

Abstract
Alternative splicing is highly regulated in tissue-specific and development-specific patterns, and it has been estimated that 15% of disease-causing point mutations affect pre-mRNA splicing. In this review, we consider the cis-acting splice site and trans-acting splicing factor mutations that affect pre-mRNA splicing and contribute to retinal degeneration. Numerous splice site mutations have been identified in retinitis pigmentosa and various cone-rod dystrophies. For example, mutations in alternatively spliced retina-specific exons of the widely expressed RPGR and COL2A1 genes lead primarily to X-linked retinitis pigmentosa and ocular variants of Stickler Syndrome, respectively. Furthermore, mutations in general pre-mRNA splicing factors, such as PRPF31, PRPF8, and PRPF3, predominantly cause autosomal dominant retinitis pigmentosa. These findings suggest an important role for pre-mRNA splicing in retinal homeostasis and the pathogenesis of retinal degenerative diseases. The development of novel therapeutic strategies to modulate aberrant splicing, including small molecule based therapies, has the potential to lead to the development of new treatments for retinal degenerative diseases.
Keywords: alternative splicing, retinal degeneration, retinitis pigmentosa, small molecules
Retinitis pigmentosa (RP) is a heterogeneous group of inherited retinal degenerative disorders that affects approximately 1 in 4,000 in the United States (1). Patients initially experience night blindness and peripheral vision loss, with central retinal involvement generally becoming prominent only later in life. Clinical assessment reveals atrophy of the retinal pigment epithelium (RPE) with bone spicule pigmentation in the peripheral retina, waxy pallor of the optic disc, and retinal vessel attenuation. Mutations in more than 45 genes that can cause nonsyndromic RP have been identified (https://sph.uth.edu/retnet/), including rhodopsin, affected in 25% of autosomal dominant RP (adRP), USH2A, affected in 20% of autosomal recessive RP (arRP), and RPGR, affected in 70% of X-linked RP (XLRP) (2). Although nonsyndromic RP, by definition, has an exclusively ocular phenotype, not all of the disease-causing genes are preferentially or exclusively expressed in the retina. As one example, relevant to the focus of this review, mutations in factors that are ubiquitously expressed and important for the general process of pre-mRNA splicing have been identified as causes of adRP.
Eukaryotic pre-mRNA splicing is a process by which intervening intronic sequences are removed and the remaining exonic segments are ligated to form mature mRNA molecules (Fig. 1a) (3). Splicing occurs in the spliceosome, a complex of 5 small nuclear ribonucleoproteins (snRNPs) and additional factors, which catalyzes the two transesterification reactions needed to join each donor splice site, located at the 5’ end of the intron, to its corresponding acceptor splice site, located at the 3’ end of the intron. A branch point adenosine residue, located within the intron, serves as the nucleophile for the first transesterification reaction. The U1 snRNP binds the 5’ splice site, the U2 snRNP binds the branch point adenosine, and U2AF binds the intronic polypyrimidine tract of the 3’ splice site. The U4/U6/U5 tri-snRNP is then recruited, and a series of conformational changes occurs, allowing joining of adjacent exons and the removal of the intron lariat.
Figure 1.
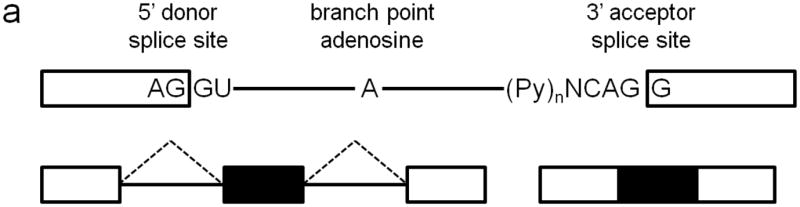
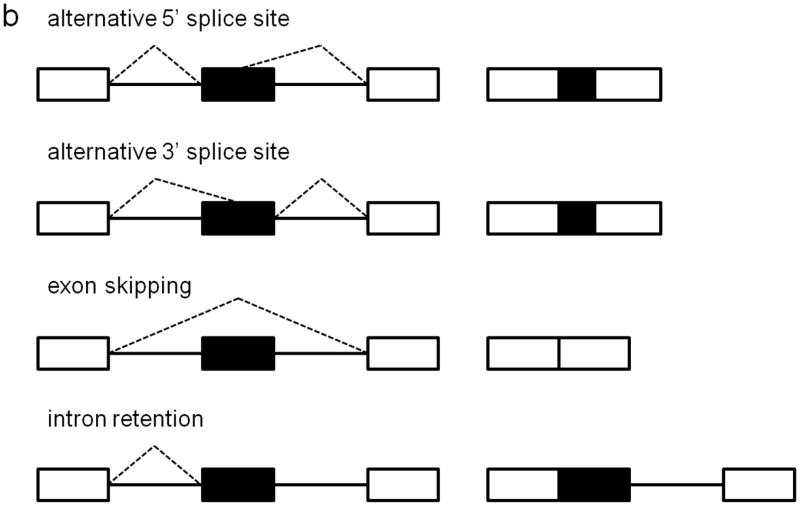
Approximately 80% of human exons are less than 200 bases in length (4), and they are separated by introns that may be up to many thousands of bases long, but despite this complexity, pre-mRNA splicing nevertheless occurs with high fidelity. Splice site selection and recognition is mediated by the strength of the splice site sequence, the presence of cis-acting regulatory elements, and the availability of trans-acting splicing factors (5). Exon definition allows the splice sites flanking each exon to be recognized as paired, via exon-bridging interactions between snRNPs and other splicing factors. Non-splice site RNA sequences can also enhance or repress spliceosome activity. Serine-argine (SR) proteins are exonic splicing enhancers that bind purine-rich exonic splicing enhancer element sequences to promote both constitutive and regulated splicing activity by enhancing usage of weak upstream 3’ splice sites, stabilizing intron-spanning interactions, and promoting exon-bridging interactions by binding to both U2AF and U1 (6). A number of other factors have been identified as enhancer or repressor splicing regulators, and the combinatorial binding of these different splicing factors, whose expression levels are also regulated, leads to high fidelity spatiotemporal exon/intron definition (7, 8). Alternative splicing can result from alternative 5’ splice site usage, alternative 3’ splice site usage, exon skipping, or intron retention, and these mechanisms allow for a single gene to encode multiple protein products that can have vastly different functional properties (Fig. 1b). It is estimated that at least 74% of human genes are alternatively spliced (9), and of these, at least 10-30% have tissue-specific splice isoforms (10). Alternative splicing is highly regulated in tissue-specific and development-specific patterns, and neurons have a particularly high abundance of differentially spliced genes (11). It is estimated that 15% of disease-causing point mutations affect pre-mRNA splicing (12). In this review, we consider the role of cis-acting splice site mutations and mutations in trans-acting splicing factors in the pathogenesis of retinal degenerative disease.
Cis-acting splice site mutations
A myriad of splice site mutations have been identified in patients with retinitis pigmentosa, Usher syndrome, cone rod dystrophy, and other retinal degenerative diseases, and assays have been performed to characterize the functional effects of some of these splice site mutations in vitro. Most of the mutations disrupt a consensus splice site sequence and cause exon skipping, but some result in intron inclusion, novel exon inclusion, or the usage of cryptic upstream or downstream splice sites. The resultant insertions or deletions in the protein sequence, often accompanied by frameshift and premature termination, disrupt conserved or functional protein domains and result in retinal degeneration. A detailed discussion of these cis-acting splice site mutations is included in the Supplement. Additionally, although they will not be considered further in this review, there are also a number of rodent models of retinal degeneration that result from cis-acting splicing defects or large deletions in the genomic DNA that result in exon skipping, including the rd16 mouse, in which exons 35-39 of Cep290 are skipped (13); the RCS rat, in which exon 2 of Mertk is skipped (14); the rd6 mouse, in which exon 4 of Mfrp is skipped (15); and the rd7 mouse, in which exons 4-5 of Nr2e3 are skipped (16).
Alternative splice isoforms
Stickler syndrome type I, an autosomal dominant disease caused by mutations in COL2A1, is characterized by acquired radial perivascular retinal degeneration, congenital vitreous syneresis, high myopia, early-onset cataract, glaucoma, junvenile osteoarthritis, sensorineural hearing loss, and craniofacial abnormalities. In the vitreous, COL2A1 includes exon 2, whereas exon 2 is absent in most other tissues, including cartilage (17). In a large family from the Southwestern United States with Stickler syndrome type I, the causative mutation was Cys86X, a premature stop codon in the alternatively spliced exon 2 (18). Interestingly, as this mutated transcript is expressed specifically in the eye, only 4 of 100 people in this family showed any of the extraocular manifestations characteristic of Stickler syndrome. A frameshift mutation in exon 2 leading to premature termination, Cys57X, was also identified in a French-Canadian family with a similar autosomal dominant vitreoretinal degeneration (19).
The retinitis pigmentosa GTPase regulator (RPGR) localizes to the connecting cilium of photoreceptors and plays a role in microtubule organization and cellular transport (20). RPGR undergoes extensive alternative splicing and has two main transcripts, a widely expressed RPGRexon1-19 form and a retina-specific RPGRORF15 form. Mutations in RPGR have been identified as the cause of 72% of XLRP, and 80% of these mutations occur in the purine-rich ORF15 (21). Many XLRP mutations, including splice site mutations (22–25), have been identified throughout the RPGRORF15 transcript, suggesting that each of the contained exons is necessary for retinal function, but interestingly, no mutations have been identified in exons 16-19 (26). The ratio of RPGRexon1-19 to RPGRORF15 is important to the integrity of the adult retina in mouse, and overexpression of RPGRexon1-19 leads to severe retinal degeneration (27). It has also been shown that certain truncated forms of RPGR can have dominant gain-of-function effects (28). Another alternatively spliced exon, exon 9a, was identified 418 base pairs downstream of the 5’ splice site of intron 9 and is 136 bases long. This exon is present in approximately 4% of retinal transcripts and is enriched in cone inner segments. An intronic G to A substitution between exon 9 and exon 9a was identified in a family with XLRP and increases the percentage of transcripts containing exon 9a (29). Mutations in tissue-specific exons and mutations that affect the relative prevalence of tissue-specific transcripts permit mutations in ubiquitously expressed genes to result in primarily ocular disease (30).
Splicing factor mutations
PRPF31 encodes a homologue to the yeast pre-mRNA splicing factor Prp31p, and mutations in this gene have been identified as a cause of adRP (31). In S. cerevisiae, Prp31p is not necessary for the formation of the U4/U6/U5 tri-snRNP but is instead responsible for recruiting the tri-SNP to the spliceosome. PRPF31 in humans is required for tri-SNP formation and spliceosome activity.(32) Seven PRPF31 mutations have been identified in British families with adRP, including two intronic mutations that disrupt the 5’ and 3’ splice sites of intron 6, Ala216Pro and Ala194Glu mutations in exon 7, two frameshift mutations leading to premature termination, and an in-frame insertion of 11 amino acids (33). A 12 base pair deletion in exon 5 causing an in-frame deletion of His111Lys112Phe113Ile114, which includes the highly conserved His111 residue, has also been identified in a Chinese family with adRP (34). A G to A substitution in the last base of intron 5 disrupts the 3’ splice site, causes a one base pair deletion in the first codon of exon 6, frameshift, and premature termination in another large Chinese family with adRP (35). Three nonsense mutations in exon 8 have also been identified in Spanish families with adRP (36). In a cohort of French adRP patients, it was found that 6.7% have mutations in PRPF31 (37).
Studies to evaluate the effects of PRPF31 mutations on pre-mRNA splicing have shown a range of results. The AD5 and SP117 PRPF31 mutants, which have an 11 base pair deletion after amino acid 371 and a single base pair insertion after amino acid 256, respectively, were co-expressed with minigene constructs for RDS and FSCN2. Both mutants showed impaired splicing of RDS intron 1, but only the AD5 mutant showed impaired splicing of FSCN2 intron 3 (38). The PRPF31 mutants containing the N-terminal 371 or 256 amino acids showed reduced splicing of intron 3 of rhodopsin, and in primary retinal cell cultures, led to reduced rhodopsin protein expression and apoptosis (39). In contrast, PRPF31 Ala194Glu and Ala216Pro mutants showed only mild effects on in vitro splicing function (40). Nevertheless, it has been hypothesized that more significant deficiencies may manifest in the setting of high splicing activity demand.
Mutations in PRPF8 have also been implicated in severe, early-onset adRP (41). PRPF8 is a component of the U5 snRNP and can interact with the 5’ splice site, the branch point adenosine, and the 3’ splice site in pre-mRNA, suggesting it maybe a cofactor in the catalytic domain of the spliceosome (42). Seven missense mutations (His2309Pro, His2309Arg, Arg2310Lys, Pro2301Thr, Phe2304Leu, Arg2310Gly, and Phe2314Leu) have been identified in South African families (41). Three frameshift mutations, a mutation disrupting the stop codon, and the Arg2310Gly missense mutation have also been reported in Spanish families (36). Interestingly, all of these mutations lie in PRPF8 exon 42.
Several adRP-causing mutations have also been identified in PRPF3, which encodes a U4/U6 associated splicing factor required for spliceosome assembly and U4/U6/U5 tri-snRNP stability (43, 44). Three missense mutations have been identified: Ala489Asp (45), Pro493Ser, and Thr494Met (43). Whereas wildtype PRPF3 is not especially abundant in photoreceptors, T494M mutant PRPF3 has been shown to form large aggregates of mislocalized proteins in photoreceptors in vitro that cause apoptosis, a phenomenon not observed in epithelial or fibroblast cell lines, perhaps due to an inability to recycle the PRPF3 splicing factor (44). Notably, all of these mutations cluster in a conserved C-terminal region flanked by binding sites for other splicing factors, including PAP-1 (46).
Two adRP-causing missense mutations, His137Leu and Asp170Gly, have also been reported in PAP-1, another component of the U4/U6/U5 tri-snRNP (47). PRPF6 is an additional splicing factor that interacts with components of the U4/U6/U5 snRNP, and the Arg729Trp mutation has been identified in a patient with adRP (48). Immortalized lymphoblasts from this patient showed intranuclear aggregates of PRPF6, suggesting that the mutation leads to a defect in tri-snRNP assembly or recycling, and in vitro splicing assays showed impaired splicing activity. The Ser1087Leu (49) and Arg1090Leu (50) mutations in SNRNP2000 have also been identified in Chinese families with adRP, and they impair the ability of SNRNP200 to unwind U4/U6 snRNAs.
There have been a number of proposed models for how mutations in ubiquitously expressed splicing factors, all of which are associated with the U4/U6/U5 snRNP, can lead to a retina-specific autosomal dominant disease, including haploinsufficiency, impaired spliceosome assembly, impaired interaction with a photoreceptor-specific splicing cofactor, or gain-of-function toxicity (51). The Prpf3-Thr494Met, Prpf8-His2309Pro, and Prpf31-knockout mouse models of splicing factor adRP all demonstrate some aspects of retinal degeneration, with a relatively late onset (52). In the mouse models, the primary cell affected appears to be the RPE, but whether this is the case in humans is unclear. One study indicated that adRP-causing splicing factor mutations disrupt the relative ratios of snRNAs, the composition of the U4/U6/U5 snRNPs, and spliceosome assembly and therefore are likely to have systemic effects on pre-mRNA splicing that, for reasons that remain to be elucidated, are tolerated in other tissues but not in the retina (53). The expression levels of PRPF3, PRPF31, PRPC8, and the 5 snRNPs are higher in the retina than in other tissues in normal adult mice, suggesting that the retina, one of the most metabolically active tissues in the body, may have higher basal splicing requirements than other tissues (54). Splicing factor mutations may manifest uniquely in the retina as adRP due to pre-mRNA splicing deficiencies that only occur in the setting of high demand.
Small molecules to modulate pre-mRNA splicing
Aberrant pre-mRNA splicing contributes profoundly to human disease, and the development of novel therapeutic strategies to modulate pre-mRNA splicing is of great clinical interest. Lentiviral delivery of U1snRNA engineered to bind mutated donor splice sites has shown moderate efficacy in rescuing splicing of BBS1 (55) and RPGR (24) in fibroblasts derived from patients with Bardet-Biedl syndrome and XLRP, respectively. siRNA strategies have been investigated to target variant splice isoforms resulting from adRP-associated splice site mutations in rhodopsin (56). Antisense oligonucleotide and related approaches are also being developed to modulate splicing in Duchenne muscular dystrophy, spinal muscular atrophy (SMA), Hutchinson-Gilford progeria syndrome, and myotonic dystrophy (57, 58).
Some notable successes have been achieved using small molecules therapies to modulate splicing. SMA, an autosomal recessive disease characterized by lower motor neuron degeneration leading to progressive proximal muscle atrophy and weakness, is caused by loss of function mutations in SMN1. SMN2 encodes a nearly identical protein, but its mRNA transcript generally skips exon 7 and leads to the production of a truncated protein product that cannot compensate for the decreased levels of functional SMN1 (59). In a small molecule screen of Epstein-Barr virus-transformed lymphoid cells derived from SMA patients, sodium butyrate was identified as a modulator of SMN2 splicing that increased exon 7 inclusion and the production of full-length SMN2 transcripts and protein (60). In the same study, sodium butyrate treatment in a murine model of SMA improved tail muscle tone and prolonged survival. The anthracycline aclarubicin (61) and the tetracycline-like compound PTK-SMA1 (62) have also been shown to increase inclusion of exon 7. The bacteria-derived pladienolides (63), FR901464, and herboxidiene are inhibitors of the spliceosome component SF3b and prevent binding of U2 snRNA to the branch point adenosine, leading to aberrant splicing and apoptosis in cancer cells (64). Derivatives such as sudemycins also demonstrate anti-tumor activity in human tumor cell lines and xenograft models (65).
Tau is a microtubule-associated protein that is alternatively spliced in humans, and mutations that promote the inclusion of exon 10 are associated with frontotemporal dementia and parkinsonism. Cdc2-like kinases 1-4 (CLK 1-4) phosphorylate SR proteins and promote exon 10 skipping in tau, suggesting that using small molecules to modulate the phosphorylation of SR proteins or the kinases and phosphatases that regulate them may also be a viable therapeutic strategy to target aberrant pre-mRNA splicing (66, 67). A benzothiazole compound named TG003 is a CLK family inhibitor and blocks the activity of the SR protein SF2/ASF (68). Inhibition of CLK1 by TG003 reduces influenza virus replication by inhibiting splicing of the viral M2 pre-mRNA (69). TG003 can also promote skipping of exon 31 of the dystrophin gene in myoblasts derived from patients with premature stop codons in this exon (70). Vascular endothelial growth factor (VEGF) has a pro-angiogenic form, VEGF165, and an anti-angiogenic form, VEGF165b, that are produced as alternative splice isoforms. SRPIN340, an inhibitor of the SR protein kinase SRPK1/2, promotes the anti-angiogenic form and can inhibit angiogenesis in a mouse model of retinal neovascularization (71). Finally, a screen of small molecule splicing modulators in a human hepatocellular carcinoma cell line revealed that the potassium sparing diuretic amiloride was able to revert oncogenic splicing of BCL-X, HIPK3, and RON/MISTR1 (72).
Concluding remarks
It is clear that abnormalities of alternative splicing can play an important role in the development of retinitis pigmentosa and other retinal degenerative diseases. The advent of next-generation sequencing technologies and massively parallel high-throughput RNA sequencing (RNA-seq) opens unprecedented opportunities to study tissue-specific alternative splicing, characterize disease-relevant cis-acting splice variants, and assess the impact of trans-acting splicing factor mutations across the transcriptome. A more thorough understanding of the contribution of aberrant splicing to retinal degeneration is crucial to the development of novel therapeutic strategies. Small molecule and other modulators of alternative splicing have shown promise in the context of a variety of human diseases, and provide hope that it will be possible to develop analogous approaches for the treatment of retinal degenerations and other forms of ophthalmic disease that are due to abnormalities of RNA splicing.
Supplementary Material
Table 1.
Splice site mutations associated with retinal degeneration
| Retinitis Pigmentosa | |||
|---|---|---|---|
| Gene | Mutation | Effect | Reference |
| Rhodopsin | IVS4+2 G>T | Skipping of exon 4 due to disruption of donor splice site | (S1–S5) |
| IVS4-1 G>A | Deletion of exon 5 and 143 bases of 3’ UTR | (S6, S7) | |
| IVS4-1 G>T | (S8) | ||
| 30 bp deletion including acceptor splice site of intron 4 and first 7 bases of exon 5, 150 bp insertion | (S9) | ||
| β-PDE | IVS2-1 G>T | Multiple alternatively spliced transcripts: intron 2 inclusion, alternative 3’ splice site in intron 3 | (S10, S11) |
| IVS3+1 G>T | (S12) | ||
| TULP1 | IVS2+1 G>A | (S13) | |
| IVS14+1 A>G | (S14) | ||
| BBS8 | IVS1-2 A>G | Skipping of photoreceptor-specific alternatively spliced exon 2a | (S21) |
| EYS | IVS4+6 A>T | (S22) | |
| IVS12+1 G>C | (S22) | ||
| MERTK | IVS16+1 G>T | Skipping of exon 16, frameshift, premature termination in exon 17 | (S23) |
| RBP4 | IVS2+1 G>A | (S24) | |
| ABCR | IVS30+1 G>T | (S25) | |
| IVS40+5 G>A | (S25) | ||
| OFD1 | IVS9+706 A>G | Insertion of cryptic exon between exons 9 and 10, frameshift | (S26) |
| Usher syndrome | |||
| Gene | Mutation | Effect | Reference |
| USH1C | IVS5-2 delA | (S27) | |
| IVS1+1 G>T | (S28) | ||
| IVS5+1 G>A | (S28) | ||
| IVS14+6 T>G | Skipping of exon 14; 31 base deletion from exon 14 | (S32) | |
| CDH23 | IVS29-1 G>T | Skipping of exon 30; 51 base deletion from exon 30 | (S34) |
| c.4488 G>C | Alternative 5’ splice site within intron 35, 80 amino acid insertion, premature termination | (S29) | |
| IVS45-9 G>A | Insertion of 7 bases of intron 45 | (S34) | |
| IVS51+5 G>A | Skipping of exon 51 | (S29) | |
| c.7872 G>A | Insertion of 86 bases of intron 54 | (S32) | |
| USH2A | IVS2-14 G>A | Insertion of 12 bases of intron 2 | (S32) |
| c.949 C>A | 193 base deletion of exon 6 | (S34) | |
| IVS10-2 A>G | Skipping of exon 11 | (S30) | |
| IVS12+5 G>A | Skipping of exon 12; alternative 5’ splice site within exon 12, 30 base pair deletion | (S30) | |
| IVS12-1 G>C | 7 base deletion of exon 13 | (S34) | |
| c.2993G>A | Arg998Lys; skipping of exon 14 | (S32) | |
| IVS17+3 A>T | Skipping of exon 17 | (S32) | |
| c.4576-4579dupGGGT | 50 base deletion of exon 21 | (S34) | |
| IVS25-6 T>A | Skipping of exon 26 | (S32) | |
| IVS26+1 G>C | Skipping of exon 26 | (S30) | |
| IVS40-8 C>G | 7 base insertion of intron 40 | (S32) | |
| IVS40-3 C>G | 2 base insertion of intron 40 | (S32) | |
| IVS43-17 A>G | 39 base deletion of exon 44 | (S32) | |
| IVS53+3 A>G | 82 base deletion from exon 53 | (S32) | |
| IVS66+39 C>T | Alternative 5’ splice site within intron 66, 37 base insertion | (S32) | |
| MYO7A | c.592 G>A | Ala198Thr; skipping of exon 6 | (S32) |
| IVS13-8 C>G | 7 base insertion of intron 13; skipping of exon 14 | (S32) | |
| c.1935 G>A | Met645Ile; skipping of exon 16 | (S31) | |
| IVS19-1 G>T | Skipping of exon 20 | (S30) | |
| IVS22-9 A>G | 8 base insertion of intron 22 | (S32) | |
| c.3503 G>C | Arg1168Pro; skipping of exon 27 | (S32) | |
| c.5856 G>A | Skipping of exon 42 | (S30) | |
| c.5944 G>A | Gly1982Arg; skipping of exon 43 | (S32) | |
| Cone rod dystrophy and additional retinal degenerative diseases | |||
| Gene | Mutation | Effect | Reference |
| RLBP1 | c.342 G>A | (S35) | |
| IVS3+2 T>C | (S35, S36) | ||
| ABCA4 | IVS2+2 T>C | (S38) | |
| IVS8-6 T>A | (S38) | ||
| IVS9-2 A>G | (S38) | ||
| IVS15+1 G>A | (S38) | ||
| IVS19-2 A>G | (S38) | ||
| IVS23+5 delG | (S38) | ||
| IVS23-1 G>A | (S38) | ||
| IVS25-2 A>G | (S38) | ||
| IVS29+1 G>A | (S38) | ||
| IVS29-15del23 | Skipping of exons 28 and 29; skipping of exon 29; skipping of exon 29 and first 114 bases of exon 30 | (S37) | |
| IVS31-1 G>T | (S38) | ||
| IVS37+1 G>A | (S38) | ||
| IVS41-2 A>C | (S38) | ||
| IVS46-1 G>T | (S38) | ||
| IVS47+1 G>A | (S38) | ||
| IVS47+1 G>C | (S38) | ||
| CDHR1 | IVS13+2 T>G | (S39) | |
| C8orf38 | IVS1-2 A>G | (S40) | |
| CACNA1F | IVS24+1 G>A | Premature stop in intron 24 | (S42) |
| IVS28-1 GCGTC>TGG | Multiple aberrantly spliced products | (S41) | |
| IVS40-2 A>G | Skipping of exon 41, premature stop in exon 42 | (S42) | |
| CEP290 | IVS26+1655 A>G | Insertion of cryptic exon between exons 26 and 27 | (S44, S45) |
| c.451 C>T | Skipping of exon 7; skipping of exon 7 and 8 | (S45) | |
| MERTK | IVS1+1 G>A | (S46) | |
IVSX+n, nth base of intron X; IVS –n, nth base from the end of intron X; c.n, nth base of CDS.
Table 2.
Alternative splice isoforms associated with retinal degeneration
| Gene | Mutation | Effect | Reference |
|---|---|---|---|
| COL2A1 | Cys86X | Premature termination in alternatively spliced exon | (18) |
| Cys57X | Premature termination in alternatively spliced exon | (19) | |
| RPGR | ORF15, various | Various | (21-25) |
| IVS9+363 G>A | Increased inclusion of exon 9a | (29) |
IVSX+n, nth base of intron X; IVS –n, nth base from the end of intron X; c.n, nth base of CDS.
Table 3.
Splicing factor mutations associated with retinal degeneration
| PRPF31 | c.269-273 del | Frameshift, premature termination | (37) |
| c.331-342 del | Deletion of His111Lys112Phe113Ile114 | (34) | |
| IVS5-1 G>A | 1 base deletion of exon 6, frameshift, premature termination | (35) | |
| IVS6+2 T>C | (37) | ||
| IVS6+3 A>G | Inactivate 5’ splice site | (33) | |
| IVS6-3 to IVS5-45 del | Inactivate 3’ splice site | (33) | |
| c.580-581 dup 33 bases | Frameshift, in-frame 11 amino acid insertion | (33) | |
| c.581 C>A | Ala194Glu | (33) | |
| c.666 dup | Frameshift, premature termination | (37) | |
| c.646 G>C | Ala216Pro | (33) | |
| c.709-734 dup | Frameshift, premature termination | (37) | |
| c.732-737 delins 20 bases | Frameshift, premature termination | (36) | |
| c.769-770 ins A (SP117) | Frameshift, premature termination; impaired splicing of RDS intron 1; impaired splicing of rhodopsin intron 3 | (33, 36, 38, 39) | |
| c.828-829 delCA | Frameshift, premature termination | (36) | |
| c.873-897 dup | Frameshift, premature termination | (37) | |
| c.997 delG | Frameshift, premature termination | (37) | |
| c.1115-1125 del (AD5) | Frameshift, premature termination; impaired splicing of RDS intron 1 and FSCN2 intron 3; impaired splicing of rhodopsin intron 3 | (33, 38, 39) | |
| PRPF8 | c.6901 C>A | Pro2301Thr | (41) |
| c.6912 C>G | Phe2304Leu | (41) | |
| c.6926 A>C | His2309Pro | (41) | |
| c.6926 A>G | His2309Arg | (41) | |
| c.6928 A>G | Arg2310Gly | (36, 41) | |
| c.6929 G>A | Arg2310Lys | (41) | |
| c.6942 C>A | Phe2314Leu | (41) | |
| c.6943-6944 delC | Frameshift, premature termination | (36) | |
| c.6974-6994 del21 | Frameshift, premature termination | (36) | |
| c.6893-6896 delins7 | Frameshift, premature termination | (36) | |
| c.7006 T>C | Disruption of stop codon, protein extension | (36) | |
| PRPF3 | c.1466 C>A | Ala489Asp | (45) |
| c.1477 C>T | Pro493Ser | (43) | |
| c.1481 C>T | Thr494Met | (43, 44) | |
| PAP1 | c.410 A>T | His137Leu | (47) |
| c.509 A>G | Asp170Gly | (47) | |
| PRPF6 | c.2185 C>T | Arg729Trp; defect in tri-snRNP assembly or recycling | (48) |
| SNRNP2000 | c.3260 C>T | Ser1087Leu | (49) |
| c.3269 G>T | Arg1090Leu | (50) |
IVSX+n, nth base of intron X; IVS –n, nth base from the end of intron X; c.n, nth base of CDS.
Acknowledgments
This work was supported by research grants from the National Institutes of Health (Medical Scientist Training Program Grant T32GM007309 (M.M.L); R01EY009769 (D.J.Z.); core grant P30EY001765), Foundation Fighting Blindness, and unrestricted funds from Research to Prevent Blindness, Inc., and generous gifts from Mr. and Mrs. Robert and Clarice Smith and the Guerrieri Family Foundation.
Footnotes
Conflicts of interest:
Melissa M. Liu, none. Donald J. Zack, none.
References
- 1.Bunker CH, Berson EL, Bromley WC, et al. Prevalence of retinitis pigmentosa in Maine. Am J Ophthalmol. 1984;97:357–365. doi: 10.1016/0002-9394(84)90636-6. [DOI] [PubMed] [Google Scholar]
- 2.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 3.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 4.Sakharkar MK, Chow VTK, Kangueane P. Distributions of exons and introns in the human genome. In Silico Biol. 2004;4:387–393. [PubMed] [Google Scholar]
- 5.Black DL. Finding splice sites within a wildnerness of RNA. RNA. 1995;1:763–771. [PMC free article] [PubMed] [Google Scholar]
- 6.Blencowe BJ. Exonic splicing enhancers: mechanism of action, diversity and role in human genetic diseases. Trends Biochem Sci. 2000;25:106–110. doi: 10.1016/s0968-0004(00)01549-8. [DOI] [PubMed] [Google Scholar]
- 7.Smith CWJ, Valcárcel J. Alternative pre-mRNA splicing : the logic of combinatorial control. Trends Biochem Sci. 2000;25:381–388. doi: 10.1016/s0968-0004(00)01604-2. [DOI] [PubMed] [Google Scholar]
- 8.Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009;10:741–754. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson JM, Castle J, Garrett-Engele P, et al. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003;302:2141–2144. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- 10.Xu Q, Modrek B, Lee C. Genome-wide detection of tissue-specific alternative splicing in the human transcriptome. Nucleic Acids Res. 2002;30:3754–3766. doi: 10.1093/nar/gkf492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Q, Lee JA, Black DL. Neuronal regulation of alternative pre-mRNA splicing. Nat Rev Neurosci. 2007;8:819–831. doi: 10.1038/nrn2237. [DOI] [PubMed] [Google Scholar]
- 12.Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 13.Chang B, Khanna H, Hawes N, et al. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum Mol Genet. 2006;15:1847–1857. doi: 10.1093/hmg/ddl107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D’Cruz PM, Yasumura D, Weir J, et al. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet. 2000;9:645–651. doi: 10.1093/hmg/9.4.645. [DOI] [PubMed] [Google Scholar]
- 15.Kameya S, Hawes NL, Chang B, et al. Mfrp, a gene encoding a frizzled related protein, is mutated in the mouse retinal degeneration 6. Hum Mol Genet. 2002;11:1879–1886. doi: 10.1093/hmg/11.16.1879. [DOI] [PubMed] [Google Scholar]
- 16.Haider NB, Naggert JK, Nishina PM. Excess cone cell proliferation due to lack of a functional NR2E3 causes retinal dysplasia and degeneration in rd7/rd7 mice. Hum Mol Genet. 2001;10:1619–1626. doi: 10.1093/hmg/10.16.1619. [DOI] [PubMed] [Google Scholar]
- 17.Bishop PN, Reardon AJ, McLeod D, et al. Identification of alternatively spliced variants of type II procollagen in vitreous. Biochem Biophys Res Commun. 1994;203:289–295. doi: 10.1006/bbrc.1994.2180. [DOI] [PubMed] [Google Scholar]
- 18.Parma ES, Korkko J, Hagler WS, et al. Key to the Clinical Diagnosis of an Ocular Variant of Stickler Syndrome with Minimal or No Systemic Manifestations. Am J Ophthalmol. 2002;134:728–734. doi: 10.1016/s0002-9394(02)01646-x. [DOI] [PubMed] [Google Scholar]
- 19.Gupta SK, Leonard BC, Damji KF, et al. A Frame shift mutation in a tissue-specific alternatively spliced exon of collagen 2A1 in Wagner’s vitreoretinal degeneration. Am J Ophthalmol. 2002;133:203–210. doi: 10.1016/s0002-9394(01)01339-3. [DOI] [PubMed] [Google Scholar]
- 20.Khanna H, Hurd TW, Lillo C, et al. RPGR-ORF15, which is mutated in retinitis pigmentosa, associates with SMC1, SMC3, and microtubule transport proteins. J Biol Chem. 2005;280: 33580–33587. doi: 10.1074/jbc.M505827200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vervoort R, Lennon A, Bird AC, et al. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat Genet. 2000;25:462–466. doi: 10.1038/78182. [DOI] [PubMed] [Google Scholar]
- 22.Fujita R, Buraczynska M, Gieser L, et al. Analysis of the RPGR gene in 11 pedigrees with the retinitis pigmentosa type 3 genotype: paucity of mutations in the coding region but splice defects in two families. Am J Hum Genet. 1997;61:571–580. doi: 10.1086/515523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demirci FYK, Radak AL, Rigatti BW, et al. A Presumed Missense Mutation of RPGR Causes Abnormal RNA Splicing With Exon Skipping. Am J Ophthalmol. 2004;138:504–505. doi: 10.1016/j.ajo.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 24.Glaus E, Schmid F, Da Costa R, et al. Gene therapeutic approach using mutation-adapted U1 snRNA to correct a RPGR splice defect in patient-derived cells. Mol Ther. 2011;19:936–941. doi: 10.1038/mt.2011.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmid F, Glaus E, Cremers FPM, et al. Mutation- and tissue-specific alterations of RPGR transcripts. Invest Ophthalmol Vis Sci. 2010;51:1628–1635. doi: 10.1167/iovs.09-4031. [DOI] [PubMed] [Google Scholar]
- 26.Vervoort R, Wright AF. Mutations of RPGR in X-linked retinitis pigmentosa (RP3) Hum Mutat. 2002;19:486–500. doi: 10.1002/humu.10057. [DOI] [PubMed] [Google Scholar]
- 27.Wright RN, Hong DH, Perkins B. Misexpression of the constitutive Rpgr(ex1-19) variant leads to severe photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2011;52:5189–5201. doi: 10.1167/iovs.11-7470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong DH, Pawlyk BS, Adamian M, et al. Dominant, Gain-of-Function Mutant Produced by Truncation of RPGR. Invest Ophthalmol Vis Sci. 2004;45:36–41. doi: 10.1167/iovs.03-0787. [DOI] [PubMed] [Google Scholar]
- 29.Neidhardt J, Glaus ÃE, Barthelmes D, et al. Identification and Characterization of a Novel RPGR Isoform in Human Retina. Human Nutation. 2007;28:797–807. doi: 10.1002/humu.20521. [DOI] [PubMed] [Google Scholar]
- 30.Donoso LA, Edwards AO, Frost AT, et al. Clinical Variability of Stickler Syndrome. Surv Ophthalmol. 2003;48:191–203. doi: 10.1016/s0039-6257(02)00460-5. [DOI] [PubMed] [Google Scholar]
- 31.Weidenhammer EM, Ruiz-Noriega M, Woolford JL. Prp31p promotes the association of the U4/U6 x U5 tri-snRNP with prespliceosomes to form spliceosomes in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:3580–3588. doi: 10.1128/mcb.17.7.3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Makarova OV, Makarov EM, Liu S, et al. Protein 61K, encoded by a gene (PRPF31) linked to autosomal dominant retinitis pigmentosa, is required for U4/U6*U5 tri-snRNP formation and pre-mRNA splicing. EMBO J. 2002;21:1148–1157. doi: 10.1093/emboj/21.5.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vithana EN, Abu-Safieh L, Allen MJ, et al. A human homolog of yeast pre-mRNA splicing gene, PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11) Mol Cell. 2001;8:375–381. doi: 10.1016/s1097-2765(01)00305-7. [DOI] [PubMed] [Google Scholar]
- 34.Wang L, Ribaudo M, Zhao K, et al. Novel Deletion in the Pre-mRNA Splicing Gene PRPF31 Causes Autosomal Dominant Retinitis Pigmentosa in a Large Chinese Family. Am J Med Genet A. 2006;121:235–239. doi: 10.1002/ajmg.a.20224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia K, Zheng D, Pan Q, et al. A novel PRPF31 splice-site mutation in a Chinese family with autosomal dominant retinitis pigmentosa. Mol Vis. 2004;10:361–365. [PubMed] [Google Scholar]
- 36.Martinez-Gimeno M, Gamundi MJ, Hernan I, et al. Mutations in the Pre-mRNA Splicing-Factor Genes PRPF3, PRPF8, and PRPF31 in Spanish Families with Autosomal Dominant Retinitis Pigmentosa. Invest Ophthalmol Vis Sci. 2003;44:2171–2177. doi: 10.1167/iovs.02-0871. [DOI] [PubMed] [Google Scholar]
- 37.Audo I, Bujakowska K, Mohand-Said S, et al. Prevalence and novelty of PRPF31 mutations in French autosomal dominant rod-cone dystrophy patients and a review of published reports. BMC Med Genet. 2010;11:145–153. doi: 10.1186/1471-2350-11-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mordes D, Yuan L, Xu L, et al. Identification of photoreceptor genes affected by PRPF31 mutations associated with autosomal dominant retinitis pigmentosa. Neurobiol Dis. 2007;26:291–300. doi: 10.1016/j.nbd.2006.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan L, Kawada M, Havlioglu N, et al. Mutations in PRPF31 Inhibit Pre-mRNA Splicing of Rhodopsin Gene and Cause Apoptosis of Retinal Cells. J Neurosci. 2007;25:748–757. doi: 10.1523/JNEUROSCI.2399-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deery EC, Vithana EN, Newbold RJ, et al. Disease mechanism for retinitis pigmentosa (RP11) caused by mutations in the splicing factor gene PRPF31. Hum Mol Genet. 2002;11:3209–3219. doi: 10.1093/hmg/11.25.3209. [DOI] [PubMed] [Google Scholar]
- 41.McKie AB, McHale JC, Keen TJ, et al. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13) Hum Mol Genet. 2001;10:1555–1562. doi: 10.1093/hmg/10.15.1555. [DOI] [PubMed] [Google Scholar]
- 42.Grainger RJ, Beggs JD. Prp8 protein : At the heart of the spliceosome. RNA. 2005;11:533–557. doi: 10.1261/rna.2220705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chakarova CF, Hims MM, Bolz H, et al. Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2002;11:87–92. doi: 10.1093/hmg/11.1.87. [DOI] [PubMed] [Google Scholar]
- 44.Comitato A, Spampanato C, Chakarova C, et al. Mutations in splicing factor PRPF3, causing retinal degeneration, form detrimental aggregates in photoreceptor cells. Hum Mol Genet. 2007;16:1699–1707. doi: 10.1093/hmg/ddm118. [DOI] [PubMed] [Google Scholar]
- 45.Gamundi MJ, Hernan I, Muntanyola M, et al. Transcriptional expression of cis-acting and trans-acting splicing mutations cause autosomal dominant retinitis pigmentosa. Hum Mutat. 2008;29:869–878. doi: 10.1002/humu.20747. [DOI] [PubMed] [Google Scholar]
- 46.Maita H, Kitaura H, Ariga H, et al. Association of PAP-1 and Prp3p, the products of causative genes of dominant retinitis pigmentosa, in the tri-snRNP complex. Exp Cell Res. 2005;302:61–68. doi: 10.1016/j.yexcr.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 47.Keen TJ, Hims MM, McKie AB, et al. Mutations in a protein target of the Pim-1 kinase associated with the RP9 form of autosomal dominant retinitis pigmentosa. Eur J Hum Genet. 2002;10:245–249. doi: 10.1038/sj.ejhg.5200797. [DOI] [PubMed] [Google Scholar]
- 48.Tanackovic G, Ransijn A, Ayuso C, et al. A missense mutation in PRPF6 causes impairment of pre-mRNA splicing and autosomal-dominant retinitis pigmentosa. Am J Hum Genet. 2011;88:643–649. doi: 10.1016/j.ajhg.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao C, Bellur DL, Lu S, et al. Autosomal-dominant retinitis pigmentosa caused by a mutation in SNRNP200, a gene required for unwinding of U4/U6 snRNAs. Am J Hum Genet. 2009;85:617–627. doi: 10.1016/j.ajhg.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li N, Mei H, MacDonald IM, et al. Mutations in ASCC3L1 on 2q11.2 are associated with autosomal dominant retinitis pigmentosa in a Chinese family. Invest Ophthalmol Vis Sci. 2010;51:1036–1043. doi: 10.1167/iovs.09-3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mordes D, Luo X, Kar A, et al. Review Pre-mRNA splicing and retinitis pigmentosa. Mol Vis. 2006;12:1259–1271. [PMC free article] [PubMed] [Google Scholar]
- 52.Graziotto JJ, Farkas MH, Bujakowska K, et al. Three gene-targeted mouse models of RNA splicing factor RP show late-onset RPE and retinal degeneration. Invest Ophthalmol Vis Sci. 2011;52:190–198. doi: 10.1167/iovs.10-5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tanackovic G, Ransijn A, Thibault P, et al. PRPF mutations are associated with generalized defects in spliceosome formation and pre-mRNA splicing in patients with retinitis pigmentosa. Hum Mol Genet. 2011;20:2116–2130. doi: 10.1093/hmg/ddr094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cao H, Wu J, Lam S, et al. Temporal and tissue specific regulation of RP-associated splicing factor genes PRPF3, PRPF31 and PRPC8--implications in the pathogenesis of RP. PLoS One. 2011;6:e15860. doi: 10.1371/journal.pone.0015860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schmid F, Glaus E, Barthelmes D, et al. U1 snRNA-mediated gene therapeutic correction of splice defects caused by an exceptionally mild BBS mutation. Hum Mutat. 2011;32:815–824. doi: 10.1002/humu.21509. [DOI] [PubMed] [Google Scholar]
- 56.Hernan I, Gamundi MJ, Planas E, et al. Cellular expression and siRNA-mediated interference of rhodopsin cis-acting splicing mutants associated with autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2011;52:3723–3729. doi: 10.1167/iovs.10-6933. [DOI] [PubMed] [Google Scholar]
- 57.Singh RK, Cooper Ta. Pre-mRNA splicing in disease and therapeutics. Trends Mol Med. 2012;18:472–482. doi: 10.1016/j.molmed.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 2012;11:125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371:2120–2133. doi: 10.1016/S0140-6736(08)60921-6. [DOI] [PubMed] [Google Scholar]
- 60.Chang JG, Hsieh-Li HM, Jong YJ, et al. Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci U S A. 2001;98:9808–9813. doi: 10.1073/pnas.171105098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Andreassi C, Jarecki J, Zhou J, et al. Aclarubicin treatment restores SMN levels to cells derived from type I spinal muscular atrophy patients. Hum Mol Genet. 2001;10:2841–2849. doi: 10.1093/hmg/10.24.2841. [DOI] [PubMed] [Google Scholar]
- 62.Hastings ML, Berniac J, Liu YH, et al. Tetracyclines That Promote SMN2 Exon 7 Splicing as Therapeutics for Spinal Muscular Atrophy. Sci Transl Med. 2010;1:5ra12. doi: 10.1126/scitranslmed.3000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kotake Y, Sagane K, Owa T, et al. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol. 2007;3:570–575. doi: 10.1038/nchembio.2007.16. [DOI] [PubMed] [Google Scholar]
- 64.Webb TR, Joyner AS, Potter PM. The development and application of small molecule modulators of SF3b as therapeutic agents for cancer. Drug Discov Today. 2013;18:43–49. doi: 10.1016/j.drudis.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fan L, Lagisetti C, Edwards CC, et al. Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing. ACS Chem Biol. 2012;6:582–589. doi: 10.1021/cb100356k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hartmann AM, Rujescu D, Giannakouros T, et al. Regulation of alternative splicing of human tau exon 10 by phosphorylation of splicing factors. Mol Cell Neurosci. 2001;18:80–90. doi: 10.1006/mcne.2001.1000. [DOI] [PubMed] [Google Scholar]
- 67.Ninomiya K, Kataoka N, Hagiwara M. Stress-responsive maturation of Clk1/4 pre-mRNAs promotes phosphorylation of SR splicing factor. J Cell Biol. 2011;195:27–40. doi: 10.1083/jcb.201107093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Muraki M, Ohkawara B, Hosoya T, et al. Manipulation of alternative splicing by a newly developed inhibitor of Clks. J Biol Chem. 2004;279:24246–24254. doi: 10.1074/jbc.M314298200. [DOI] [PubMed] [Google Scholar]
- 69.Karlas A, Machuy N, Shin Y, et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. 2010;463:818–822. doi: 10.1038/nature08760. [DOI] [PubMed] [Google Scholar]
- 70.Nishida A, Kataoka N, Takeshima Y, et al. Chemical treatment enhances skipping of a mutated exon in the dystrophin gene. Nat Commun. 2011;2:1–8. doi: 10.1038/ncomms1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nowak DG, Amin EM, Rennel ES, et al. Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: a novel therapeutic strategy for angiogenesis. J Biol Chem. 2010;285:5532–5540. doi: 10.1074/jbc.M109.074930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chang JG, Yang DM, Chang WH, et al. Small molecule amiloride modulates oncogenic RNA alternative splicing to devitalize human cancer cells. PLoS One. 2011;6:e18643. doi: 10.1371/journal.pone.0018643. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.