

Abstract
Nitric oxide produced by inducible nitric oxide synthase (iNOS) contributes to ischemic brain injury, but the cell types expressing iNOS and mediating tissue damage have not been elucidated. To examine the relative contribution of iNOS in resident brain cells and peripheral leukocytes infiltrating the ischemic brain, we used bone marrow (BM) chimeric mice in which the middle cerebral artery was occluded and infarct volume was determined 3 days later. iNOS−/− mice engrafted with iNOS+/+ BM exhibited larger infarcts (44±2 mm3; n=13; Mean±SE) compared to autologous transplanted iNOS−/− mice (24±3 mm3; n=10; p<0.01), implicating blood-borne leukocytes in the damage. Furthermore, iNOS+/+ mice transplanted with iNOS−/− BM had large infarcts (39±6 mm3; n=13), similar to those of autologous transplanted iNOS+/+ mice (39±4 mm3; n=14), indicating the resident brain cells also play a role. Flow cytometry and cell sorting revealed that iNOS is highly expressed in neutrophils and endothelium, but not microglia. Surprisingly, post-ischemic iNOS expression was enhanced in the endothelium of iNOS+/+ mice transplanted with iNOS−/− BM, and in leukocytes of iNOS−/− mice with iNOS+/+ BM, suggesting that endothelial iNOS suppresses iNOS expression in leukocytes and vice-versa. To provide independent evidence that neutrophils mediate brain injury, neutrophils were isolated and transferred to mice 24 hours after stroke. Consistent with the result in chimeric mice, transfer of iNOS+/+, but not iNOS−/−, neutrophils into iNOS−/−mice increased infarct volume. The findings establish that iNOS both in neutrophils and endothelium mediates tissue damage and identify these cell types as putative therapeutic targets for stroke injury.
Introduction
Inflammation is a key component in the pathophysiology of ischemic stroke (1), a leading cause of death and disability with limited treatment options (2, 3). Numerous experimental approaches have explored the therapeutic potential of modulating post-ischemic inflammation (4). Despite promising attempts, such strategies have not been successful in clinical trials (5). One reason for this failure might be the poor understanding of the cellular mechanisms of the injury attributable to post-ischemic inflammation (6). For instance, the respective contributions to the damage of resident brain cells (microglia, endothelial cells) and peripheral blood-borne cells infiltrating the post-ischemic brain (leukocytes), which can express the same inflammatory mediators, has not been established (6).
Inducible nitric oxide synthase (iNOS or NOS2) is a key player in the post-ischemic inflammatory cascade (7, 8), and NO produced by de novo expression of iNOS (9) contributes to cerebral ischemic injury (10, 11). In rodents, iNOS is expressed from 12 hours to several days after middle cerebral artery occlusion (MCAo) (12–14). Furthermore, in rodents as in humans, post-ischemic iNOS immunoreactivity is present in inflammatory cells and blood vessels in brain (10, 15–17). iNOS is an attractive therapeutic target since its inhibition has an extended therapeutic window, induces long-lasting protection, and is observed both with permanent and transient ischemia, as well as in animals with stroke risk factors (10, 15, 18). However, it remains to be established whether the cells expressing iNOS and causing the damage are intrinsic or extrinsic to the brain, and whether the reduction in injury afforded by iNOS inhibition results from preventing the brain infiltration of blood-borne inflammatory cells. These issues have translational relevance since focusing treatments to the specific iNOS-expressing cells involved in the damage would minimize adverse effects associated with systemic iNOS inhibition, such as immunosuppression.
Therefore, a major goal of this study was to determine whether the iNOS expressing cells that contribute to ischemic injury are intrinsic to the brain or blood derived. We used iNOS bone marrow (BM) chimeras and adoptive transfer-based approaches to investigate the cellular source(s) of iNOS expression and their pathogenic relevance. We found that iNOS both in infiltrating neutrophils and cerebral endothelial cells contributes to ischemic brain injury, and that there is a previously unrecognized reciprocal interaction between iNOS expression in endothelial cells and blood leukocytes that has an impact on the damage. These data provide new insight into the cellular bases of the deleterious effects of post-ischemic iNOS expression and suggest that harnessing the therapeutic potential of iNOS inhibition would require targeting iNOS both in endothelial cells and neutrophils.
Material & Methods
Mice
All procedures were approved by the institutional animal care and use committee of Weill Cornell Medical College. Experiments were performed in 7–8 week-old male iNOS−/− mice on a C57Bl/6J background originally developed by MacMicking et al. (19) and obtained from an in-house colony (20). Age-matched wild type mice (C57Bl/6J, Jackson Laboratory, Bar Harbor, ME) served as controls.
Middle Cerebral Artery occlusion
Transient focal cerebral ischemia was induced using the intraluminal filament model of middle cerebral artery occlusion (MCAo), as described previously (21). Under isoflurane anesthesia (maintenance 1.5–2%), a heat-blunted nylon suture (6/0) was inserted into the right external carotid artery of anesthetized mice and advanced until it obstructed the MCA. This was confirmed by cerebral blood flow (CBF) measured using transcranial laser Doppler flowmetry (Periflux System 5010, Perimed, King Park, NY) in the territory irrigated by the right MCA (2 mm posterior, 5 mm lateral to bregma). The filament was left in place for 35 min (naïve mice) or 30 min (chimeric mice), and then withdrawn. Ischemia time was reduced in chimeric mice because in a pilot study we found high mortality of iNOS−/− mice transplanted with iNOS+/+ bone marrow (BM) subjected to 35 min MCAO (iNOS−/− BM−/−, all survived (n=6); iNOS−/− BM+/+, only 4 of 8 mice survived, chi-square p<0.05). Only animals that exhibited a reduction in CBF of >85% during MCA occlusion and in which CBF recovered by >80% after 10 min of reperfusion, were included in the study (21). Rectal temperature was monitored and kept constant (37.0 ± 0.5°C) during the surgical procedure and in the recovery period until the animals regained full consciousness.
Measurement of infarct volume
As described in detail elsewhere (21), infarct volume, corrected for swelling, was quantified 3 days after ischemia using cresyl violet staining on 30–μm-thick coronal brain sections and image analysis software (MCID, Imaging Research, UK ).
Bone marrow transplantation
Whole-body irradiation was performed on 7 week-old male mice (iNOS+/+ and iNOS−/−) with a lethal dose of 9.5 Gy of γ radiation using a 137Cs source (Nordion Gammacell® 40 Exactor, Theratronics, Ottawa, Canada). Eighteen hours later, irradiated mice were transplanted with BM cells (2×106, i.v.) isolated from donor iNOS+/+ or iNOS−/− mice. Therefore, 4 groups of chimeric mice were obtained: irradiated iNOS+/+ with iNOS+/+ BM transplant (iNOS+/+ BM+/+); irradiated iNOS−/− with iNOS−/− BM transplant (iNOS−/− BM−/−); irradiated iNOS−/− with iNOS+/+ BM transplant (iNOS−/− BM+/+) and irradiated iNOS+/+ with iNOS−/− BM transplant (iNOS+/+ BM−/−). Donor BM cells were flushed out with 2–3 ml of PBS from the femur and tibias, filtered through a 40-μm-nylon mesh and counted for viability before injection. Transplanted mice were housed in cages with sulfamethoxazole (0.12%; w/v) and trimethoprim (0.024%) antibiotic added to the drinking water for the first 2 weeks. Reconstitution of BM cells was verified 5 weeks after irradiation by testing the percentage of positive iNOS genomic DNA in isolated blood leukocytes. To this end, 300 μl of blood was withdrawn by cardiac puncture from mice under deep anesthesia, immediately before sacrifice. Erythrolysis was performed three times using 5 ml of erythrocyte lysis buffer (ELB, 0.15 M NH4Cl, 1 mM KHCO3, 0.1 mM Na-EDTA; 5 min, RT). Afterwards, DNA was purified from leukocytes using DNeasy Blood & Tissue Kit (QIAGEN 69504, Germantown, MD) according to the manufacturer’s instructions. Reference primers sequences were: m_ICAM1_prom.3 5′-GGACTCACCTGCTGGTCTCT-3′and m_ICAM1_prom.4 5′-GAACGAGGGCTTCGGTATTT-3′; target primers sequences were: NOS2_gt_1 5′-GAGACAGATGGCTCAGGAGTG-3′ and NOS2_gt_2 5′-TAGGGACCGGGTAAAGAGAAA-3′, all purchased from Invitrogen Life Technologies (Grand Island, NY). qRT-PCR was conducted with 20 ng of DNA, in duplicate 15 μl reactions using the Maxima SYBR Green/ROX qPCR Master Mix (2X) (Thermo Scientific, Pittsburgh, PA). The reactions were incubated at 50°C for 2 min and then at 95°C for 10 min. A polymerase chain reaction cycling protocol consisting of 15 sec at 95°C and 1 min at 60°C for 45 cycles was used for quantification. iNOS relative expression levels were calculated by 2(−ΔΔ C(T)) method (22).
Brain cell isolation
Mice were transcardially perfused under deep anesthesia with 30 ml of heparinized saline to exclude blood cells. The ischemic hemisphere was separated from the cerebellum and olfactory bulb and gently triturated in Hepes-HBSS buffer (138 mM NaCl, 5 mM KCl, 0.4 mM Na2HPO4, 0.4 mM KH2PO4, 5 mM D-Glucose, 10 mM Hepes) using Gentle MACS dissociator (Miltenyi Biotec, Auburn, CA) following the manufacturer’s instructions. The suspension was digested with 62.5 μg/ml Liberase DH (Roche Diagnostics, Indianapolis, IN) and 50 U/ml DNAse I, at 37°C for 45 min in an orbital shaker at 160 rpm. Cells were washed and subjected to discontinuous 70%/25% Percoll (GE Healthcare, Piscataway, NJ) density gradient centrifugation (25 min, 800g, 4°C). Enriched-mononuclear cells were collected from the interphase, washed with Hepes-HBSS buffer and resuspended in 2% fetal bovine serum in PBS for staining and FACS analysis.
FACS analysis and cell sorting
Rat monoclonal antibodies used for FACS analysis include: CD45-APC (clone 30F-11), CD11b-PE (clone M1/70), Gr1-PerCP-Cy5.5 (clone RB6-865), Ly6C-FITC (clone HK1.4), CD54-FITC (ICAM-1, clone AF647) and CD106-PE (VCAM-1, clone 429/MVCAM.A) from BioLegend (San Diego, California). Isolated brain cells were stained at 4°C with predetermined optimal concentrations of antibodies for 20 min and analyzed on an Accuri C6 flow cytometer (BD Biosciences, San Jose, California). Dead cells and debris were gated out by forward scatter (FSC) and side scatter (SSC) properties. Cell populations were separated based on CD45 and Ly6C expression and further sorted by CD11b and Gr1 expression. Infiltrating leukocytes were identified as CD45hi and further gated by CD11b and Gr1 markers. Neutrophils were identified as CD45hi/CD11bhi/Gr1hi. CD45hi/CD11bhi/Gr1lo sorting identifies monocytes/macrophages. Non-myeloid leukocytes were considered as CD45hi/CD11blo/Gr1lo cells. Microglia were identified by intermediate CD45 expression as CD45int/CD11bhi/Ly6Clo. Endothelial cells were identified as CD45lo/Ly6Chi (23) and expression of CD54 (ICAM-1) and CD106 (VCAM-1) adhesion molecules on EC surface was also analyzed. Appropriate isotype control Abs, ‘fluorescence minus one’ staining and staining of negative populations were use to establish sorting parameters. Absolute cell numbers and frequencies were recorded. Additionally, in other experiments, the cell fractions of neutrophils, monocytes/macrophages, microglia and endothelial cells, identified using the same gating strategy as detailed above, were sorted on a FACSVantage cytometer (BD Bioscience) for mRNA analysis.
Quantitative Real-Time PCR
Cell fractions were collected in TRIzol® LS (Invitrogen Life Technologies) and RNA was extracted according to the manufacturer’s instructions. RNA samples were treated with Rnase free DnaseI (Roche, Indianapolis, IN) to remove DNA contamination. cDNA was produced from mRNA samples by using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific). Quantitative determination of gene expression for endothelial-leukocyte adhesion molecule 1 (Elam-1), Ly6G and iNOS genes were performed on a Chromo 4 Detector (Bio-Rad, Hercules, CA) using a two-step cycling protocol. Hypoxanthine-guanine phosphoribosyltransferase (HPRT) was used to normalize gene expression. Primer sequences, purchased from Invitrogen Life Technologies, were: HPRT: 5′-AGTGTTGGATACAGGCCAGAC-3′ and 5′-CGTGATTCAAATCCCTGAAGT-3′; Elam-1, 5′-CTCACTCCTGACATCGTCCTC-3′ and 5′-ACGTTGTAAGAAGGCACATGG-3′; Ly6G 5′-CCTGAGACTTCCTGCAACACA-3′ and 5′-TGCTTCTGTGAGAGTCCACAAT-3′; iNOS 5′-TCACCACAAGGCCACATCGGATT-3′ and 5′-AGCTCCTCCAGAGGGGTAGGCT-3′. qRT-PCR was conducted with cDNA in duplicate 15 μl reactions using the Maxima SYBR Green/ROX qPCR Master Mix (2X) (Thermo Scientific). The reactions were incubated at 50°C for 2 min and then at 95°C for 10 min. A polymerase chain reaction cycling protocol consisting of 15 sec at 95°C and 1 min at 60°C for 45 cycles was used for quantification. Elam-1 and Ly6G relative expression levels were calculated by 2(−ΔΔ C(T)) method (22). The number of both HPRT and iNOS gene copies in cell fractions was calculated from their respective CT (cycle threshold), using an equation derived from a HPRT-cDNA or iNOS-cDNA plasmid standard curve, respectively, amplified in parallel. Data were expressed as iNOS mRNA copies relative to HPRT mRNA copies for each cell fraction.
Western blot
C57BL6 wild type mice (n=12) were subjected to transient MCAO and 3 days later, their brains where processed for flowmetric cell sorting of neutrophils, monocytes/macrophages, microglia and endothelial cells, as described above. The cell fractions were pooled (n=12 mice/cell fraction) and resolved on a 7% PAGE-SDS gel. Separated proteins were transferred onto a PVDF membrane and iNOS was immunodetected using a polyclonal-rabbit anti-iNOS antibody (1/100; Abcam #15323; Cambridge, MA). Immunoreactive bands were visualized using SuperSignal West Femto reagent (Thermo Scientific) on a chemiluminescent imager (LI-COR C-DiGit Blot Scanner, LI-COR, Lincoln, NE). Lysates from Raw264.7 cells stimulated with LPS (10 ng/ml) and IFNγ (100 U/ml) were used as a positive control of iNOS protein expression.
Adoptive cell transfer
Neutrophils were purified from BM of iNOS+/+ or iNOS−/−mice by immunomagnetic negative enrichment. Whole BM was flushed out from femurs and tibias using a 26-gauge needle attached to a 10 ml PBS-filled syringe, and cells were dispersed by slow aspiration through a 20-gauge needle. After centrifugation (300g, 10min, RT), the supernatant was removed by aspiration and erythrolysis was performed using 5 ml of ELB 1 min, RT). Subsequently, 15 ml PBS was added and the suspension was passed through a 40-μm cell strainer (BD Falcon Cat#: 352340). The strainer was then washed with 10 mL PBS. BM cells were centrifuged (300g, 10 min, RT) and resuspended in MACS buffer (PBS supplemented with 2% FBS, 2 mM EDTA; 100μl/107 cells). An appropriate volume of biotinylated-antibody cocktail (see table 1) and cells were purified with Anti-Biotin-Microbeads according to the manufacturers suggestions (Miltenyi Biotech). Diluted and centrifuged cell preparations contained >90% neutrophils as determined by flow cytometry (anti-Ly6G-PerCP-Cy5.5, 1A8 clone, Biolegend). Afterwards, cells were washed, resuspended in sterile physiological saline (5×105 cell/ 100 μl) and administered by injection into the right retro-orbital sinus in anesthetized mice 24 hours after MCAo (100 μl/mouse). Control animals received 100 μl saline.
Table 1.
Antibody cocktail for neutrophil purification.
| Antigen | Label | Clone | Isotype | Supplier |
|---|---|---|---|---|
| CD117 | biotin | 2B8 | rat IgG2b, κ | Biolegend |
| CD45R (B220) | biotin | RA3-6B2 | rat IgG2a, κ | Biolegend |
| CD49 | biotin | HMa2 | armenian hamster IgG | eBioscience |
| TER 119 | biotin | TER-119 | rat IgG2b, κ | Biolegend |
| CD5 | biotin | 3 7.3 | rat IgG2a, κ | Biolegend |
| CD115 | biotin | FS98 | rat IgG2a | Biolegend |
| F4/80 | biotin | BM8 | rat IgG2a, κ | Biolegend |
Data analysis
Data are expressed as mean ± SE. Statistical differences in mortality were assayed by the Fisher’s exact test and Chi-square test. Intergroup differences in infarct volume, flow analysis and mRNA expression were analyzed using a Student unpaired t-test or one-way ANOVA with Dunnett’s post-hoc analysis, as appropriate. Differences were considered statistically significant for p<0.05.
Results
Mice groups and mortality
A total of 229 mice were used in this study. Of these, 52 naïve mice were used for determination of infarct volume or FACS analysis of brain cells after MCAo. For adoptive transfer experiments, 25 mice were used as neutrophil donors and 40 mice were used as recipients, which were subjected to MCAo for infarct volume measurement. Six iNOS−/− mice were excluded due to inadequate ischemia-reperfusion. Three mice (1 iNOS+/+ mouse and 2 iNOS−/− mice) died before 72 hours and no differences were found between groups (Fisher’s test, p=1). To generate chimeric animals, 10 mice were used as BM donors to obtain 102 chimeric mice, which were subjected to MCAO and used for infarct volume determination or FACS sorting. Four chimeric mice were excluded from the study due to inadequate ischemia-reperfusion (1 iNOS+/+BM+/+ mouse, 2 iNOS−/−BM+/+ mice, and 1 iNOS−/−BM−/− mouse). Four chimeric mice (2 iNOS−/−BM+/+ mice and 2 iNOS+/+BM−/− mice) died before 72 hours. No differences were found in mortality rate among groups (chi-square, p=0.4).
The protective effect of iNOS deletion is not due to reduced post-ischemic leukocyte infiltration
First, we sought to confirm that iNOS deficiency results in less severe ischemic injury after MCAo, as previously reported (11). iNOS−/− mice had smaller infarcts than iNOS+/+ mice 72 hours after MCAo (figure 1A), a finding not due to differences in intraischemic CBF, which was similar in both groups (figure 1B). Since iNOS may influence the migration of inflammatory cells (24), we next examined whether the protection observed in iNOS−/− mice was the result of reduced leukocyte infiltration. To this end, we performed FACS analysis of brain cells in iNOS+/+ and iNOS−/− mice 48 hours after MCAo (figure 2), the earliest time point of leukocyte infiltration after ischemia/reperfusion (figure 2B). FACS analysis showed no differences in the total number of infiltrating leukocytes (CD45hi cells) between iNOS+/+ and iNOS−/− mice, or in monocytes/macrophages and neutrophils (figure 2C). Similarly, no differences in the number of microglia and endothelial cells were observed between iNOS+/+ and iNOS−/−mice. Furthermore, ICAM-1 and VCAM-1 expression in brain endothelial cells, which mediates post-ischemic neutrophil and monocyte/lymphocyte endothelial adhesion (25, 26), was similar in both groups 48 hours after MCAo (figure 2C). Therefore, the protection afforded by iNOS deficiency is not secondary to reduced leukocyte infiltration after stroke.
Figure 1. Ischemic injury is attenuated in iNOS−/− mice.
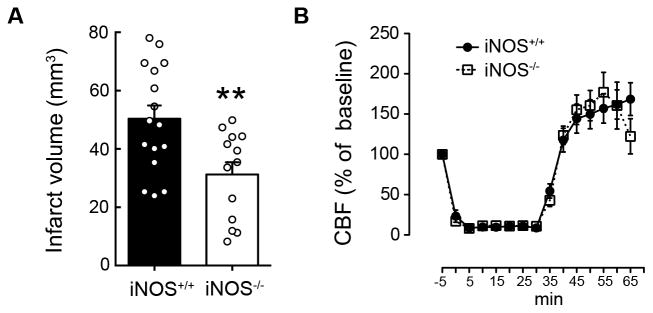
A. iNOS−/− mice have smaller infarcts than iNOS+/+ 72 hours after MCAo; values are mean±SE, n=13–16/group. **p<0.01. B. Cerebral blood flow (CBF) changes during ischemia and reperfusion were similar in both groups (mean±SE, n=13–16/group).
Figure 2. Brain leukocyte infiltration after MCAo is similar in both iNOS+/+ and iNOS−/− mice.

A. Gating strategy and flow cytometric analysis of isolated brain cells stained for CD45, Ly6C, CD11b and Gr1 cell surface markers. Four different populations were identified: CD45hi/CD11bhi/Gr1lo(monocytes), CD45hi/CD11bhi/Gr1hi (neutrophils), CD45int/CD11bhi/Gr1lo/Ly6Clo(microglia) and CD45lo/CD11blo/Gr1lo/Ly6Chi (endothelial cells, EC). B. Time course of infiltrating leukocytes in the ischemic brain of iNOS+/+ mice showed an increase of both monocytes and neutrophils as early as 48 hours after MCAo. Values are mean±SE, n=5–7/group. **P<0.01, vs. naïve (N) and sham (S) groups. C. Total infiltrating leukocytes (CD45hi), monocytes and neutrophils showed similar levels in either iNOS+/+ or iNOS−/− 48 hours after MCAo. No changes were also detected in the number of microglia or EC cells. In addition, the % of EC expressing ICAM-1 and VCAM-1 was similar in both groups. Values are mean±SE, n=6/group. ***p<0.001, *p<0.5 vs. iNOS+/+ sham group.
iNOS expression in blood-borne leukocytes and resident brain cells contributes to brain injury after MCAo
To investigate the relative contribution to ischemic injury of iNOS in resident brain cells or BM-derived cells, we studied iNOS chimeric mice. Successful induction of chimerism in BM transplanted mice was verified by genomic analysis for the presence of the intact iNOS (NOS2) gene in peripheral blood leukocytes (figure 3A). Chimeric mice were subjected to MCAo and infarct volume was determined 72 hours later (figure 3B). The infarct volume in iNOS−/− mice transplanted with iNOS−/− BM (iNOS−/−BM−/−) was smaller than that of iNOS+/+ mice transplanted with iNOS+/+ BM (iNOS+/+BM+/+), indicating that irradiation and BM transplant did not influence the neuroprotection associated with the iNOS−/− genotype. However, in iNOS−/−BM+/+ chimeric mice, in which iNOS is present in peripheral leukocytes but not in resident brain cells, infarcts were larger than in iNOS−/− BM−/−(p<0.01) and not different from those of iNOS+/+BM+/+ mice (p>0.05) (figure 3B). Surprisingly, in iNOS+/+ mice transplanted with iNOS−/− BM (iNOS+/+BM−/−) the infarcts were not different from those of iNOS+/+BM+/+ mice (figure 3B), implicating also iNOS in resident brain cells in the damage. CBF changes during ischemia-reperfusion did not differ among groups (figure 3C), although a trend for increased hyperemia during early reperfusion was seen in iNOS+/+BM−/− mice. These data suggest that iNOS in BM-derived cells contributes to brain injury after MCAo, but that iNOS in resident brain cells may also play a role.
Figure 3. iNOS expression either in blood-borne leukocytes or resident brain cells reconstitute infarct volume after MCAo.
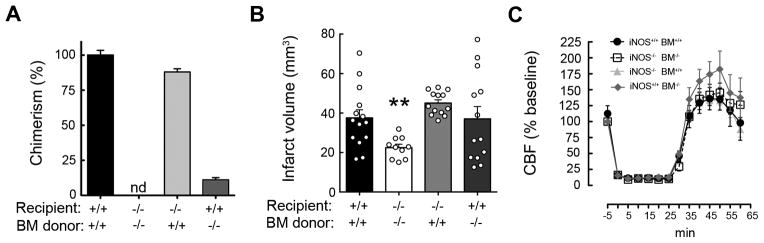
A. Percentage of chimerism in blood leukocytes assayed by genomic qRT-PCR; nd, not detected. Values are mean±SE. B. Transplanted wild type mice (iNOS+/+BM+/+, n=14) had larger infarcts than transplanted iNOS null mice (iNOS−/−BM−/−, n=10) 72 hours after MCAo. iNOS−/− mice expressing iNOS only in peripheral leukocytes (iNOS−/−BM+/+, n=13) or iNOS+/+ mice expressing iNOS only in resident brain cells (iNOS+/+BM−/−; n=13) had similar infarcts to iNOS+/+BM+/+. Values are mean±SE. **p<0.01, vs. iNOS+/+BM+/+. C. No statistical significances in cerebral blood flow (CBF) during ischemia and reperfusion were observed among groups.
iNOS is expressed in infiltrating leukocytes and in endothelial cells, but not in microglia, after MCAo
The data presented above indicate that iNOS both in BM-derived and resident brain cells contribute to ischemic brain injury. We next sought to determine the cell-types expressing iNOS after MCAo which may be involved in the damage. Microglia and endothelial cells, resident brain cells with the greatest potential for iNOS expression (15, 27, 28), and infiltrating leukocytes (monocytes/macrophages and neutrophils) were isolated by FACS sorting from chimeric mice 72 hours after MCAo, when post-ischemic iNOS expression is highest (29). Cell fractions were processed for qRT-PCR and iNOS mRNA expression was analyzed in each population (figure 4A). The purity of the sorted populations was evaluated by the expression of Elam-1 in EC and of Ly6G in neutrophils (figure 4B). In wild type BM-transplanted mice, iNOS mRNA was induced in neutrophils, monocytes and endothelial cells, while iNOS expression was not observed in microglia. In iNOS−/− mice transplanted with iNOS+/+ BM, iNOS expression was enhanced in neutrophils and monocytes compared to iNOS+/+BM+/+ mice, but it was not detected in EC or microglia (figure 4A). Conversely, iNOS+/+ mice transplanted with iNOS−/− BM exhibited a significantly higher iNOS upregulation in EC compared to controls, whereas iNOS expression was not detected in microglia or infiltrating leukocytes (figure 4A). Additionally, cell fractions isolated from wild type mice 72 hours after MCAO were analyzed for iNOS expression at the protein level by western blotting (figure 4C). In agreement with mRNA data, iNOS protein expression was observed in monocytes/macrophages, neutrophils and endothelial cells, but not in microglia.
Figure 4. iNOS is induced either in brain infiltrating cells or endothelium.

Flow cytometric sorting was performed to purify neutrophils, monocytes, microglia and endothelial cells (EC) after staining for CD45, Ly6C, CD11b and Gr1 cell surface markers as described in figure 2. A. Flow-sorted cell fractions from ischemic brains of chimera mice were processed for qRT-PCR to quantify iNOS mRNA expression. In iNOS+/+BM+/+ mice, iNOS mRNA was detected in all cell populations except for microglia (not shown). In iNOS−/− mice transplanted with WT BM cells (iNOS−/−BM+/+), iNOS mRNA was detected in both neutrophils and monocytes, while iNOS mRNA was detected in EC from iNOS+/+ mice transplanted with iNOS−/− BM (iNOS+/+BM−/−). Values are mean±SE, n=3 pool/group, 4 samples pooled; iNOS mRNA expression in chimeric mice vs. iNOS+/+BM+/+ mice, *p<0.05, nd, not detected. B. Ly6G and Elam-1 mRNA was assessed by qRT-PCR in cell fractions to verify purity of separated cell populations. Values are mean±SE, n=3 pool/group, 4 samples pooled. C. iNOS protein levels in neutrophils, monocytes, microglia and endothelial cells (EC) purified by flow cytometric sorting (n=12 pooled mice/cell fraction). The number of cells that corresponds to the amount of lysate loaded into each lane is indicated for each cell population. LPS/IFNγ stimulated (+) or un-stimulated (−) Raw264.7 cells were used as a positive control for iNOS expression. The dash line indicates different exposure time for chemiluminescent detection on the left (short) and right (long) of the blot. Molecular weight, (MW). Non-specific band (NS) served as a loading control.
Adoptive transfer of iNOS+/+ neutrophils worsens ischemic injury in iNOS−/− mice
The data presented above indicate that iNOS is highly expressed in neutrophils infiltrating the ischemic brain. To determine whether iNOS in neutrophils contributes to tissue damage, we adoptively transferred iNOS+/+ or iNOS−/− neutrophils (5×105 cells, iv) into recipient mice 24 hours after MCAo (figure 5). iNOS−/− mice receiving iNOS−/−neutrophils had smaller infarcts than iNOS+/+ mice receiving iNOS+/+ neutrophils. However, infarct volumes were larger in iNOS−/− mice receiving iNOS+/+ neutrophils and not statistically different from those of iNOS+/+ mice receiving iNOS+/+ neutrophils (p>0.05).
Figure 5. Adoptive transfer of iNOS+/+ neutrophils worsens ischemic injury in iNOS−/− mice.
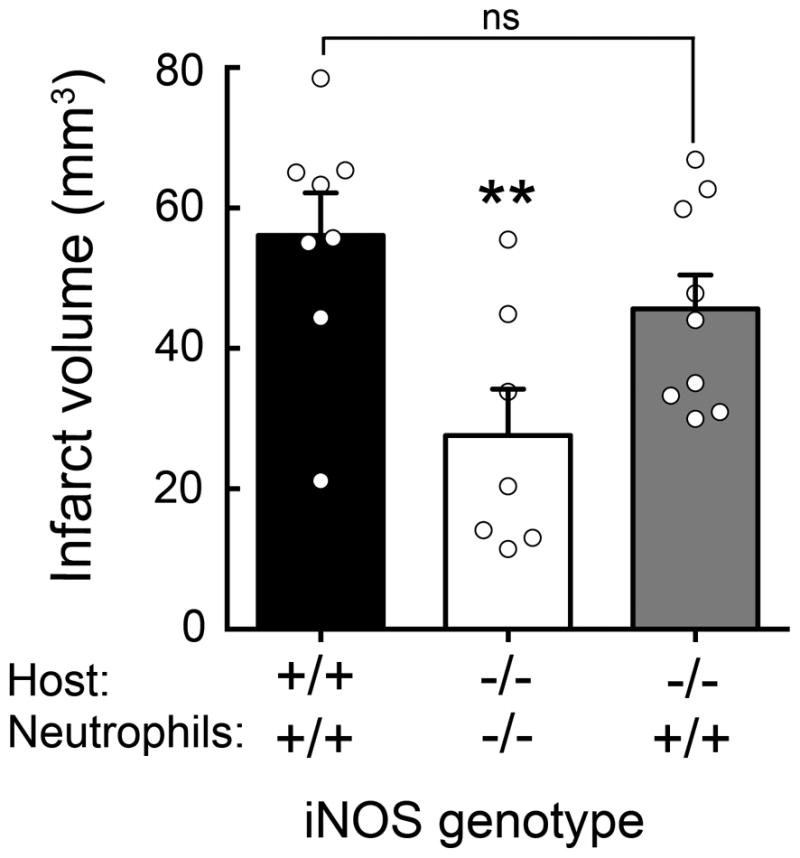
Adoptive transfer of iNOS+/+ (5×105 cells) but not iNOS−/− neutrophils into iNOS−/− mice 24 hours after MCAO increased infarct volume at 72 hours; values are mean±SE, n=9–8 group. **p<0.05.
Discussion
This study provides several new findings. First, we demonstrated that the protection from ischemic injury observed in iNOS−/− mice is not due to reduced infiltration of blood-borne inflammatory cells, assessed quantitatively by FACS analysis. Second, using chimeric mice, we found that both resident brain cells (endothelial cells) and infiltrating leukocytes expressing iNOS contribute to post-ischemic brain injury. Third, we demonstrated that adoptive transfer of iNOS+/+ neutrophils is sufficient to offset in large part the protection in iNOS−/− mice, highlighting the pathogenic role of neutrophil iNOS in ischemic brain injury. Fourth, we found that suppression of iNOS expression in leukocytes enhances iNOS expression in endothelial cells, and vice-versa, unveiling a previously unappreciated reciprocal interaction regulating post-ischemic iNOS expression. These observations collectively provide new insight into the cellular expression of iNOS after ischemic stroke and its role in the ensuing brain damage.
iNOS is upregulated at the transcriptional level by the activity of NF-κB and Stat1α transcription factors, among others (30). Accordingly, iNOS levels are normally very low, but after ischemia, iNOS transcription is induced resulting in the generation of large amounts of NO (31). Reacting with superoxide (O2−), a free radical generated in the brain after ischemia/reperfusion (32), NO gives rise to the powerful oxidant peroxynitrite resulting in significant nitrosative damage of biological molecules, which, in turn, contributes to ischemic injury (33). However, another aspect of iNOS-derived NO function is its ability to regulate leukocyte recruitment to inflammatory sites (34). Although it is well established that inhibition (35, 36) or genomic deletion (37) of iNOS enhances endothelial leukocyte adhesion, in hepatic ischemic/reperfusion injury iNOS-derived NO facilitates leukocyte transmigration (24). Therefore, we first investigated whether the neuroprotection afforded by iNOS deficiency was due to reduced leukocyte trafficking into the injured brain. However, we did not find differences in the number of infiltrating leukocytes or changes in the level of discrete leukocyte populations involved in post-ischemic inflammation, such as neutrophils and monocytes (38). Furthermore, iNOS deletion did not affect the expression of endothelial ICAM-1 and VCAM-1, adhesion molecules involved in the post-ischemic transmigration of leukocytes across the blood-brain barrier (25, 26). Accordingly, our findings indicate that the deleterious effect of iNOS is not secondary to enhancement of leukocyte infiltration into the ischemic brain, but to a direct action iNOS-expressing cells on the ischemic tissue.
iNOS immunoreactivity has been observed in microvessels and neutrophils after experimental stroke (12, 15, 16) and post-mortem in stroke patients (17), but the pathogenic significance of iNOS in these cells was not established. The present study is the first to provide evidence in support of a cell-specific pathogenic effect of endothelial and neutrophil iNOS on the post-ischemic brain. We found that both brain infiltrating neutrophils and monocytes express iNOS and reconstitution of an iNOS−/− host with iNOS+/+ BM was sufficient to counteract the protection and restore the injury to levels observed in WT mice. These findings, collectively, demonstrate that BM-derived cells, specifically neutrophils, represent a major source of the iNOS responsible for brain damage. Polymorphonuclear cells have long been implicated in the brain damage induced by post-ischemic inflammation, through a wide variety of mechanisms (39–42). Our data suggest that iNOS-derived NO, which promotes lipid peroxidation, protein nitration, DNA damage and BBB breakdown (43), is a major neurotoxic molecule produced by neutrophils (44, 45). To provide further evidences that expression of iNOS in infiltrating leukocytes exacerbates ischemic injury, we adoptively transferred neutrophils into iNOS−/− mice. We chose to transfer neutrophils because they express higher levels of iNOS than monocytes. Doing so was sufficient to increase the infarct volume to similar levels observed in iNOS+/+ mice, confirming that neutrophil iNOS is a key player in ischemic brain damage. Our data show that neither transfer of iNOS−/− neutrophils into iNOS−/−mice, nor transfer of iNOS+/+ neutrophils into iNOS+/+ mice affected the infarct volume when compared to control iNOS−/− and iNOS+/+ mice, respectively. These findings indicate that the increased brain damage in iNOS−/− mice following iNOS+/+ neutrophil transfer is most likely mediated by direct effects of iNOS-derived NO, rather than by an increase in the number of circulating neutrophils after adoptive transfer or microvascular clogging (46). Moreover, the number of transferred neutrophils (5×105 cells) is well below the total number of circulating neutrophils in C57Bl/6 mice (1×106 cells) and can thus be considered to be within physiological limits (47).
We did not observe iNOS expression in microglia. iNOS expression can be induced in cultured microglia in the presence of LPS or inflammatory cytokines (48, 49) and, in vivo, after cerebral ischemia (50). However, iNOS expression in vivo has usually been attributed to microglia on the bases of immunohistochemical markers that cannot differentiate microglia from infiltrating macrophages (51). In contrast, FACS analysis, as used in this study, allows to more clearly differentiate microglia from monocytes/macrophages based on quantitative assessment of their CD45 expression level (52). Using this approach we were able to document iNOS expression in monocytes/macrophages but not in microglia. However, this observation does not exclude that possibility that microglial cells play a role in ischemic injury by producing other mediators (1, 53, 54). iNOS immunoreactivity has also been reported in astrocytes after ischemia, in the post-ischemic period but the role of astrocytic iNOS in the acute phase of the damage remains unclear since the expression occurs in the late post-ischemic period (3 days after ischemia) (16, 55).
Unexpectedly, we observed that iNOS+/+ mice transplanted with iNOS−/− BM developed large infarcts despite of iNOS deficiency in infiltrating leukocytes. This effect was correlated with increased cerebral endothelial iNOS expression in the host, thus compensating for the lack of NO generation by infiltrating leukocytes. Therefore, it is conceivable that NO-generating leukocytes may limit endothelial iNOS expression after brain ischemia. Although the reasons for the increased endothelial iNOS expression in mice lacking leukocyte iNOS remain unclear, there is evidence for a negative feed-back loop by which iNOS-derived NO suppresses iNOS induction by inhibiting NF-κB activity (56–58), providing a potential mechanisms for this effect. This finding has therapeutic implications in that it suggests that suppression of iNOS expression or activity in blood-borne leukocytes is not sufficient to confer protection from ischemic injury, and that cerebral endothelial cells have to be targeted at the same time.
In conclusion, we have demonstrated that the harmful effects of iNOS in the ischemic brain are not mediated by enhancing the accumulation of inflammatory cells in the injured brain. Rather, iNOS in neutrophils infiltrating the ischemic brain and endothelial cells is required for the expression of its destructive effects. We also found that iNOS deletion only in leukocytes or endothelial cells is not sufficient to confer protection, because lack of iNOS in one cell type enhances iNOS expression in the other. These findings provide further insight into the cellular bases through which iNOS exerts its deleterious effects on the brain, and suggest specific cellular targets for therapeutic approaches harnessing the protective potential of iNOS inhibition.
Footnotes
Supported by NIH Grants NS34179 and NS081179. LG-B was recipient of a Sara Borrell grant from the Spanish Ministry of Health (Instituto de Salud Carlos III).
References
- 1.Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB American Heart Association Statistics Committee and Stroke Statistics Subcommittee. . Heart disease and stroke statistics--2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fonarow GC, Smith EE, Saver JL, Reeves MJ, Bhatt DL, Grau-Sepulveda MV, Olson DM, Hernandez AF, Peterson ED, Schwamm LH. Timeliness of Tissue-Type Plasminogen Activator Therapy in Acute Ischemic Stroke: Patient Characteristics, Hospital Factors, and Outcomes Associated With Door-to-Needle Times Within 60 Minutes. Circulation. 2011;123:750–758. doi: 10.1161/CIRCULATIONAHA.110.974675. [DOI] [PubMed] [Google Scholar]
- 4.Shichita T, Ago T, Kamouchi M, Kitazono T, Yoshimura A, Ooboshi H. Novel therapeutic strategies targeting innate immune responses and early inflammation after stroke. J Neurochem. 2012;123:29–38. doi: 10.1111/j.1471-4159.2012.07941.x. [DOI] [PubMed] [Google Scholar]
- 5.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denes A, Thornton P, Rothwell NJ, Allan SM. Inflammation and brain injury: acute cerebral ischaemia, peripheral and central inflammation. Brain Behav Immun. 2010;24:708–723. doi: 10.1016/j.bbi.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 7.del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol. 2000;10:95–112. doi: 10.1111/j.1750-3639.2000.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 9.Nathan C. Inducible nitric oxide synthase: what difference does it make? J Clin Invest. 1997;100:2417–2423. doi: 10.1172/JCI119782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iadecola C, Zhang F, Xu X. Inhibition of inducible nitric oxide synthase ameliorates cerebral ischemic damage. Am J Physiol. 1995;268:R286–92. doi: 10.1152/ajpregu.1995.268.1.R286. [DOI] [PubMed] [Google Scholar]
- 11.Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci. 1997;17:9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iadecola C, Zhang F, Xu S, Casey R, Ross ME. Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. J Cereb Blood Flow Metab. 1995;15:378–384. doi: 10.1038/jcbfm.1995.47. [DOI] [PubMed] [Google Scholar]
- 13.Iadecola C, Xu X, Zhang F, el-Fakahany EE, Ross ME. Marked induction of calcium-independent nitric oxide synthase activity after focal cerebral ischemia. J Cereb Blood Flow Metab. 1995;15:52–59. doi: 10.1038/jcbfm.1995.6. [DOI] [PubMed] [Google Scholar]
- 14.Grandati M, Verrecchia C, Revaud ML, Allix M, Boulu RG, Plotkine M. Calcium-independent NO-synthase activity and nitrites/nitrates production in transient focal cerebral ischaemia in mice. Br J Pharmacol. 1997;122:625–630. doi: 10.1038/sj.bjp.0701427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iadecola C, Zhang F, Casey R, Clark HB, Ross ME. Inducible nitric oxide synthase gene expression in vascular cells after transient focal cerebral ischemia. Stroke. 1996;27:1373–1380. doi: 10.1161/01.str.27.8.1373. [DOI] [PubMed] [Google Scholar]
- 16.Niwa M, Inao S, Takayasu M, Kawai T, Kajita Y, Nihashi T, Kabeya R, Sugimoto T, Yoshida J. Time course of expression of three nitric oxide synthase isoforms after transient middle cerebral artery occlusion in rats. Neurol Med Chir (Tokyo) 2001;41:63–72. doi: 10.2176/nmc.41.63. discussion 72–3. [DOI] [PubMed] [Google Scholar]
- 17.Forster C, Clark HB, Ross ME, Iadecola C. Inducible nitric oxide synthase expression in human cerebral infarcts. Acta Neuropathol. 1999;97:215–220. doi: 10.1007/s004010050977. [DOI] [PubMed] [Google Scholar]
- 18.Zhao X, Haensel C, Araki E, Ross ME, Iadecola C. Gene-dosing effect and persistence of reduction in ischemic brain injury in mice lacking inducible nitric oxide synthase. Brain Res. 2000;872:215–218. doi: 10.1016/s0006-8993(00)02459-8. [DOI] [PubMed] [Google Scholar]
- 19.MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 20.Kunz A, Park L, Abe T, Gallo EF, Anrather J, Zhou P, Iadecola C. Neurovascular Protection by Ischemic Tolerance: Role of Nitric Oxide and Reactive Oxygen Species. J Neurosci. 2007;27:7083–7093. doi: 10.1523/JNEUROSCI.1645-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jackman K, Kunz A, Iadecola C. Methods Mol Biol Methods in Molecular Biology. Vol. 793. Humana Press; Totowa, NJ: 2011. Modeling Focal Cerebral Ischemia In Vivo; pp. 195–209. [DOI] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Jutila MA, Kroese FG, Jutila KL, Stall AM, Fiering S, Herzenberg LA, Berg EL, Butcher EC. Ly-6C is a monocyte/macrophage and endothelial cell differentiation antigen regulated by interferon-gamma. Eur J Immunol. 1988;18:1819–1826. doi: 10.1002/eji.1830181125. [DOI] [PubMed] [Google Scholar]
- 24.Hamada T, Duarte S, Tsuchihashi S, Busuttil RW, Coito AJ. Inducible nitric oxide synthase deficiency impairs matrix metalloproteinase-9 activity and disrupts leukocyte migration in hepatic ischemia/reperfusion injury. Am J Pathol. 2009;174:2265–2277. doi: 10.2353/ajpath.2009.080872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Connolly ES, Winfree CJ, Springer TA, Naka Y, Liao H, Yan SD, Stern DM, Solomon RA, Gutierrez-Ramos JC, Pinsky DJ. Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J Clin Invest. 1996;97:209–216. doi: 10.1172/JCI118392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Becker K, Kindrick D, Relton J, Harlan J, Winn R. Antibody to the alpha4 integrin decreases infarct size in transient focal cerebral ischemia in rats. Stroke. 2001;32:206–211. doi: 10.1161/01.str.32.1.206. [DOI] [PubMed] [Google Scholar]
- 27.Tran EH, Hardin-Pouzet H, Verge G, Owens T. Astrocytes and microglia express inducible nitric oxide synthase in mice with experimental allergic encephalomyelitis. J Neuroimmunol. 1997;74:121–129. doi: 10.1016/s0165-5728(96)00215-9. [DOI] [PubMed] [Google Scholar]
- 28.Ono K, Suzuki H, Sawada M. Delayed neural damage is induced by iNOS-expressing microglia in a brain injury model. Neurosci Lett. 2010;473:146–150. doi: 10.1016/j.neulet.2010.02.041. [DOI] [PubMed] [Google Scholar]
- 29.Kunz A, Abe T, Hochrainer K, Shimamura M, Anrather J, Racchumi G, Zhou P, Iadecola C. Nuclear Factor- B Activation and Postischemic Inflammation Are Suppressed in CD36-Null Mice after Middle Cerebral Artery Occlusion. J Neurosci. 2008;28:1649–1658. doi: 10.1523/JNEUROSCI.5205-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pautz A, Art J, Hahn S, Nowag S, Voss C, Kleinert H. Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide. 2010;23:75–93. doi: 10.1016/j.niox.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 31.Forman LJ, Liu P, Nagele RG, Yin K, Wong PY. Augmentation of nitric oxide, superoxide, and peroxynitrite production during cerebral ischemia and reperfusion in the rat. Neurochem Res. 1998;23:141–148. doi: 10.1023/a:1022468522564. [DOI] [PubMed] [Google Scholar]
- 32.Chen H, Song YS, Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J Cereb Blood Flow Metab. 2009;29:1262–1272. doi: 10.1038/jcbfm.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pacher P, Beckman JS, Liaudet L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hickey MJ. Role of inducible nitric oxide synthase in the regulation of leucocyte recruitment. Clin Sci. 2001;100:1–12. [PubMed] [Google Scholar]
- 35.Peng HB, Spiecker M, Liao JK. Inducible nitric oxide: an autoregulatory feedback inhibitor of vascular inflammation. J Immunol. 1998;161:1970–1976. [PubMed] [Google Scholar]
- 36.Binion DG, Fu S, Ramanujam KS, Chai YC, Dweik RA, Drazba JA, Wade JG, Ziats NP, Erzurum SC, Wilson KT. iNOS expression in human intestinal microvascular endothelial cells inhibits leukocyte adhesion. Am J Physiol. 1998;275:G592–603. doi: 10.1152/ajpgi.1998.275.3.G592. [DOI] [PubMed] [Google Scholar]
- 37.Hickey MJ, Sharkey KA, Sihota EG, Reinhardt PH, MacMicking JD, Nathan C, Kubes P. Inducible nitric oxide synthase-deficient mice have enhanced leukocyte-endothelium interactions in endotoxemia. FASEB J. 1997;11:955–964. doi: 10.1096/fasebj.11.12.9337148. [DOI] [PubMed] [Google Scholar]
- 38.Yilmaz G, Granger DN. Leukocyte Recruitment and Ischemic Brain Injury. Neuromol Med. 2009;12:193–204. doi: 10.1007/s12017-009-8074-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Justicia C, Panés J, Solé S, Cervera A, Deulofeu R, Chamorro A, Planas AM. Neutrophil infiltration increases matrix metalloproteinase-9 in the ischemic brain after occlusion/reperfusion of the middle cerebral artery in rats. J Cereb Blood Flow Metab. 2003;23:1430–1440. doi: 10.1097/01.WCB.0000090680.07515.C8. [DOI] [PubMed] [Google Scholar]
- 40.Allen C, Thornton P, Denes A, McColl BW, Pierozynski A, Monestier M, Pinteaux E, Rothwell NJ, Allan SM. Neutrophil Cerebrovascular Transmigration Triggers Rapid Neurotoxicity through Release of Proteases Associated with Decondensed DNA. J Immunol. 2012;189:381–392. doi: 10.4049/jimmunol.1200409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang XN, Zheng Z, Giffard RG, Yenari MA. Significance of marrow-derived nicotinamide adenine dinucleotide phosphate oxidase in experimental ischemic stroke. Ann Neurol. 2011;70:606–615. doi: 10.1002/ana.22476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matsuo Y, Onodera H, Shiga Y, Nakamura M, Ninomiya M, Kihara T, Kogure K. Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat. Effects of neutrophil depletion. Stroke. 1994;25:1469–1475. doi: 10.1161/01.str.25.7.1469. [DOI] [PubMed] [Google Scholar]
- 43.Chen XM, Chen HS, Xu MJ, Shen JG. Targeting reactive nitrogen species: a promising therapeutic strategy for cerebral ischemia-reperfusion injury. Acta Pharmacol Sin. 2013;34:67–77. doi: 10.1038/aps.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakazawa H, Fukuyama N, Takizawa S, Tsuji C, Yoshitake M, Ishida H. Nitrotyrosine formation and its role in various pathological conditions. Free Radic Res. 2000;33:771–784. doi: 10.1080/10715760000301291. [DOI] [PubMed] [Google Scholar]
- 45.Coeroli L, Renolleau S, Arnaud S, Plotkine D, Cachin N, Plotkine M, Ben-Ari Y, Charriaut-Marlangue C. Nitric oxide production and perivascular tyrosine nitration following focal ischemia in neonatal rat. J Neurochem. 1998;70:2516–2525. doi: 10.1046/j.1471-4159.1998.70062516.x. [DOI] [PubMed] [Google Scholar]
- 46.del Zoppo GJ, Schmid-Schönbein GW, Mori E, Copeland BR, Chang CM. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke. 1991;22:1276–1283. doi: 10.1161/01.str.22.10.1276. [DOI] [PubMed] [Google Scholar]
- 47.The Jackson Laboratory. Mouse Phenome Database web site. Bar Harbor, Maine USA: The Jackson Laboratory; 2014. Hematological survey of 11 inbred strains of mice. MPD:22910. See http://phenome.jax.org. [Google Scholar]
- 48.Possel H, Noack H, Putzke J, Wolf G, Sies H. Selective upregulation of inducible nitric oxide synthase (iNOS) by lipopolysaccharide (LPS) and cytokines in microglia: in vitro and in vivo studies. Glia. 2000;32:51–59. doi: 10.1002/1098-1136(200010)32:1<51::aid-glia50>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 49.Simmons ML, Murphy S. Induction of nitric oxide synthase in glial cells. J Neurochem. 1992;59:897–905. doi: 10.1111/j.1471-4159.1992.tb08328.x. [DOI] [PubMed] [Google Scholar]
- 50.Han HS, Qiao Y, Karabiyikoglu M, Giffard RG, Yenari MA. Influence of mild hypothermia on inducible nitric oxide synthase expression and reactive nitrogen production in experimental stroke and inflammation. J Neurosci. 2002;22:3921–3928. doi: 10.1523/JNEUROSCI.22-10-03921.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guillemin GJ, Brew BJ. Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol. 2004;75:388–397. doi: 10.1189/jlb.0303114. [DOI] [PubMed] [Google Scholar]
- 52.Sedgwick JD, Schwender S, Imrich H, Dörries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci USA. 1991;88:7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wood PL. Microglia as a unique cellular target in the treatment of stroke: potential neurotoxic mediators produced by activated microglia. Neurol Res. 1995;17:242–248. doi: 10.1080/01616412.1995.11740321. [DOI] [PubMed] [Google Scholar]
- 54.Patel AR, Ritzel R, McCullough LD, Liu F. Microglia and ischemic stroke: a double-edged sword. Int J Physiol Pathophysiol Pharmacol. 2013;5:73–90. [PMC free article] [PubMed] [Google Scholar]
- 55.Bidmon HJ, Wu J, Buchkremer-Ratzmann I, Mayer B, Witte OW, Zilles K. Transient changes in the presence of nitric oxide synthases and nitrotyrosine immunoreactivity after focal cortical lesions. Neurosci. 1998;82:377–395. doi: 10.1016/s0306-4522(97)00275-3. [DOI] [PubMed] [Google Scholar]
- 56.Park SK, Lin HL, Murphy S. Nitric oxide regulates nitric oxide synthase-2 gene expression by inhibiting NF-kappaB binding to DNA. Biochem J. 1997;322(Pt 2):609–613. doi: 10.1042/bj3220609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Taylor BS, Kim YM, Wang Q, Shapiro RA, Billiar TR, Geller DA. Nitric oxide down-regulates hepatocyte-inducible nitric oxide synthase gene expression. Arch Surg. 1997;132:1177–1183. doi: 10.1001/archsurg.1997.01430350027005. [DOI] [PubMed] [Google Scholar]
- 58.Chang K, Lee SJ, Cheong I, Billiar TR, Chung HT, Han JA, Kwon YG, Ha KS, Kim YM. Nitric oxide suppresses inducible nitric oxide synthase expression by inhibiting post-translational modification of IkappaB. Exp Mol Med. 2004;36:311–324. doi: 10.1038/emm.2004.42. [DOI] [PubMed] [Google Scholar]