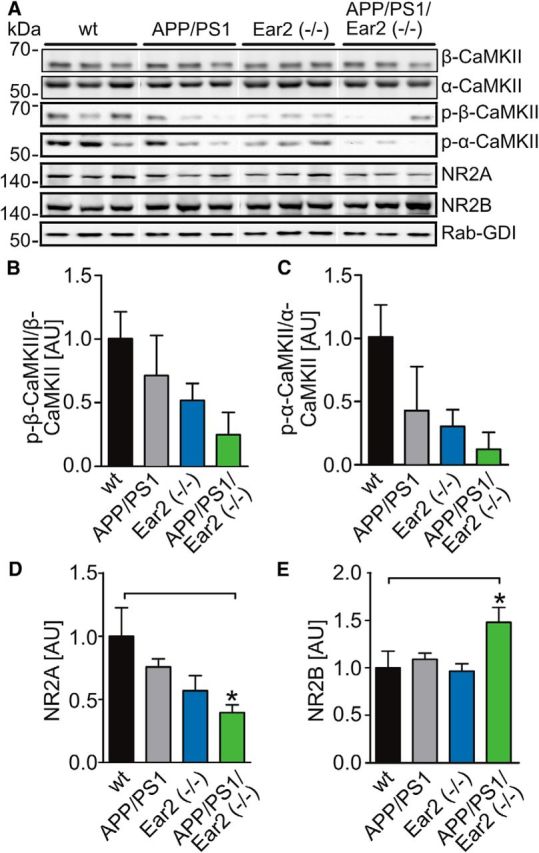
Changes of learning-associated proteins in Ear2(−/−) and APP/PS1 Ear2(−/−) mice. A, Western blot of forebrain synaptosomal preparations from wt, Ear2(−/−), APP/PS1, and APP/PS1 Ear2(−/−) mice for the learning-associated proteins with antibodies against α-/β-CaMKII, p-Thr286-α-/p-Thr286-β-CaMKII, NR2A, and NR2B. Rab-GDI was used as a loading control. Pictures were derived from the same blot exposure and were rearranged for presentation purposes. B, Densitometric evaluation of the phosphorylation of Thr286 of β-CaMKII normalized to total β-CaMKII levels. C, Densiometric evaluation of the phosphorylation of Thr286 of α-CaMKII normalized to total α-CaMKII levels NR2A (D) and of NR2B (E) calculated with respect to the expression of the specific target in the wild-type group (mean of n = 4 ± SEM; one-way ANOVA followed by Newman–Keuls post hoc test, *p < 0.05).
DOPS partially rescues spatial memory impairment caused by loss of Ear2
To test whether acute NA restoration can revert spatial memory impairment in APP/PS1 Ear2(−/−) mice, the NA precursor DOPS was coadministered with benserazide, an inhibitor of the peripheral AADC. Since benserazide does not pass the blood–brain barrier, but DOPS, NA production was restricted to the brain (Thomas et al., 1998). MWM sessions were performed 5 h after DOPS treatment, a time when cerebral NA levels peak (Thomas et al., 1998). DOPS-treated APP/PS1 Ear2(−/−) mice showed improved performance in the MWM concerning distance compared with untreated APP/PS1 Ear2(−/−) littermates (Fig. 5A,B).
Figure 5.

DOPS partially rescues spatial memory impairment induced by noradrenaline depletion. The Morris water maze test after DOPS treatment is described. A, B, Latency (A) and distance (B) to escape to a hidden platform on 8 consecutive days. The bar graphs represent the mean of time and the mean of distance to the platform for each genotype from day 5 to day 8 (mean of n = 14 ± SEM; Student's t test, *p < 0.05). C, Western blot analysis of forebrain synaptosomal preparations from APP/PS1 Ear2(−/−) treated with DOPS versus APP/PS1 Ear2(−/−) for changes in α-CamKII and β-CamKII and for their phosphorylation state at Thr286. Pictures were derived from the same blot exposure and were rearranged for presentation purposes. D, Densitometric evaluation of C (mean of n = 4 ± SEM; Student's t test, *p < 0.05).
DOPS treatment increases phosphorylation of CaMKII in APP/PS1 Ear2(−/−) mice
To determine whether NA restoration changes β-p-CaMKII and NR2A/NR2B levels, we investigated potential alterations in APP/PS1 Ear2(−/−) mice treated with DOPS compared with APP/PS1 Ear2(−/−) mice. We observed that phosphorylation of β-CaMKII at threonine 286 was increased after DOPS treatment in APP/PS1 Ear2(−/−) mice (Fig. 5C,D).
Aβ deposition is not influenced by Ear2(−/−)
Because Aβ have been reported to affect synaptic function, LTP, and memory performance (Selkoe, 2008) and LC ablation by the neurotoxin DSP-4 increases Aβ levels (Heneka et al., 2006), APP processing and Aβ levels were analyzed at 4 months of age (before Aβ deposition in APP/PS1 mice; Fig. 6A–F) and at 12 months of age (an age at which Aβ deposition is high in APP/PS1 mice; Fig. 6H–L). APP expression levels were unchanged between wild-type and Ear2(−/−) mice and APP/PS1 and APP/PS1 Ear2(−/−) mice (Fig. 6B). There were no changes in the steady-state levels of β-CTF or α-CTF in the APP/PS1 transgenic groups (Fig. 6C,D). We did observe an increase in α-CTF between the wild-type and the Ear2(−/−) mice (Fig. 6C). Aβ levels were unaffected by the loss of Ear2(−/−) in the RIPA- and SDS-soluble pool in the APP/PS1 transgenic mice (Fig. 6F,G). Since Aβ levels are unchanged, another LC-derived factor might be responsible for the memory loss caused by Ear2(−/−). We therefore determined the expression of BDNF, an important factor for memory formation, in these mice. Interestingly, we found that BDNF levels reduced in the APP/PS1 Ear2(−/−) mice (Fig. 6G).
Figure 6.
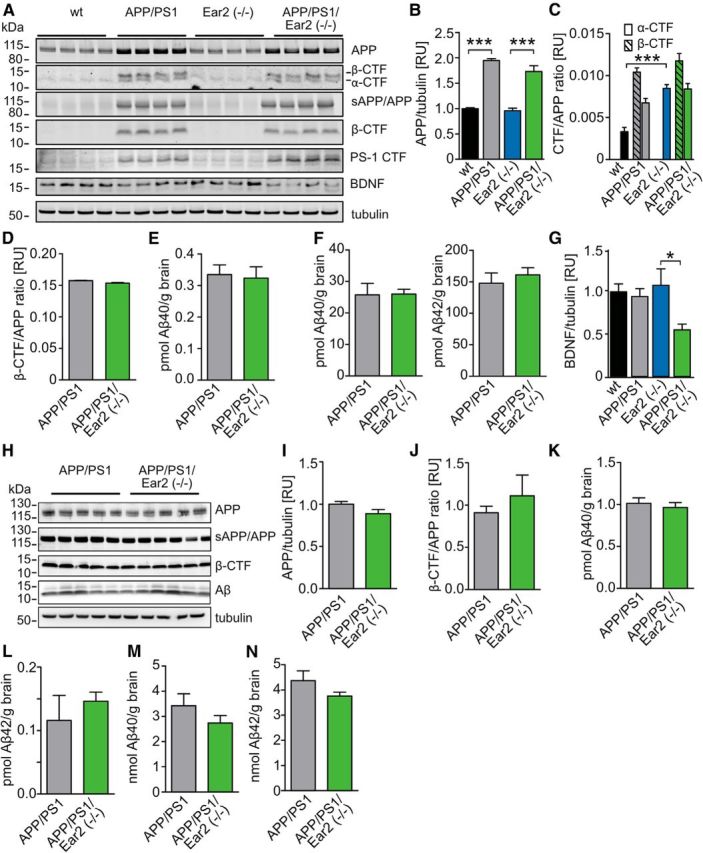
Analysis of APP processing in 4- and 12-month-old mice. A, Four-month-old wt, Ear2(−/−), APP/PS1, and APP/PS1 Ear2(−/−) mice were analyzed by Western blot using antiserum 140 for APP and CTF, antibody 6E10 for sAPP/APP and β-CTF, antiserum 3109 for PS1, antiserum sc-546 for BDNF, and antibody E7 for tubulin. B, Quantification of the APP levels from A normalized to tubulin (mean of n = 4 ± SEM; one-way ANOVA, ***p < 0.001). C, Quantification of the CTF/APP ratios from A using APP C-terminal antiserum 140 (mean of n = 4 ± SEM; one-way ANOVA, ***p < 0.001). D, Quantification of the β-CTF/APP ratios from A using antibody 6E10 (mean of n = 4 ± SEM; Student's t test). E, F, ELISA analysis of RIPA-extractable Aβ40 (E) and SDS-extractable (F) Aβ40 and Aβ42 (mean of n = 6 ± SEM; Student's t test). G) Quantification of the BDNF levels from A normalized to tubulin (mean of n = 4 ± SEM; one-way ANOVA, ***p < 0.05). H, Twelve-month-old wt, APP/PS1, Ear2(−/−), and APP/PS1 Ear2(−/−) mice were analyzed by Western blot using antiserum 140 for APP, antibody 6E10 for sAPP/APP and β-CTF, and antibody E7 for tubulin. I) Quantification of the APP levels from H normalized to tubulin (mean of n = 5–6 ± SEM; Student's t test). J, Quantification of the β-CTF/APP ratios from H (mean of n = 5–6 ± SEM; Student's t test). K, L, ELISA analysis of RIPA-extractable Aβ40 (K) and Aβ42 (L; mean of n = 5–6 ± SEM; Student's t test). M, N, ELISA analysis of SDS-extractable Aβ40 (M) and Aβ42 (N; mean of n = 5–6 ± SEM; Student's t test).
Analysis in 12-month-old animals revealed no changes in APP expression (Fig. 6H,I), CTF/APP ratio (Fig. 6H,J), and Aβ levels in the RIPA- and SDS-soluble fractions (Fig. 6K–N) either. These results suggest that the increase in Aβ levels observed previously in DSP-4-treated mice is caused by pathways initiated in response to the acute loss of LC neurons or NA, a phenomenon that has not been mimicked by the present models, possibly because of compensatory mechanisms in genetic models of chronic NA deficiency.
Discussion
NA is involved in the behavioral changes in response to imperatives in the environment by modulating most stages of memory. We therefore hypothesized that perturbation of the NA system, caused by loss of LC neurons, could interfere with synaptic plasticity and memory formation (Sara, 2009). Previously, the selective destruction of the LC by the neurotoxin DSP-4 has been used to model LC degeneration in murine models of Alzheimer's disease (Wenk et al., 2003; Heneka et al., 2006; Kalinin et al., 2007). DSP-4-induced LC degeneration has the disadvantage that the resulting neuronal cell death may create an inflammatory reaction within the LC and in the projection areas. We therefore aimed to generate a genetic model that mimics early neuronal LC loss in a mouse model of Alzheimer's disease by cross-breeding APP/PS1 with Ear2(−/−) mice. In this model, we observed that loss of Ear2 causes a severe drop of NA in all investigated brain regions. In addition, we observed increased expression of GFAP in APP/PS1 Ear2(−/−) mice compared with APP/PS1 mice in the frontal cortex even before the onset of plaque deposition, suggesting that Ear2(−/−) affects the inflammatory status of astrocytes in LC projection areas. This increase of GFAP expression is in accordance with what has been observed for DSP-4 lesions in the APP23 model (Heneka et al., 2006).
Our behavioral data provide evidence that before plaque deposition, mice carrying the APP/PS1 transgene or lacking Ear2 were modestly impaired in the MWM, whereas APP/PS1 Ear2(−/−) mice, reflecting the AD patient's situation, showed an additive effect and therefore a robust loss of spatial memory formation. Loss of spatial memory in mouse models based on APP overexpression has been described in detail previously (Zhang et al., 2006) and may be explained by findings that soluble Aβ oligomers interfere with cognition even before plaque deposition starts (Cleary et al., 2005; Comery et al., 2005; Saura et al., 2005). The results of the present study indicate that LC degeneration and the subsequent reduction of NA levels in its projection areas may worsen spatial memory, either on its own or in combination with Aβ. Spatial memory deficits caused by Ear2 deficiency were partially rescued by NA supplementation with l-threo DOPS (Thomas et al., 1998; Heneka et al., 2010). These results indicate that the deficits were caused, at least in part, by the lack of NA at the time of the test, rather than by alterations of other factors generated by the LC. These data are in line with previous reports demonstrating a DSP-4-induced increase in working and reference memory errors observed in APP23 transgenic mice (Sara, 2009).
We found that both Ear2(−/−) and the presence of APP/PS1 individually impaired LTP. In line with the behavioral observations, both factors additively impaired hippocampal LTP, which was almost completely abolished in APP/PS1 Ear2(−/−) mice. In addition, we observed similar effects on LTP in a model of complete-loss NA (Hammerschmidt et al., 2013), which implies that NA acts as the critical factor that modulates these events. We are aware that the LC produces several other factors that might influence memory formation (e.g., BDNF). Nevertheless, our results indicate a critical role of the LC for synaptic plasticity, particularly in the presence of AD-like neuropathology.
Effects of Ear2 deficiency on spatial memory and synaptic plasticity were already observed in preplaque, 4-month-old APP/PS1 mice. We hypothesize that reduced NA levels, along with soluble Aβ species, cause alterations of learning-associated synaptic proteins at this age and thereby account for the impaired synaptic plasticity. Since paired-pulse facilitation was unaffected by both factors, APP/PS1 transgene expression and Ear2 deficiency, we conclude that postsynaptic, but not presynaptic, protein changes are responsible for the reduction of LTP.
Analyzing postsynaptic proteins of APP/PS1 mice, we found that APP/PS1 Ear2(−/−) mice showed decreased NR2A levels and increased NR2B levels. These subunits take part in the assembly of tetrameric NMDARs consisting of two obligatory NR1 subunits and two other subunits, mostly heteromeric or homomeric NR2A and NR2B (Yashiro and Philpot, 2008). These data imply that Ear2 deficiency may alter NMDAR composition, which is critical for receptor gating and conductance. NR1/NR2B channels have greater conductance (Zhang et al., 2006) and a higher affinity to bind CaMKII compared with NR2A-containing receptors (Lisman et al., 2002). Interestingly, impaired LTP in CaMKII mutant mice was accompanied by a selective reduction of NR2A at the synapse without affecting NR2B localization (Park et al., 2008).
We also found that absence of Ear2 decreased CaMKII subunit phosphorylation at threonine 286; even so, these data did not reach the level of statistical significance. CaMKII phosphorylation at threonine 286 represents a critical step for LTP induction (Yashiro and Philpot, 2008), since CaMKII threonine 286 point mutations abolish hippocampal LTP formation in mice (Lamsa et al., 2007). Interestingly, restoring NA by DOPS treatment improved behavior and was associated with increased β-CaMKII phosphorylation, suggesting that replenishment of cerebral NA levels can partly reverse the negative impact of LC deficits.
In summary, we speculate that the LTP and memory deficits observed in APP/PS1 Ear2(−/−) mice are caused, at least in part, by a reduction in CaMKII phosphorylation and a shift from NR2A- to NR2B-containing NMDA receptors. However, one should be careful to link these two events too tightly, since DOPS treatment increased phosphorylation of β-CaMKII and partly rescued spatial memory in APP/PS1 Ear2(−/−) without any impact on NMDAR subunit composition. The loss of NA innervations will likely cause multiple changes at the synapse; nevertheless, a reduction of phosphorylated CaMKII levels has been found in frontal cortices and hippocampi of AD patients (Amada et al., 2005).
We and others have previously shown that acute reductions of forebrain NA levels increased Aβ deposition (Heneka et al., 2006; Hammerschmidt et al., 2013). Surprisingly, we did not observe any changes of APP processing or Aβ levels in 4- or 12-month-old APP/PS1 Ear2(−/−) mice. Multiple differences between the DSP-4 and the Ear2(−/−) models may account for this observation. Thus, the genetic LC loss may have induced changes within the affected LC projection areas that compensate for the loss of NA. Alternatively, DSP-4-caused acute LC cell death may lead to the induction of inflammatory brain responses, which increases Aβ deposition and/or impairs its clearance.
Additional studies are needed to elucidate when and why LC dysfunction and degeneration occurs in AD. If memory decline in AD is aggravated by LC degeneration and subsequent loss of NA, replenishment of NA levels may provide therapeutic benefit.
Footnotes
This work was supported by grants from the INMiND project and the Cluster of Excellence “ImmunoSensation” to M.T.H., the Interdisciplinary Center of Clinical Research (IZKF; HEN3/003/06) to M.T.H. and H.-C.P., the Institute for the Study of Aging to D.W., and a pilot grant from the Emory University Alzheimer's Disease Research Center (PHS AG025688) to D.W., M.T.H. is a member of the DFG Cluster of Excellence “ImmunoSensation.” We thank the Dainippon Sumitomo Pharmaceutical Company for the generous gift of l-threo DOPS. We also thank C. Jerome and C. Hülsmann for excellent technical assistance.
The authors declare no competing financial interests.
References
- Amada N, Aihara K, Ravid R, Horie M. Reduction of NR1 and phosphorylated Ca2+/calmodulin-dependent protein kinase II levels in Alzheimer's disease. Neuroreport. 2005;16:1809–1813. doi: 10.1097/01.wnr.0000185015.44563.5d. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Comery TA, Martone RL, Aschmies S, Atchison KP, Diamantidis G, Gong X, Zhou H, Kreft AF, Pangalos MN, Sonnenberg-Reines J, Jacobsen JS, Marquis KL. Acute gamma-secretase inhibition improves contextual fear conditioning in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2005;25:8898–8902. doi: 10.1523/JNEUROSCI.2693-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forno LS. Pathology of parkinsonism. J Neurosurg. 1966;24:266–271. [Google Scholar]
- Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM. Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer's disease. Neurobiol Aging. 2007;28:327–335. doi: 10.1016/j.neurobiolaging.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Hammerschmidt T, Kummer MP, Terwel D, Martinez A, Gorji A, Pape HC, Rommelfanger KS, Schroeder JP, Stoll M, Schultze J, Weinshenker D, Heneka MT. Selective loss of noradrenaline exacerbates early cognitive dysfunction and synaptic deficits in APP/PS1 mice. Biol Psychiatry. 2013;73:454–463. doi: 10.1016/j.biopsych.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Ramanathan M, Jacobs AH, Dumitrescu-Ozimek L, Bilkei-Gorzo A, Debeir T, Sastre M, Galldiks N, Zimmer A, Hoehn M, Heiss WD, Klockgether T, Staufenbiel M. Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J Neurosci. 2006;26:1343–1354. doi: 10.1523/JNEUROSCI.4236-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Nadrigny F, Regen T, Martinez-Hernandez A, Dumitrescu-Ozimek L, Terwel D, Jardanhazi-Kurutz D, Walter J, Kirchhoff F, Hanisch UK, Kummer MP. Locus ceruleus controls Alzheimer's disease pathology by modulating microglial functions through norepinephrine. Proc Natl Acad Sci U S A. 2010;107:6058–6063. doi: 10.1073/pnas.0909586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurko O, Boudonck K, Gonzales C, Hughes ZA, Jacobsen JS, Reinhart PH, Crowther D. Ablation of the locus coeruleus increases oxidative stress in tg-2576 transgenic but not wild-type mice. Int J Alzheimers Dis. 2010;2010:864625. doi: 10.4061/2010/864625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/S1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Jardanhazi-Kurutz D, Kummer MP, Terwel D, Vogel K, Dyrks T, Thiele A, Heneka MT. Induced LC degeneration in APP/PS1 transgenic mice accelerates early cerebral amyloidosis and cognitive deficits. Neurochem Int. 2010;57:375–382. doi: 10.1016/j.neuint.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Kalinin S, Gavrilyuk V, Polak PE, Vasser R, Zhao J, Heneka MT, Feinstein DL. Noradrenaline deficiency in brain increases beta-amyloid plaque burden in an animal model of Alzheimer's disease. Neurobiol Aging. 2007;28:1206–1214. doi: 10.1016/j.neurobiolaging.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Kummer MP, Hermes M, Delekarte A, Hammerschmidt T, Kumar S, Terwel D, Walter J, Pape HC, König S, Roeber S, Jessen F, Klockgether T, Korte M, Heneka MT. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron. 2011;71:833–844. doi: 10.1016/j.neuron.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Lamsa K, Irvine EE, Giese KP, Kullmann DM. NMDA receptor-dependent long-term potentiation in mouse hippocampal interneurons shows a unique dependence on Ca(2+)/calmodulin-dependent kinases. J Physiol. 2007;584:885–894. doi: 10.1113/jphysiol.2007.137380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Marien MR, Colpaert FC, Rosenquist AC. Noradrenergic mechanisms in neurodegenerative diseases: a theory. Brain Res Brain Res Rev. 2004;45:38–78. doi: 10.1016/j.brainresrev.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Murchison CF, Zhang XY, Zhang WP, Ouyang M, Lee A, Thomas SA. A distinct role for norepinephrine in memory retrieval. Cell. 2004;117:131–143. doi: 10.1016/S0092-8674(04)00259-4. [DOI] [PubMed] [Google Scholar]
- O'Neil JN, Mouton PR, Tizabi Y, Ottinger MA, Lei DL, Ingram DK, Manaye KF. Catecholaminergic neuronal loss in locus coeruleus of aged female dtg APP/PS1 mice. J Chem Neuroanat. 2007;34:102–107. doi: 10.1016/j.jchemneu.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CS, Elgersma Y, Grant SG, Morrison JH. Alpha-isoform of calcium-calmodulin-dependent protein kinase II and postsynaptic density protein 95 differentially regulate synaptic expression of NR2A- and NR2B-containing N-methyl-d-aspartate receptors in hippocampus. Neuroscience. 2008;151:43–55. doi: 10.1016/j.neuroscience.2007.09.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prager K, Wang-Eckhardt L, Fluhrer R, Killick R, Barth E, Hampel H, Haass C, Walter J. A structural switch of presenilin 1 by glycogen synthase kinase 3β-mediated phosphorylation regulates the interaction with β-catenin and its nuclear signaling. J Biol Chem. 2007;282:14083–14093. doi: 10.1074/jbc.M608437200. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Sara SJ. The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci. 2009;10:211–223. doi: 10.1038/nrn2573. [DOI] [PubMed] [Google Scholar]
- Saura CA, Chen G, Malkani S, Choi SY, Takahashi RH, Zhang D, Gouras GK, Kirkwood A, Morris RG, Shen J. Conditional inactivation of presenilin 1 prevents amyloid accumulation and temporarily rescues contextual and spatial working memory impairments in amyloid precursor protein transgenic mice. J Neurosci. 2005;25:6755–6764. doi: 10.1523/JNEUROSCI.1247-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson LW. The locus coeruleus: a cytoarchitectonic, Golgi and immunohistochemical study in the albino rat. Brain Res. 1976;110:39–56. doi: 10.1016/0006-8993(76)90207-9. [DOI] [PubMed] [Google Scholar]
- Thomas SA, Marck BT, Palmiter RD, Matsumoto AM. Restoration of norepinephrine and reversal of phenotypes in mice lacking dopamine beta-hydroxylase. J Neurochem. 1998;70:2468–2476. doi: 10.1046/j.1471-4159.1998.70062468.x. [DOI] [PubMed] [Google Scholar]
- Warnecke M, Oster H, Revelli JP, Alvarez-Bolado G, Eichele G. Abnormal development of the locus coeruleus in Ear2(Nr2f6)-deficient mice impairs the functionality of the forebrain clock and affects nociception. Genes Dev. 2005;19:614–625. doi: 10.1101/gad.317905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinshenker D. Functional consequences of locus coeruleus degeneration in Alzheimer's disease. Curr Alzheimer Res. 2008;5:342–345. doi: 10.2174/156720508784533286. [DOI] [PubMed] [Google Scholar]
- Wenk GL, McGann K, Hauss-Wegrzyniak B, Rosi S. The toxicity of tumor necrosis factor-alpha upon cholinergic neurons within the nucleus basalis and the role of norepinephrine in the regulation of inflammation: implications for Alzheimer's disease. Neuroscience. 2003;121:719–729. doi: 10.1016/S0306-4522(03)00545-1. [DOI] [PubMed] [Google Scholar]
- Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaylaoglu MB, Titmus A, Visel A, Alvarez-Bolado G, Thaller C, Eichele G. Comprehensive expression atlas of fibroblast growth factors and their receptors generated by a novel robotic in situ hybridization platform. Dev Dyn. 2005;234:371–386. doi: 10.1002/dvdy.20441. [DOI] [PubMed] [Google Scholar]
- Zarow C, Lyness SA, Mortimer JA, Chui HC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol. 2003;60:337–341. doi: 10.1001/archneur.60.3.337. [DOI] [PubMed] [Google Scholar]
- Zhang Y-J, Xu YF, Liu YH, Yin J, Li HL, Wang Q, Wang JZ. Peroxynitrite induces Alzheimer-like tau modifications and accumulation in rat brain and its underlying mechanisms. FASEB J. 2006;20:1431–1442. doi: 10.1096/fj.05-5223com. [DOI] [PubMed] [Google Scholar]