

Abstract
Morphine-like analgesics act on μ opioid receptors in the CNS to produce highly effective pain relief, but the same class of receptors also mediates non-therapeutic side effects. The analgesic properties of morphine were recently shown to require the activity of a brain neuronal cytochrome P450 epoxygenase, but the significance of this pathway for opioid side effects is unknown. Here we show that brain P450 activity is not required for three of morphine’s major side effects (respiratory depression, constipation, and locomotor stimulation). Following systemic or intracerebroventricular administration of morphine, transgenic mice with brain neuron–specific reductions in P450 activity showed highly attenuated analgesic responses as compared with wild-type (control) mice. However, brain P450-deficient mice showed normal morphine-induced side effects (respiratory depression, locomotor stimulation, and inhibition of intestinal motility). Pretreatment of control mice with the P450 inhibitor CC12 similarly reduced the analgesia, but not these side effects of morphine. Because activation of brain μ opioid receptors produces both opioid analgesia and opioid side effects, dissociation of the mechanisms for the therapeutic and therapy-limiting effects of opioids has important consequences for the development of analgesics with reduced side effects and/or limited addiction liability.
Keywords: analgesia, cytochrome P450, brain, respiration, opioid, locomotor activity
1. Introduction
Opioids such as morphine activate μ opioid receptors in the CNS to reduce pain perception (Yaksh and Wallace, 2010). Millions of pain patients obtain relief with μ opioids, yet the side effects of these drugs limit therapeutic efficacy (Stannard, 2011). Furthermore, the rewarding properties of opioids have led to epidemic increases in prescription opioid misuse, with a concomitant increased risk for lethal overdose (Stannard, 2011). The therapeutic, side effects, and rewarding effects of these drugs are all mediated by μ opioid receptors (Kieffer, 2000). Transduction mechanisms for these receptors include inhibition of adenylate cyclase, blockade of voltage-sensitive calcium channels, stimulation of voltage-gated potassium channels, and opening of G protein-gated inwardly rectifying channels (Williams et al., 2013; Law, 2011). The possibility that opioid transduction mechanisms for pain relief might be distinct from those mediating side effects has remained largely untested.
Morphine analgesia results from stimulation of brain stem circuits which inhibit spinal nociceptive transmission (Yaksh and Wallace, 2010). In the periaqueductal gray, morphine produces many of the above changes, but opening of voltage-gated potassium channels in pre-synaptic GABAergic terminals (reducing GABA release) best accounts for the activation of descending, pain-relieving circuits (Vaughan et al., 1997). Activation of this conductance is mimicked by arachidonic acid (AA) or an AA metabolite (Vaughan et al., 1997). AA metabolism is complex, and can occur through lipoxygenase, cyclooxygenase, and epoxygenase mechanisms. Epoxygenase pathways are catalyzed by several cytochrome P450 mono-oxygenases (P450, Spector, 2009).
In support of an AA epoxygenase mechanism, we recently reported that transgenic mice lacking functional brain neuronal P450 activity showed highly attenuated analgesic responses to morphine (Conroy et al., 2010). Taken with earlier literature, an epoxygenase model of opioid analgesia was proposed wherein opioids act in the brain to relieve pain through sequential activation of μ opioid receptors, phospholipase Cγ, inositol-1,4,5-triphosphate receptors, calcium-dependent phospholipase A2, AA release, P450-catalyzed epoxidation of AA, and formation of epoxyeicosatrienoic acids (EETs, Conroy et al., 2010). EETs may open voltage-gated potassium channels in the periaqueductal gray, but this has not yet been established. More recent in vivo (Conroy et al., 2013) and in vitro (Zhang and Pan, 2012) findings add support for the epoxygenase mechanism of μ action in the brain stem. An alternative hypothesis, that opioid effects in the PAG are mediated by the AA 12-lipoxygenase pathway (Vaughan et al., 1997) has mixed support (Walters et al., 2003; Nigam and Zafiriou, 2005; Conroy et al., 2010; Zhang and Pan, 2012).
The findings outlined above support a brain P450 epoxygenase mechanism for μ opioid analgesia, but the importance of this mechanism in μ opioid side effects has not been assessed. Toward this goal, we presently measured analgesic responses and non-analgesic side effects of morphine in brain neuronal P450-deficient mice and in normal mice treated with a P450 inhibitor. Results confirm the importance of brain P450 activity for morphine analgesia, but not for three important side effects of morphine (respiratory depression, inhibition of intestinal motility, and locomotor stimulation).
2. Materials and methods
2.1. Materials
Morphine sulfate (Sigma-Aldrich, St. Louis, MO) and CC12 hydrochloride (prepared by the Curragh Chemistries method, Hough et al., 2007) were dissolved in saline. Carmine Red (Acros, Fair Lawn, NJ) was suspended in a 0.5% methylcellulose solution (Fisher, Fair Lawn, NJ) for oral administration.
2.2. Animals
Although the mouse genome contains over 100 functional P450 genes (Nelson et al., 2004), all microsomal P450 activity requires cytochrome P450 reductase, encoded by the Cpr gene (also known as Por). Brain neuron-specific P450-deficient mice (designated here as Null) were generated by targeted deletion of the loxP-flanked Cpr gene in Cre-expressing brain neurons (via Camk2a-cre, under control of the Camk2a promoter) as described (Conroy et al., 2010). Null (Cre +/− Cprlox/lox) and wild-type control (Cre −/− Cprlox/lox) adults (greater than 10 weeks of age) of either sex were used for all studies. Animals were maintained on a 12-h light/dark cycle (lights on from 0700 to 1900), provided with food and water and housed in groups of 3–5 until the time of surgery. Genotypes were verified by PCR. All animal experiments were approved by the Institutional Animal Care and Use Committee of Albany Medical College.
2.3. Research design
Separate experiments measured five dependent variables following morphine treatment in mice (nociceptive threshold, respiratory rate, intestinal motility, locomotor activity, and rectal temperature). For each end point, two kinds of experiments were performed, each designed to detect one effect: 1) in naïve (non-cannulated) mice, genotype differences (Null vs. wild-type) were studied following morphine treatment (data in Figs. 1A, 2, 3A, 4A, 5A, 5C). For each effect of morphine in these experiments, the null hypothesis was that there are no genotype differences. 2) In surgically cannulated, wild-type mice, the effects of the P450 inhibitor CC12 (given by intracerebroventricular [i.c.v.] injection) were studied on morphine responses (Figs. 1C, 3B, 4B, 5B, 5D). In these experiments, the null hypothesis was that, for each effect of morphine, CC12 pretreatment had no effect as compared with saline pretreatment. In one additional experiment, i.c.v. morphine was given to wild-type and Null mice to confirm genotype differences (Fig. 1B). Sample sizes were at least 5 subjects per group.
Figure 1.

Significance of brain P450 activity in morphine antinociception. A) Control (WT) and Null mice of either sex were tested for tail-immersion nociceptive responses (time zero = baseline), received saline or morphine sulfate (Mor, 20 mg/kg, s.c.) and were re-tested at the indicated times (abscissa, min). Ordinate shows latencies (sec, mean ± S.D.) for the n values in parentheses. Data from both genders were pooled. B) WT and Null mice of either sex were baseline tested, received an i.c.v. injection of either saline or Mor (10 μg) and re-tested as in A. For A and B, **, ++P < 0.01 versus WT Saline, WT Mor, respectively, at the same time. C) Following BL testing, WT mice of either sex received the P450 inhibitor CC12 (200 nmol, i.c.v.) or saline. Fifteen min later, they were re-tested (Post), injected with Mor (5.6 mg/kg, s.c.) and re-tested at the indicated times. +, ++P < 0.05, 0.01, respectively (Bonferroni), versus Saline/Mor at the same time.
Figure 2.
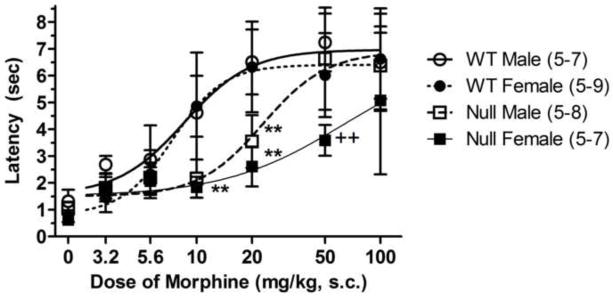
Dose-response curve for morphine antinociception in brain P450-deficient and control mice. Control (WT) and Null mice of either sex received the indicated dose of morphine (abscissa, log scale), and were tested exactly as described in Fig. 1A. Ordinate shows peak latencies (sec, mean ± S.D.) for each dose of morphine for the number of subjects in parentheses. Peak effects occurred at 50 min after 3.2 and 5.6 mg/kg doses, and at 90 min after all other doses. **P < 0.01 for genotype difference within the same gender (Bonferroni), ++P < 0.01 for gender difference within the same genotype (Bonferroni).
Figure 3.

Morphine-induced respiratory depression in brain P450-deficient and control mice. A) Control (WT) and Null mice of either sex received saline or the specified dose of morphine sulfate, and were placed in individual plethysmographic chambers for 150 min. Average breathing rates (breaths per min, averaged over 30–90 min, ordinate, mean ± S.D.) are shown for the number of subjects specified in each bar. **P < 0.01 significant effect of morphine versus saline within same genotype. B) WT mice of either sex were placed in plethysmographic chambers for 33 min, then received CC12 (200 nmol, i.c.v.) or saline. Fifteen min later, they received the indicated doses of morphine, and were placed back in the chambers for an additional 150 min. Breathing rates shown as in A. **P < 0.01 significant effect of morphine versus saline within the same i.c.v. treatment group (Bonferroni).
Figure 4.

Morphine-induced suppression of intestinal motility in brain P450-deficient and control mice. GI motility (ordinate, percent of the small intestine traversed in 20 min by orally-administered dye, mean ± S.D.) is shown for the number of subjects in each bar. Dye was administered 70 min after morphine. A) Control (WT) and Null mice of either sex received the indicated dose of morphine. **P < 0.01 significant effect of morphine versus saline within same genotype. B) WT mice of either sex received CC12 (200 nmol, i.c.v.) or saline, followed 15 min later by the indicated dose of morphine. **P < 0.01 significant effect of morphine versus saline within the same i.c.v. treatment group (Bonferroni).
Figure 5.

Locomotor and thermoregulatory effects of morphine: significance of brain P450 activity. A, C) Control (WT) and Null mice of either sex were tested for baseline rectal temperature, received saline or the designated dose of morphine, and were re-tested as described below. B, D) Cannulated WT mice of either sex were tested for baseline rectal temperature, then received CC12 (200 nmol, i.c.v.) or saline. Fifteen min later, they received the indicated dose of morphine and were re-tested as described below. Ambulatory counts (A, B, ordinate, total photocell beam breaks in ten min, mean ± S.D. for the number of subjects specified, sexes pooled) are shown 40–50 min after morphine. Body temperature changes from baseline (C, D, ordinate, °C, mean ± S.D. for number of subjects specified, sexes pooled) are shown 30 min after morphine. *,**P <0.05, 0.01, respectively, significant effect of morphine versus saline within the same genotype (Bonferroni); +P<0.05 significant difference between genotypes within the same dose of morphine (Bonferroni).
2.4. Brain cannulations and drug injections
As described above, mice were chronically cannulated for subsequent i.c.v. drug administration (Conroy et al., 2010). Following anesthesia with pentobarbital sodium (60 mg/kg, i.p., supplemented with isoflurane), stainless steel guide cannulae were stereotaxically inserted (AP −0.5, ML−1.0, DV−2.0 mm, Paxinos and Franklin, 2001) into the right lateral ventricle and anchored to the skull with stainless steel screws and cranioplast cement. After surgery, subjects were individually housed and were allowed to recover for at least 5 to 7 days before testing. To administer drugs by i.c.v. injection, animals were gently secured using a laboratory pad, the cannula stylet removed, and the injection cannula inserted. The injection cannula extended 1 mm beyond the guide to penetrate the lateral ventricle. All i.c.v. injections were made in a total volume of 2 μl administered over a 1 min period. One min after the end of the infusion, the injection cannula was clipped approximately 2 mm above the juncture with the guide cannula. Successful injections were verified by following the movement of an air bubble in the tubing. After testing, animals received pentobarbital sodium (100 mg/kg, i.p.) and India Ink (5 μl, i.c.v.). Proper distribution of the ink in the cerebroventricular system indicated successful injections. Data from animals with poor placements or unsuccessful injections were excluded. Each animal was used for a single experiment.
2.5. Nociceptive testing
Morphine analgesia was measured in the hot water tail immersion test (Sewell and Spencer, 1976). Subjects were restrained in a conical polypropylene tube, the tail immersed (2–3 cm) into a 55°C water bath, and latency to sudden tail movement or removal of the tail from the water was recorded, with a cutoff latency of 8s. For genotype experiments (Figs. 1A, 1B, 2), subjects were tested for baseline responses, immediately received either an i.c.v. or s.c. injection of morphine or saline, and were re-tested as specified. For the CC12 experiments (Figs. 1C), subjects were baseline tested, immediately received an i.c.v. injection, were re-tested 15 min later, then received a single s.c. injection of morphine and were re-tested as described.
2.6. Upper gastrointestinal transit
For genotype experiments (Fig. 4A), mice received the specified dose of morphine or saline (s.c.). Seventy min later, they received 0.2 cc of a Carmine Red suspension orally (3 g in 50 ml 0.5% methylcellulose). Twenty min later, animals were euthanized with pentobarbital (100 mg/kg, i.p.), the abdomen opened, and the small intestine from the pyloric juncture to the cecal end removed. Percentage of gastrointestinal transit was calculated as the distance traveled by the head of the dye divided by the total length of the intestine (Carai et al., 2006). For the CC12 experiments (Fig. 4B), mice received CC12 or saline (i.c.v.), followed by the specified dose of morphine 15 min later. The Carmine Red procedure was then followed 70 min later as described.
2.7. Body temperature and locomotor activity
In experiments from Fig. 5, nociceptive latencies, rectal temperature and locomotor activity were measured. Temperature was recorded by inserting a lubricated probe (Physitemp, Clifton, NJ) two cm into the rectum. In genotype experiments (Fig. 5A, 5C), rectal temperature was measured immediately after tail immersion testing at baseline, and at 30 and 50 min after s.c. morphine or saline injections. Locomotor activity was recorded by placing individual subjects into a locomotor chamber (Med Associates, St. Albans, VT); total ambulatory counts were quantified over a 10 min period (40 – 50 min post-s.c.). An identical procedure was followed in the CC12 experiments (Figs. 5B, 5D), except that i.c.v. injections were performed immediately after baseline testing, and morphine was administered 15 min later.
2.8. Respiration
Respiratory function was assessed by plethysmography (Whole Body Plethysmograph, Buxco Electronics, Sharon, CT). The system consisted of Model PLY3211 whole body chambers, Max II 2270 Preamp, Bias Flow Regulators, and Biosystem XA software. Prior to testing, each subject was placed in a recording chamber for 30 min a day for three consecutive days. On day 4, animals from Fig. 3A were placed in each chamber for 30 min and baseline respiratory responses recorded. Animals were then removed from the chamber, injected with morphine or saline (s.c.), and returned to each chamber for respiratory monitoring over the next 150 min. In the Fig. 3B experiments, animals on day 4were placed in chambers for 33 min for baseline recording. They were then removed, received an i.c.v. injection, were returned to the chamber and responses recorded for the next 10 min. They were again removed, received a s.c. injection of morphine or saline, and were returned to the chamber where responses were recorded for the next 150 min.
2.9. Statistical analysis
All data were analyzed by analysis of variance (ANOVA, Statistica, StatSoft, Tulsa, OK). For the genotype experiments (Figs. 1A, 1B, 2, 3A, 4A, 5A, 5C), between-group factors included dose of morphine, genotype, and, in some cases, gender. When time course data were recorded (e.g. Figs. 1A, 1B), ANOVAs also included time as a within-group (repeated measures) factor. For the CC12 experiments (Figs. 1C, 3B, 4B, 5B, 5D), between-group factors were dose of morphine and i.c.v. treatment (CC12/saline). For each type of experiment, the structure of the ANOVA performed is explained in the Results section. In addition, F statistics, degrees of freedom, and P values are given for all significant main terms and interaction terms. Effects of morphine, genotype or CC12 were identified as significant when a main effect or one or more relevant interaction terms from the ANOVA yielded a P value of less than 0.05. In such cases, post-hoc analyses were performed by the Bonferroni test. In the absence of such P values, the null hypothesis was concluded to be correct and no post-hoc testing was performed. Significant effects seen from post-hoc testing are labeled in the graphs and explained in each legend. Dose-response curves were generated for morphine by non-linear regression with Prism 5.0 software (Graphpad, San Diego, CA).
3. Results
3.1. Morphine analgesia (systemic administration)
Systemic administration of morphine (20 mg/kg, s.c.) to control mice produced a robust, long-lasting increase in nociceptive latencies which was reduced by approximately 50% in Null subjects (Fig. 1A). ANOVA of the data in Fig. 1A (between groups: morphine, genotype; within groups [repeated measures]: time) found significant main effects of morphine (F1,28=194.8, P<0.0001), genotype (F1,28=30.2, P<0.0001), and time (F5,140 =26.1, P<0.0001), with significant morphine by genotype (F1,28=21.7, P<0.0001) and morphine by genotype by time (F5,140 =3.7, P<0.01) interaction terms.
Complete dose-response curves with systemically-administered morphine were obtained in both genders in the two genotypes (Fig. 2). Results revealed a clear deficit in morphine analgesia in male and female Null mice after doses of 10 and 20 mg/kg, as compared with respective wild-type control subjects. After 50 mg/kg, the genotype difference was seen only in female mice. No significant genotype differences were noted after the highest dose of morphine (100 mg/kg). ANOVA of the data in Fig. 2 (between groups: dose of morphine, genotype, and gender) found significant main effects of dose (F4,90=59.7, P<0.0001), genotype (F1,90=47.5, P<0.0001), and gender (F1,90 =9.3, P<0.01), with a significant dose by genotype (F4,90=5.7, P<0.001) interaction.
3.2. Morphine analgesia (i.c.v. administration)
To verify that the genotype differences following systemically-administered morphine are attributable to differences in brain responses, we also measured nociceptive responses following i.c.v. administration of morphine. I.c.v. morphine (10 μg) produced a large, long-lasting, antinociceptive effect in control subjects which was attenuated by more than 50% in Null mice (Fig. 1B). ANOVA of the data in Fig. 1B (between groups: morphine, genotype, gender; within groups [repeated measures]: time) found no significant gender-related factors. After collapsing genders, ANOVA found significant main effects of morphine (F1,25=57.6, P<0.0001), genotype (F1,25=18.3, P<0.0001), and time (F5,125 =29.8, P<0.0001), with significant morphine by genotype (F1,25=17.7, P<0.0001) and morphine by genotype by time (F5,125 =5.6, P<0.0002) interaction terms. We also confirmed that i.c.v. pretreatment of control mice with the P450 inhibitor CC12 blocked morphine analgesia (Fig. 1C). ANOVA of Fig. 1C data found main effects of CC12 (F1,14=8.2, P<0.02), time (F4,56=51.9, P<0.0001), with a significant CC12 by time interaction (F4,56=6.0, P<0.001).
3.3. Respiratory depression
To study opioid-induced respiratory depression, we administered morphine to control and Null mice, and monitored breathing rates. Systemic administration of morphine (20 mg/kg, s.c.) suppressed average breathing frequencies in all mice by approximately 40%. No genotype differences were detected in breathing rates following saline or morphine (Fig. 3A). ANOVA of the data in Fig. 3A (morphine, genotype, gender) found a highly significant main effect of morphine (F1,32=57.1, P<0.0001 ), with no significant genotype-related factors. P values were 0.21 and 0.80 for main effect of genotype and genotype by morphine interaction, respectively. Breathing rates in males were slightly, but significantly lower than in females in all groups (main effect of gender: F1,32=4.5, P<0.05, data not shown ), with no significant drug by gender or genotype by gender interactions
To further study P450 function on respiration, we also blocked brain P450 activity with CC12 in control mice. I.c.v. pre-treatment with CC12 did not affect respiratory rates following saline or morphine (Fig. 3B). In i.c.v.-cannulated mice, pilot studies found no respiratory depression by 20 mg/kg of morphine (data not shown), and a dose of 50 mg/kg of morphine was required to match the respiratory depression achieved by 20 mg/kg in non-cannulated controls (Fig. 3A). ANOVA of the data of Fig. 2B (dose of morphine, CC12) found a significant main effect of morphine (F2,34=26.1, P<0.0001), with no significant CC12-related factors.
3.4. Intestinal transit
Opioids act on brain and peripheral μ opioid receptors to reduce intestinal motility, thereby causing constipation, a therapy-limiting side effect of these medications. To assess a possible role for brain P450 activity in mediating this opioid effect, we measured the intestinal transit of an orally-administered dye after morphine treatment in control and Null mice. As compared with saline responses, morphine (20 mg/kg, s.c.) suppressed intestinal transit by 79% and 67% in control and Null mice, respectively (Fig. 4A), not a significant genotype difference. Genotype differences were also not observed in the saline treatment groups. ANOVA of the transit data in Fig. 4A (morphine, genotype, gender) found a highly significant main effect of morphine (F1,25=157.6, P<0.0001 ), with no significant genotype-related factors. P values were 0.26 and 0.44 for main effect of genotype and genotype by morphine interaction, respectively. However, females in both genotypes showed slightly (but significantly) less opioid-induced suppression of motility than did the respective male groups (data not shown, morphine by gender interaction: F1,25=7.1, P<0.02).
We also studied intestinal transit after blocking brain P450 activity. I.c.v. pre-treatment of control mice with CC12 had no effect on intestinal transit following saline or morphine administration (Fig. 4B). ANOVA (dose of morphine, CC12, gender) found a significant main effect of morphine (F1,16=74.9, P<0.0001), with no significant CC12-related or gender-related differences. It should be noted that in cannulated, i.c.v. saline-treated, wild-type mice, a dose of 5.6 mg/kg of morphine suppressed intestinal transit by 71% ( Fig. 4B), similar to the suppression (79%) seen in non-cannulated mice receiving a higher dose of morphine (20 mg/kg, Fig. 4A).
3.5. Locomotor stimulation
The motor stimulant effects of opioids do not contribute to pain relief, but are closely related to the reinforcing and addictive properties of these drugs. To investigate the role of brain P450s in opioid stimulant properties, we measured spontaneous locomotor activity following morphine treatment in control and Null mice. Two doses of morphine (20 and 50 mg/kg, s.c.) increased locomotor activity in both mouse genotypes, with no genotype differences seen after either saline or morphine (Fig. 5A). ANOVA (dose of morphine, genotype) found a highly significant main effect of dose of morphine (F2,38=28.7, P<0.0001 ), with no significant effects of genotype (P = 0.93) or genotype by dose (P=0.44) interactions. Sample sizes precluded a gender analysis of the data from these experiments.
Similar to the other experiments above, we also determined the effect of CC12 on morphine-induced locomotor stimulation in control mice. CC12 pretreatment did not affect locomotor activity following administration of saline or two doses of morphine (5.6 and 20 mg/kg, Fig. 5B). ANOVA (dose of morphine, CC12, gender) found a highly significant main effect of dose of morphine (F2,24=21.0, P<0.0001 ), with no significant CC12-related or gender-related terms.
3.6. Body temperature
Rectal temperature was also monitored during the experiments of Fig. 5. In control mice, morphine (20 mg/kg, s.c.) lowered body temperature by 2°C 30 min after administration (Fig. 5C), effects which dissipated 20 min later (data not shown). The effect was less pronounced after the higher (50 mg/kg) dose of morphine (Fig. 5C). However, no opioid-induced hypothermia was detected in Null mice (Fig. 5C). ANOVA (dose of morphine, genotype) found highly significant main effects of dose of morphine (F2,38=9.2, P<0.001), and also genotype (F1,38=8.3, P<0.01). Surprisingly, CC12 pretreatment did not antagonize morphine hypothermia (Fig. 5D). ANOVA (dose of morphine, CC12, gender) found a highly significant main effect of dose of morphine (F2,25 =34.1, P<0.0001), with no significant CC12-related or gender-related terms.
4. Discussion
Characterization of morphine responses in Null mice provides a genetic approach to the study of μ opioid receptors transduction mechanisms. It should be noted that Cpr is not deleted in every neuron of the Null mouse brain. In Null mice, the loxP-flanked Cpr gene is deleted in Cre-expressing CNS neurons via Camk2a-cre, a transgene under control of the Camk2a promoter (Conroy et al., 2010). Although Camk2a is predominantly expressed in forebrain (implying that Cpr deletion might be limited to the forebrain in these mice, Dragatsis and Zeitlin, 2000), a population of ventrolateral periaqueductal gray neurons was shown to lack cytochrome P450 reductase in the P450 Null mice (Conroy et al., 2010). Because Cpr deletion is controlled by the Camk2a promoter (not functional in all neurons), only a subset of the neurons in Null mice possess a P450 deficit. Under these circumstances, the detection of a phenotype in these mice (e.g. a deficit in morphine analgesia) is powerful evidence for the importance of brain neuronal P450. However, when Null mice do not show a deficit in morphine responses (as in the presently-observed side effects), a relevant P450 could simply be in a non-Camk2a-expressing portion of brain. To control for this possibility, all of morphine’s actions studied presently were investigated by both genetic (Null mice) and pharmacological (P450 inhibitor) approaches.
Dose-response studies (Fig. 2) showed that the deficit in morphine analgesia in Null mice was surmounted by large doses of morphine. This is likely due to activation of spinal μ opioid receptors which may not require neuronal P450 signaling. For example, G protein-gated inwardly rectifying channels play a significant role in spinal (Marker et al., 2005) but not supraspinal (Vaughan et al., 1997) opioid analgesia. Independent of spinal mechanisms, however, a small, residual analgesia was seen in Null mice following a moderate i.c.v. dose of morphine (Fig. 1B). Residual P450 activity in the brain stem (due to incomplete deletion of Cpr in all relevant Null neurons) could easily account for these results.
The present results confirm the analgesic phenotype in both sexes of Null mice (Fig. 2). Furthermore, the sex difference in morphine analgesia in Null mice (males > females, 50 mg/kg, Fig. 2) was seen previously in control C57Bl/6 mice after i.c.v. morphine (Kest et al., 1999). This gender difference was not detected in opioid responses in our control mice (Fig. 2), probably due to differing test conditions (Kest et al., 1999). The sex difference in morphine analgesia among Null mice suggests that female brains may have a greater dependence upon P450-related analgesic mechanisms. There are well-documented examples of neurochemically-distinct sexual dimorphisms in morphine analgesia (Chakrabarti et al., 2010; Mogil and Bailey, 2010).
CC12, a chemical congener of miconazole (Hough et al., 2007), was used in the present studies to inhibit brain P450 activity for several reasons: 1) other P450 inhibitors (e.g. miconazole) act at many non-P450 sites and have biphasic actions in vivo on analgesia (Hough et al., 2011), 2) CC12 in vivo produces dose-dependent inhibition of analgesia (Hough et al., 2007), 3) CC12 is potent inhibitor of a wide range of P450 isoforms. The compound is active in nanomolar concentrations on CYP1A, CYP2A, CYP2B, CYP2C, CYP2D, CYP2E, CYP3A families, as well as others (Stadel et al., 2008). Finally, 4) CC12 lacks significant activity at over 20 analgesia-relevant binding sites and enzymes (Hough et al., 2007; Conroy et al., 2010). CC12 was administered by i.c.v. injection because it does not readily penetrate the brain.
Chronic ventricular cannulation of control mice (necessary for i.c.v. CC12 administration) altered morphine potencies in several cases. Cannulated control mice receiving i.c.v. saline showed enhanced morphine effects on analgesia, locomotor stimulation, and intestinal transit as compared with non-cannulated, naïve subjects. The stress associated with surgery and/or i.c.v. injection likely explains these effects (Calcagnetti and Holtzman, 1990; Kalivas and Stewart, 1991). In order to study the effects of CC12 under conditions closest to the Null vs. control experiments (in which mice were not cannulated), the dose of morphine was adjusted in cannulated mice to give an effect equivalent to that seen with the original dose of morphine given to non-cannulated subjects. Thus, a morphine dose of 5.6 mg/kg in cannulated mice produced analgesia, locomotor stimulation, and intestinal hypomotility equivalent to the 20 mg/kg dose in naïve subjects (compare Fig. 1C vs. 1A, Fig. 5B vs. 5A, and Fig. 4B vs. 4A, respectively). Cannulation did not have consistent effects on all morphine responses, however. Cannulated mice were less sensitive to opioid-induced respiratory depression: a dose of 50 mg/kg of morphine was required to evoke an effect comparable to that of 20 mg/kg in naïve subjects (Fig. 3B vs. 3A). Morphine hypothermia was equivalent in cannulated and non-cannulated control subjects (Fig. 5D vs. Fig. 5C).
Respiratory depression, the leading cause of opioid death (Stannard, 2011), results from actions on medullary and pontine μ opioid receptors to suppress ventilation and to reduce chemosensory reflexes (Zhang et al., 2007; Pattinson, 2008). Opioids are reported to depress breathing following injections into the pre-Botzinger nucleus, other dorsal and ventral respiratory group nuclei, rostral ventromedial medulla, and caudal medial medulla (Zhang et al., 2007; Phillips et al., 2012; Hellman et al., 2009; Lalley, 2006; Manzke et al., 2003; Pattinson, 2008). There are numerous discrepancies in the literature, many of which are due to species, anesthesia, or other experimental variables. The pre-Botzinger nucleus may be particularly significant for the respiratory effects of systemically-administered opioids (Pattinson, 2008, but see also Mustapic et al., 2010; Stucke et al., 2008; and Phillips et al., 2012). The finding that morphine produces comparable respiratory depression in Null and control mice (Fig. 3) suggests that, unlike the case for analgesia, opioid-induced respiratory depression does not depend on neuronal P450 activity. Although the respiratory depression in Null mice (Fig. 3A) could be due to the absence of Cre expression in brain stem respiratory areas, inhibition of brain P450 activity in normal mice by CC12 did not prevent morphine-induced respiratory depression (Fig. 3B). Thus, both experiments of Fig. 3 suggest that P450 transduction mechanisms are not important for opioid-induced respiratory depression. Mu-mediated inhibition of adenylate cyclase has been suggested to be a transduction mechanism for respiratory depression (Manzke et al., 2003; Lalley, 2008), a hypothesis consistent with the present results.
Constipation can be a therapy-limiting side effect of opioids (Stannard, 2011). Intestinal propulsion is suppressed by both peripheral and CNS μ opioid receptors mechanisms (Shook et al., 1989). The present results in Null mice and with CC12 in normal mice (Fig. 4) found no role for brain P450 activity in mediating the central anti-motility actions of morphine. The peripheral opioid mechanisms were not tested presently.
The separation of the pain-relieving and rewarding properties of μ opioids is a critical goal for pain research,. Although the relief of ongoing pain (by any kind of medication) is a rewarding event for the brain (Navratilova et al., 2012), opioids also activate reward circuits in the absence of ongoing pain. Stimulation of μ opioid receptors on GABAergic terminals in the ventral tegmental area and on GABAergic elements of the nucleus accumbens both lead to activation of ventral pallidum, thought to account for the rewarding effects of opioids (Xi and Stein, 2002), and to explain opioid-induced locomotor stimulation (Wise and Bozarth, 1987). The lack of genotype differences in locomotor responses to moderate or large doses of morphine (Fig 5A), and the lack of effect of CC12 (Fig 5B) in control mice indicate that, unlike the case for analgesia, a brain P450 mechanism does not seem to be required for opioid-induced locomotor stimulation. Although further work is needed, the same conclusion may possibly apply to opioid reward/abuse mechanisms.
Morphine exerts biphasic actions on body temperature; experimental variables include the dose of drug, route of administration, and ambient temperature (Rawls and Benamar, 2011). Mu and kappa opioid receptors in the preoptic anterior hypothalamus are thought to mediate the hyperthermic and hypothermic effects, respectively (Xin et al., 1997). Presently, morphine-induced hyperthermia was not detected in control mice over a range of doses (Fig. 5C and data not shown). The hypothermia following morphine (20 mg/kg) was significantly blunted in Null (vs. control) mice (Fig. 5C), suggesting that kappa opioid-mediated hypothermia may utilize a brain P450 mechanism. Further studies of this hypothesis are needed, as CC12 did not alter opioid hypothermia in control mice (Fig. 5D). Such experiments might include measurement of kappa agonist-induced hypothermia in Null vs. control mice (to confirm involvement of kappa opioid vs. μ opioid receptors), as well as studies of the effects of other kinds of P450 inhibitors on opioid hypothermia (e.g. see Conroy et al., 2010).
Congruent with the P450/epoxygenase model, new classes of analgesic drugs are being developed either to mimic EET action (Brostram and Falck, 2011) or to inhibit brain EET metabolism (Wagner et al., 2013). Our present results, showing that three major μ opioid side effects (respiratory depression, locomotor stimulation, and constipation) do not require brain P450 activity, suggest that these or other epoxygenase-based pain-relieving medications may be devoid of such therapy-limiting side effects.
Acknowledgments
This work was supported by a grant from the National Institutes of Health National Institute on Drug Abuse (Grant DA027835). We thank Dr. Scott O. Zeitlin (University of Virginia School of Medicine) for providing the Camk2a-Cre mouse model (Conroy et al., 2010).
Footnotes
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aoki T, Narita M, Ohnishi O, Mizuo K, Narita M, Yajima Y, Suzuki T. Disruption of the type 1 inositol 1,4,5-trisphosphate receptor gene suppresses the morphine-induced antinociception in the mouse. Neurosci Lett. 2003;350:69–72. doi: 10.1016/s0304-3940(03)00829-2. [DOI] [PubMed] [Google Scholar]
- Brostram Lane, Falck JR. Arachidonic acid analogs and methods for analgesic treatment using same. CYTOMETIX, INC; WI, USA: 2011. pp. 1–58. PCT/US2010/058041[WO/2011/066414] [Google Scholar]
- Calcagnetti DJ, Holtzman SG. Factors affecting restraint stress-induced potentiation of morphine analgesia. Brain Res. 1990;537:157–162. doi: 10.1016/0006-8993(90)90352-c. [DOI] [PubMed] [Google Scholar]
- Carai MA, Colombo G, Gessa GL, Yalamanchili R, Basavarajappa BS, Hungund BL. Investigation on the relationship between cannabinoid CB1 and opioid receptors in gastrointestinal motility in mice. Br JPharmacol. 2006;148:1043–1050. doi: 10.1038/sj.bjp.0706824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S, Liu NJ, Gintzler AR. Formation of mu-/kappa-opioid receptor heterodimer is sex-dependent and mediates female-specific opioid analgesia. Proc Natl Acad Sci U S A. 2010;107:20115–20119. doi: 10.1073/pnas.1009923107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy JL, Fang C, Gu J, Zeitlin SO, Yang W, VanAlstine MA, Nalwalk JW, Albrecht PJ, Mazurkiewicz JE, Snyder-Keller A, Shan Z, Zhang S, Wentland MP, Behr M, Knapp BI, Bidlack JM, Zuiderveld OP, Leurs R, Ding X, Hough LB. Opioids activate brain analgesic circuits through cytochrome p450/epoxygenase signaling. Nature Neuroscience. 2010;13:284–286. doi: 10.1038/nn.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy JL, Nalwalk JW, Phillips JG, Hough LB. CC12, a P450/epoxygenase inhibitor, acts in the rat rostral, ventromedial medulla to attenuate morphine antinociception. Brain Res. 2013;1499:1–11. doi: 10.1016/j.brainres.2012.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragatsis I, Zeitlin S. CaMKIIalpha-Cre transgene expression and recombination patterns in the mouse brain. Genesis. 2000;26:133–135. doi: 10.1002/(sici)1526-968x(200002)26:2<133::aid-gene10>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Ferguson CS, Tyndale RF. Cytochrome P450 enzymes in the brain: emerging evidence of biological significance. Trends Pharmacol Sci. 2011;32:708–714. doi: 10.1016/j.tips.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda K, Kato S, Morikawa H, Shoda T, Mori K. Functional coupling of the delta-, mu-, and kappa-opioid receptors to mitogen-activated protein kinase and arachidonate release in chinese hamster ovary cells. J Neurochem. 1996;67:1309–1316. doi: 10.1046/j.1471-4159.1996.67031309.x. [DOI] [PubMed] [Google Scholar]
- Hellman KM, Mendelson SJ, Mendez-Duarte MA, Russell JL, Mason P. Opioid microinjection into raphe magnus modulates cardiorespiratory function in mice and rats. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1400–R1408. doi: 10.1152/ajpregu.00140.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough LB, Nalwalk JW, Phillips JG, Kern B, Shan Z, Wentland MP, Iwan Esch JP, Janssen E, Barr T, Stadel R. CC12, a high-affinity ligand for [3H]cimetidine binding, is an improgan antagonist. Neuropharmacology. 2007;52:1244–1255. doi: 10.1016/j.neuropharm.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough LB, Nalwalk JW, Yang J, Conroy JL, VanAlstine MA, Yang W, Gargano J, Shan Z, Zhang SZ, Wentland MP, Phillips JG, Knapp BI, Bidlack JM, Zuiderveld OP, Leurs R, Ding X. Brain P450 epoxygenase activity is required for the antinociceptive effects of improgan, a nonopioid analgesic. Pain. 2011;152:878–887. doi: 10.1016/j.pain.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Stewart J. Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res Brain Res Rev. 1991;16:223–244. doi: 10.1016/0165-0173(91)90007-u. [DOI] [PubMed] [Google Scholar]
- Kest B, Wilson SG, Mogil JS. Sex differences in supraspinal morphine analgesia are dependent on genotype. J Pharmacol Exp Ther. 1999;289:1370–1375. [PubMed] [Google Scholar]
- Kieffer BL. Opioid receptors: from genes to mice. J Pain. 2000;1:45–50. doi: 10.1054/jpai.2000.9823. [DOI] [PubMed] [Google Scholar]
- Lalley PM. Opiate slowing of feline respiratory rhythm and effects on putative medullary phase-regulating neurons. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1387–R1396. doi: 10.1152/ajpregu.00530.2005. [DOI] [PubMed] [Google Scholar]
- Lalley PM. Opioidergic and dopaminergic modulation of respiration. Respir Physiol Neurobiol. 2008;164:160–167. doi: 10.1016/j.resp.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law PY. Opioid Receptor Signal Transduction Mechanims. In: Pasternak GW, editor. The Opioid Receptors. Humana Press; New York: 2011. pp. 195–238. [Google Scholar]
- Manzke T, Guenther U, Ponimaskin EG, Haller M, Dutschmann M, Schwarzacher S, Richter DW. 5-HT4(a) receptors avert opioid-induced breathing depression without loss of analgesia. Science. 2003;301:226–229. doi: 10.1126/science.1084674. [DOI] [PubMed] [Google Scholar]
- Marker CL, Lujan R, Loh HH, Wickman K. Spinal G-protein-gated potassium channels contribute in a dose-dependent manner to the analgesic effect of mu- and delta- but not kappa-opioids. J Neurosci. 2005;25:3551–3559. doi: 10.1523/JNEUROSCI.4899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogil JS, Bailey AL. Sex and gender differences in pain and analgesia. Prog Brain Res. 2010;186:141–157. doi: 10.1016/B978-0-444-53630-3.00009-9. [DOI] [PubMed] [Google Scholar]
- Mustapic S, Radocaj T, Sanchez A, Dogas Z, Stucke AG, Hopp FA, Stuth EA, Zuperku EJ. Clinically relevant infusion rates of mu-opioid agonist remifentanil cause bradypnea in decerebrate dogs but not via direct effects in the pre-botzinger complex region. J Neurophysiol. 2010;103:409–418. doi: 10.1152/jn.00188.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Ohnishi O, Narita M, Aoki T, Suzuki M, Yajima Y, Funahashi H, Shioda S, Suzuki T. Direct evidence for the activation of phospholipase C gamma 1 by in vivo treatment with morphine in the mouse periaqueductal gray matter. Brain Res. 2003;970:140–148. doi: 10.1016/s0006-8993(03)02301-1. [DOI] [PubMed] [Google Scholar]
- Navratilova E, Xie JY, Okun A, Qu C, Eyde N, Ci S, Ossipov MH, King T, Fields HL, Porreca F. Pain relief produces negative reinforcement through activation of mesolimbic reward-valuation circuitry. Proc Natl Acad Sci U S A. 2012;109:20709–20713. doi: 10.1073/pnas.1214605109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DR, Zeldin DC, Hoffman SM, Maltais LJ, Wain HM, Nebert DW. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics. 2004;14:1–18. doi: 10.1097/00008571-200401000-00001. [DOI] [PubMed] [Google Scholar]
- Nigam S, Zafiriou MP. Hepoxilin A3 synthase. Biochem Biophys Res Commun. 2005;338:161–168. doi: 10.1016/j.bbrc.2005.09.065. [DOI] [PubMed] [Google Scholar]
- Pattinson KT. Opioids and the control of respiration. Br J Anaesth. 2008;100:747–758. doi: 10.1093/bja/aen094. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. Academic Press; San Diego: 2001. [Google Scholar]
- Phillips RS, Cleary DR, Nalwalk JW, Arttamangkul S, Hough LB, Heinricher MM. Pain-facilitating medullary neurons contribute to opioid-induced respiratory depression. J Neurophysiol. 2012;108:2393–2404. doi: 10.1152/jn.00563.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawls SM, Benamar K. Effects of opioids, cannabinoids, and vanilloids on body temperature. Front Biosci (Schol Ed) 2011;3:822–845. doi: 10.2741/190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewell RDE, Spencer PSJ. Antinociceptive activity of narcotic agonist and partial agonist analgesics and other agents in the tail-immersion test in mice and rats. Neuropharmacol. 1976;15:683–688. doi: 10.1016/0028-3908(76)90037-x. [DOI] [PubMed] [Google Scholar]
- Shook JE, Lemcke PK, Gehrig CA, Hruby VJ, Burks TF. Antidiarrheal properties of supraspinal mu and delta and peripheral mu, delta and kappa opioid receptors: inhibition of diarrhea without constipation. J Pharmacol Exp Ther. 1989;249:83–90. [PubMed] [Google Scholar]
- Spector AA. Arachidonic acid cytochrome P450 epoxygenase pathway. J Lipid Res. 2009;50(Suppl):S52–S56. doi: 10.1194/jlr.R800038-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadel R, Yang J, Nalwalk JW, Phillips JG, Hough LB. High affinity binding of [3H]-cimetidine to a heme-containing protein in rat brain. Drug Metab Dispos. 2008;36:614–621. doi: 10.1124/dmd.107.017889. Ref Type: Journal (Full) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stannard CF. Opioids for chronic pain: promise and pitfalls. Curr Opin Support Palliat Care. 2011;5:150–157. doi: 10.1097/SPC.0b013e3283458fbc. [DOI] [PubMed] [Google Scholar]
- Stucke AG, Zuperku EJ, Sanchez A, Tonkovic-Capin M, Tonkovic-Capin V, Mustapic S, Stuth EA. Opioid receptors on bulbospinal respiratory neurons are not activated during neuronal depression by clinically relevant opioid concentrations. J Neurophysiol. 2008;100:2878–2888. doi: 10.1152/jn.90620.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashvili M, Tseng LF, Wu HE, Narayanan J, Hart LM, Falck JR, Pratt PF, Harder DR. Antinociception produced by 14,15-epoxyeicosatrienoic acid is mediated by the activation of {beta}-endorphin and met-enkephalin in the rat ventrolateral periaqueductal gray. J Pharmacol Exp Ther. 2008;326:614–619. doi: 10.1124/jpet.108.136739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Ingram SL, Connor MA, Christie MJ. How opioids inhibit GABA-mediated neurotransmission. Nature. 1997;390:611–614. doi: 10.1038/37610. [DOI] [PubMed] [Google Scholar]
- Wagner K, Inceoglu B, Dong H, Yang J, Hwang SH, Jones P, Morisseau C, Hammock BD. Comparative efficacy of 3 soluble epoxide hydrolase inhibitors in rat neuropathic and inflammatory pain models. Eur J Pharmacol. 2013;700:93–101. doi: 10.1016/j.ejphar.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters CL, Wang BC, Godfrey M, Sun D, Funk CD, Blendy JA. Augmented responses to morphine and cocaine in mice with a 12-lipoxygenase gene disruption. Psychopharmacology (Berl) 2003;170:124–131. doi: 10.1007/s00213-003-1526-7. [DOI] [PubMed] [Google Scholar]
- Williams JT, Ingram SL, Henderson G, Chavkin C, von ZM, Schulz S, Koch T, Evans CJ, Christie MJ. Regulation of mu-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev. 2013;65:223–254. doi: 10.1124/pr.112.005942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA, Bozarth MA. A psychomotor stimulant theory of addiction. Psychol Rev. 1987;94:469–492. [PubMed] [Google Scholar]
- Xi ZX, Stein EA. GABAergic mechanisms of opiate reinforcement. Alcoholism (NY) 2002;37:485–494. doi: 10.1093/alcalc/37.5.485. [DOI] [PubMed] [Google Scholar]
- Xin L, Geller EB, Adler MW. Body temperature and analgesic effects of selective mu and kappa opioid receptor agonists microdialyzed into rat brain. J Pharmacol Exp Ther. 1997;281:499–507. [PubMed] [Google Scholar]
- Yaksh TL, Wallace MS. Opioids, Analgesia and Pain Management. In: Brunton LL, Chabner B, Knollman B, editors. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. McGraw-Hill Professional Publishing; New York: 2010. [Google Scholar]
- Zhang Z, Pan ZZ. Signaling cascades for delta-opioid receptor-mediated inhibition of GABA synaptic transmission and behavioral antinociception. Mol Pharmacol. 2012;81:375–383. doi: 10.1124/mol.111.076307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Xu F, Zhang C, Liang X. Activation of opioid mu receptors in caudal medullary raphe region inhibits the ventilatory response to hypercapnia in anesthetized rats. Anesthesiology. 2007;107:288–297. doi: 10.1097/01.anes.0000270760.46821.67. [DOI] [PubMed] [Google Scholar]