

Abstract
Idiopathic thrombocytopenic purpura (ITP) is defined as a hematologic disorder, characterized by isolated thrombocytopenia without a clinically apparent cause. The major causes of accelerated platelet consumption include immune thrombocytopenia, decreased bone marrow production, and increased splenic sequestration. The clinical presentation may be acute with severe bleeding, or insidious with slow development with mild or no symptoms. The initial laboratory tests useful at the first visit to predict future diagnosis were erythrocyte count, leukocyte count, anti-glycoprotein IIb/IIIa antibodies, reticulated platelets, plasma thrombopoietin level. Treatment should be restricted to those patients with moderate or severe thrombocytopenia who are bleeding or at risk of bleeding. We present a case report on ITP with clinical presentation, diagnosis and management.
Keywords: Hematoma, idiopathic thrombocytopenic purpura, petechiae, platelets
Introduction
Idiopathic thrombocytopenic purpura (ITP) is the condition of having a low platelet count (thrombocytopenia) of no known cause (idiopathic). As most causes appear to be related to antibodies against platelets, it is also known as immune thrombocytopenic purpura. Although most cases are asymptomatic, very low platelet counts can lead to a bleeding diathesis and purpura. ITP is a disorder that affects the overall number of blood platelets rather than their function. The normal platelet level in adults is between 150,000 and 450,000/mm3. Platelet counts below 50,000 mm3 increase the risk of dangerous bleeding from trauma; counts below 20,000/mm3 increase the risk of spontaneous bleeding. ITP may be either acute or chronic. The incidence of ITP is 50-100 new cases per million per year, with children accounting for half of that amount and the median age of adults at the diagnosis is 56-60.[1] In adults, ITP is considered an autoimmune disorder, which means that the body produces antibodies that damage some of its own products-in this case, blood platelets. In acute ITP cases patients usually suffer from bruising; petechiae, nosebleeds and bleeding gums may occur if the platelet count is below 20,000, compared with a normal range of 150,000-400,000/mm3.[2] In extreme cases, patients with ITP may bleed into the lungs, brain, or other vital organs, leading to subarachnoid, intracerebral hemorrhage or other internal bleeding are very serious possible complications of this disease but these complications are unlikely in patients with the platelets count above 20,000. In many cases, the pathogenesis is not actually idiopathic, but autoimmune, with antibodies against platelets being detected in approximately 60% of patients.[3] Most often these antibodies are against platelet membrane glycoproteins IIb-IIIa or Ib-IX, and are of the immunoglobulin G (IgG) type.[4] The coating of platelets with IgG renders them susceptible to opsonization and phagocytosis by splenic macrophages. Recent evidence suggests that the stimulus for autoantibody production in ITP is due to abnormal T helper cells reacting with platelet antigens on the surface of antigen presenting cells.[5] This important finding suggests that therapies directed toward T cells may be effective in treating ITP. The diagnosis of ITP is a diagnosis of exclusion. First, one has to make sure that there are no other blood abnormalities except for low platelet count and no physical signs except for signs of bleeding. Then, the secondary causes (usually 5-10% of suspected ITP cases) should be excluded. Secondary causes could be leukemia, medications (e.g., quinine, heparin), lupus erythematosus, and cirrhosis, HIV, hepatitis C, congenital causes, antiphospholipid syndrome, von Willebrand factor deficiency and others.[1,2] In approximately 1% of cases autoimmune hemolytic anemia and immune thrombocytic purpura coexist, which is a condition called Evans syndrome.[1] Complete blood count usually have platelet counts below 20,000/mm3. The size and appearance of the platelets may be abnormal. The red blood cell count and white blood cell count are usually normal, although about 10% of patients with ITP are also anemic. Bleeding time is usually prolonged. Bleeding time is prolonged in ITP patients; however, the use of bleeding time in diagnosis is discouraged by the American Society of Hematology practice guidelines as useless.[6,7] A bone marrow examination may be performed on patients over the age of 60 and people who do not respond to treatment, or when the diagnosis is in doubt. On examination of the bone marrow, an increase in the production of megakaryotes is seen and can help in determining ITP.[1] Detection of antiplatelet antibodies in the blood is considered to confirm the diagnosis of ITP.[1] Treatment should be restricted to those patients with moderate or severe thrombocytopenia who are bleeding or at risk of bleeding. It should be limited in duration unless demonstrated that symptomatic thrombocytopenia persists. Patients with mild, asymptomatic thrombocytopenia, discovered incidentally on a routine blood count, should not be treated.
Case Report
Patient reported to Department of Oral Medicine and Radiology in Tamil Nadu Government Dental College and Hospital with the complaints of bleeding gums since past 2 weeks. History of presenting illness revealed that patient noticed bleeding from his gums in lower front teeth region approximately 2 weeks before reporting to department and patient was having purpuric spots on body (legs, hands, and neck) since past 2 months. Past medical history revealed that patient was known case of epilepsy since 7 years of age and was on tablet eptoin, tablet carbamazepine for past 12 years. Patient was also the known case of ischemic heart disease for past 7 years. Patient started treatment 7 years back and was on tablet isosorbide dinitrate 10 mg, tablet atenelol 25 mg, tablet envas 5 mg and patient had discontinued aspirin 6 months back. Extraoral examination revealed petechial spots over the neck on right side [Figure 1], petechial spots over the forearms, and petechial spots over the right arm [Figure 2]. Intraoral examination revealed bleeding from gingiva in lower anterior region, which was inflamed, reddish, tender on palpation, soft in consistency [Figure 3]. Petechial spots on dorsum of tongue, petechial spot on left side hard palate and left maxillary tuberosity region. On the 2nd day after the patient reported, there was increased bleeding, hematoma and gingival enlargement in lower anterior region [Figure 4], and on the 4th day, hematoma formation was seen in lower anterior region lingually [Figure 5]. Based on the clinical findings, provisional diagnosis was made as ITP. Pertinent investigations were advised to the patient. On the day when patient reported all the hematological examination was done in which all the parameters were within normal limit except platelet count which was reduced to the level of 58,000/cu mm blood. On the 4th day, the platelet count was further reduced and it came down to 6000/cu mm blood. Other biochemical examinations, liver function tests, and ultrasonography of abdomen were normal. Bone marrow aspiration showed few fragments, megakaryocytes were absent, few erytroid/myeloid series was seen, 12% lymphocytes and 2% plasma cells were seen, marrow was diluted. Patient was admitted in Hematology Ward, Government General Hospital, Chennai on and treatment was started as described below:
Figure 1.
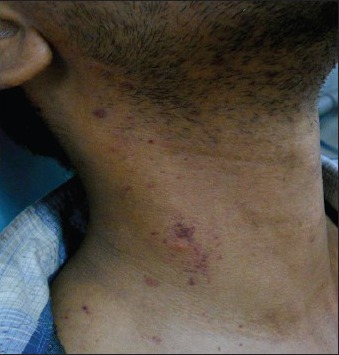
Petechial spots over the neck
Figure 2.
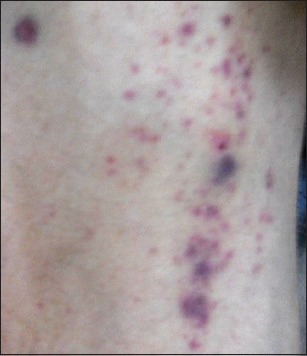
Petechial spots over the right arm
Figure 3.
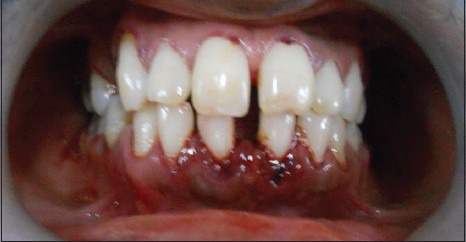
Inflamed, bleeding gingiva
Figure 4.
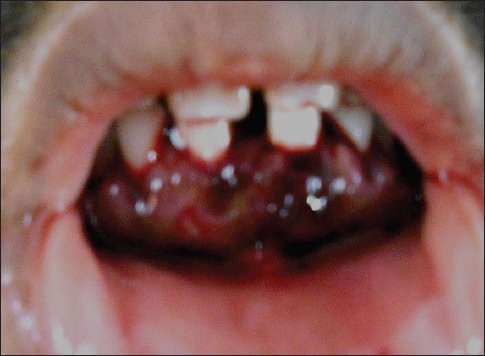
Hematoma formation in lower anterior gingiva
Figure 5.
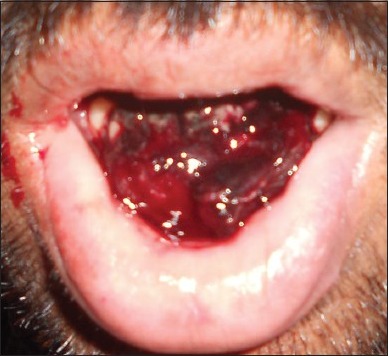
Hematoma formation lingually anteriorly
Injection methylprednisolone 1 g, intravenous (IV), OD was given for 3 days
After 3 days, injection dexamethasone 8 mg, IV, BD was given for 6 days
Injection cefotaxime 2 g, IV, BD was given for 10 days
Capsule omez - 1 capsule BD was given for 10 days.
Ferrous sulphate/folic acid 1 OD was given for 10 days
Three units platelets were transfused on the 1st day of admission
Four units platelets were transfused on the 2nd day of admission.
Patient was discharged on 10 days and was given following advises on discharge:
Tablet prednisolone 5 mg, 6 tablets OD for 1 month and dose tapering after 1 month
Tablet antacid 1 tablet OD for 1 month
To taper tablet eptoin from twice daily to once daily
To review after 1 month.
Platelet count after commencement of treatment:
Platelet count was 6000/cu mm blood on 1st day of admission
Platelet count was raised to 8000/cu mm blood on 6th day of admission
Platelet count was raised to 107,000/cu mm blood on 15th day after starting treatment.
No complications were observed and all the lesions were resolved completely. All the petechial lesions were completely resolved over the neck, arm, and forearms. Intraorally there was complete resolution of hematoma anteriorly on gingiva [Figure 6], hematoma in lower anterior lingual region [Figure 7], and petechiae over dorsum of tongue. The patient was followed-up for 2 years and there was no re-occurence reported.
Figure 6.
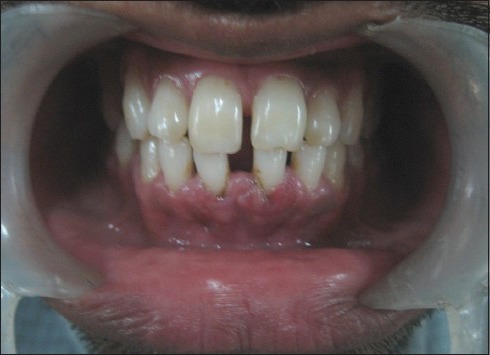
Resolved hematoma anteriorly
Figure 7.
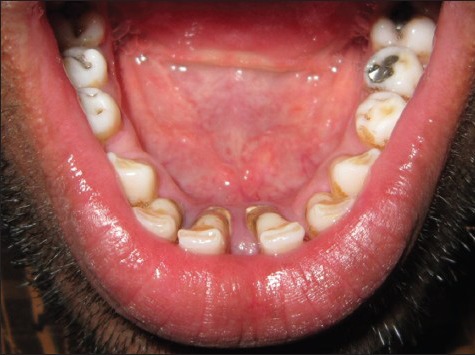
Resolved hematoma lingually
Discussion
There is marked variability in the clinical presentation of ITP. Our case was abrupt and acute in onset. The bleeding manifestations of thrombocytopenia are described as mucocutaneous to distinguish them from coagulation disorders like hemophilia. Petechia, purpura, and easy bruising are expected in ITP. Less common are epistaxis, gingival bleeding, and menorrhagia. Uncommon findings are gastrointestinal (GI) bleeding, gross hematuria and intracranial hemorrhage.[8] It is important to note that the clinical manifestations of thrombocytopenia vary with patient age. Older patients have more severe and rare bleeding manifestations, such as GI bleeding and possibly intracranial hemorrhage secondary to co-morbidities such as hypertension. Clinically important bleeding does not appear to occur in these patients unless the platelet count is less than 10,000/cu mm. However, the standard of practice among many physicians is to initiate treatment in adult patients with ITP when the platelet count is less than 30,000/cu mm. The diagnosis of ITP is in part one of exclusion, requiring that other causes of thrombocytopenia be ruled out. The major goal for treatment of ITP is to provide a safe platelet count to prevent major bleeding and avoid unnecessary treatment of asymptomatic patients with mild to moderate thrombocytopenia. Spontaneous remissions are unusual in adults. In children 70-80% have complete remission of the disease within 6 months with no specific therapy. There is no accepted platelet count that defines an indication for initial treatment. Patients with initial platelet counts above 30,000-50,000/UL, require careful follow-up but no specific initial therapy. The decision to treat ITP is based on the platelet count, degree of bleeding, and patient's lifestyle. There is no specific treatment for ITP. General care includes explaining ITP to the patient and advising him or her to watch for bruising, petechiae, or other signs of recurrence. Children should be discouraged from rough contact sports or other activities that increase the risk of trauma. Patients are also advised to avoid using aspirin or ibuprofen as pain relievers because these drugs lengthen the clotting time of blood. All medications for ITP are given either orally or IV; intramuscular injection is avoided due to the possibility of causing bleeding into the skin. Corticosteroids, typically prednisone, are the backbone of the initial treatment. The treatment begins with IV steroids (methylprednisolone or prednisone), IVIg or their combination and sometimes platelet infusions in order to raise the count quickly. After the platelet count stabilized and in the less severe cases oral prednisone (1-2 mg/kg) is used. Most cases respond during the 1st week of treatment. After several weeks of prednisone therapy, the dose is gradually reduced. However, 60-90% of patients relapse after the dose decreased below 0.25 mg/kg and stopped.[1,2] However, IVIg is expensive and the improvement is temporary (generally lasting less than a month). Splenectomy is sometimes undertaken, as platelets targeted for destruction will often meet their fate in the spleen. Splenectomy is said to be successful in 60% of cases although it is less successful in older people. A relatively new strategy is treatment with anti-D, an agent also used in mothers who have been sensitized to rhesus antigen by a Rh+ baby, but the patient must be Rh+. Other immunosuppresants, which are steroid sparing drugs like mycophenolate mofetil and azathioprine, are becoming more popular for their effectiveness. Rituximab, the monoclonal antibody against CD20+ B cells, has also been used successfully for some patients.[9,10] Extreme cases (very rare, especially rare in children) may require vincristine, a chemotherapy agent, to stop the immune system from destroying platelets. Platelet transfusion is not normally recommended and is usually unsuccessful in raising a patient's platelet count. This is because the underlying autoimmune mechanism that destroyed the patient's platelets to begin with will also destroy donor platelets. An exception to this rule is when a patient is bleeding profusely, when transfusion of platelets can quickly form a platelet plug to stop bleeding. Dapsone (also called diphenylsulfone, DDS, or avlosulfon) is an anti-infective sulfone drug. In recent years, dapsone has also proved helpful in treating lupus, rheumatoid arthritis and as a second-line treatment for ITP. The exact mechanism by which dapsone assists in ITP is unclear. However, limited studies report successful increases in platelet counts of around 40-50% of patients taking the drug.[11] AMG531 (aka Romiplostin, trade name: Nplate) is an experimental treatment for stimulating platelet production. It is a thrombopoiesis stimulating Fc-peptide fusion protein (peptibody). Initial clinical trials show it to be effective in chronic ITP.[12] The novel agent eltrombopag has been demonstrated to increase platelet counts and decrease bleeding in a dose-dependent manner.[13] Some researchers have found a possible connection between Helicobacter pylori infection and ITP. For patient with active H. pylori infection, eradication of this infection should be done.
Conclusion
The initial treatment of ITP includes: (1) Treatment should be restricted to those patients with moderate or severe thrombocytopenia who are bleeding or at risk of bleeding; (2) treatment should be limited in duration unless it is demonstrated that symptomatic thrombocytopenia persists; and (3) patients with mild, asymptomatic thrombocytopenia, discovered incidentally on a routine blood count, should not be treated.
Acknowledgment
Department of Hematology, Government General Hospital, Chennai, Tamil Nadu, India.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Cines DB, Bussel JB. How I treat idiopathic thrombocytopenic purpura (ITP) Blood. 2005;106:2244–51. doi: 10.1182/blood-2004-12-4598. [DOI] [PubMed] [Google Scholar]
- 2.Cines DB, McMillan R. Management of adult idiopathic thrombocytopenic purpura. Annu Rev Med. 2005;56:425–42. doi: 10.1146/annurev.med.56.082103.104644. [DOI] [PubMed] [Google Scholar]
- 3.Coopamah MD, Garvey MB, Freedman J, Semple JW. Cellular immune mechanisms in autoimmune thrombocytopenic purpura: An update. Transfus Med Rev. 2003;17:69–80. doi: 10.1053/tmrv.2003.50004. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz RS. Immune thrombocytopenic purpura - From agony to agonist. N Engl J Med. 2007;357:2299–301. doi: 10.1056/NEJMe0707126. [DOI] [PubMed] [Google Scholar]
- 5.Semple JW, Freedman J. Increased antiplatelet T helper lymphocyte reactivity in patients with autoimmune thrombocytopenia. Blood. 1991;78:2619–25. [PubMed] [Google Scholar]
- 6.Diagnosis and treatment of idiopathic thrombocytopenic purpura: Recommendations of the American Society of Hematology. The American Society of Hematology ITP Practice Guideline Panel. Ann Intern Med. 1997;126:319–26. doi: 10.7326/0003-4819-126-4-199702150-00010. [DOI] [PubMed] [Google Scholar]
- 7.Liesner RJ, Machin SJ. ABC of clinical haematology. Platelet disorders. BMJ. 1997;314:809–12. doi: 10.1136/bmj.314.7083.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuwana M, Okazaki Y, Satoh T, Asahi A, Kajihara M, Ikeda Y. Initial laboratory findings useful for predicting the diagnosis of idiopathic thrombocytopenic purpura. Am J Med. 2005;118:1026–33. doi: 10.1016/j.amjmed.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 9.Braendstrup P, Bjerrum OW, Nielsen OJ, Jensen BA, Clausen NT, Hansen PB, et al. Rituximab chimeric anti-CD20 monoclonal antibody treatment for adult refractory idiopathic thrombocytopenic purpura. Am J Hematol. 2005;78:275–80. doi: 10.1002/ajh.20276. [DOI] [PubMed] [Google Scholar]
- 10.Patel V, Mihatov N, Cooper N, Stasi R, Cunningham-Rundles S, Bussel JB. Long-term responses seen with rituximab in patients with ITP. Community Oncol. 2007;4:107. [Google Scholar]
- 11.Godeau B, Durand JM, Roudot-Thoraval F, Tennezé A, Oksenhendler E, Kaplanski G, et al. Dapsone for chronic autoimmune thrombocytopenic purpura: A report of 66 cases. Br J Haematol. 1997;97:336–9. doi: 10.1046/j.1365-2141.1997.412687.x. [DOI] [PubMed] [Google Scholar]
- 12.Bussel JB, Kuter DJ, George JN, McMillan R, Aledort LM, Conklin GT, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med. 2006;355:1672–81. doi: 10.1056/NEJMoa054626. [DOI] [PubMed] [Google Scholar]
- 13.Bussel JB, Cheng G, Saleh MN, Psaila B, Kovaleva L, Meddeb B, et al. Eltrombopag for the treatment of chronic idiopathic thrombocytopenic purpura. N Engl J Med. 2007;357:2237–47. doi: 10.1056/NEJMoa073275. [DOI] [PubMed] [Google Scholar]