

ABSTRACT
Mycoplasma pneumoniae synthesizes a novel human surfactant protein A (SP-A)-binding cytotoxin, designated community-acquired respiratory distress syndrome (CARDS) toxin, that exhibits ADP-ribosylating and vacuolating activities in mammalian cells and is directly linked to a range of acute and chronic airway diseases, including asthma. In our attempt to detect additional CARDS toxin-binding proteins, we subjected the membrane fraction of human A549 airway cells to affinity chromatography using recombinant CARDS toxin as bait. A 36-kDa A549 cell membrane protein bound to CARDS toxin and was identified by time of flight (TOF) mass spectroscopy as annexin A2 (AnxA2) and verified by immunoblotting with anti-AnxA2 monoclonal antibody. Dose-dependent binding of CARDS toxin to recombinant AnxA2 reinforced the specificity of the interaction, and further studies revealed that the carboxy terminus of CARDS toxin mediated binding to AnxA2. In addition, pretreatment of viable A549 cells with anti-AnxA2 monoclonal antibody or AnxA2 small interfering RNA (siRNA) reduced toxin binding and internalization. Immunofluorescence analysis of CARDS toxin-treated A549 cells demonstrated the colocalization of CARDS toxin with cell surface-associated AnxA2 upon initial binding and with intracellular AnxA2 following toxin internalization. HepG2 cells, which express low levels of AnxA2, were transfected with a plasmid expressing AnxA2 protein, resulting in enhanced binding of CARDS toxin and increased vacuolization. In addition, NCI-H441 cells, which express both AnxA2 and SP-A, upon AnxA2 siRNA transfection, showed decreased binding and subsequent vacuolization. These results indicate that CARDS toxin recognizes AnxA2 as a functional receptor, leading to CARDS toxin-induced changes in mammalian cells.
IMPORTANCE
Host cell susceptibility to bacterial toxins is usually determined by the presence and abundance of appropriate receptors, which provides a molecular basis for toxin target cell specificities. To perform its ADP-ribosylating and vacuolating activities, community-acquired respiratory distress syndrome (CARDS) toxin must bind to host cell surfaces via receptor-mediated events in order to be internalized and trafficked effectively. Earlier, we reported the binding of CARDS toxin to surfactant protein A (SP-A), and here we show how CARDS toxin uses an alternative receptor to execute its pathogenic properties. CARDS toxin binds selectively to annexin A2 (AnxA2), which exists both on the cell surface and intracellularly. Since AnxA2 regulates membrane dynamics at early stages of endocytosis and trafficking, it serves as a distinct receptor for CARDS toxin binding and internalization and enhances CARDS toxin-induced vacuolization in mammalian cells.
INTRODUCTION
Mycoplasma pneumoniae accounts for 20 to 30% of all cases of community-acquired pneumonia, causes a range of respiratory pathologies, is associated with the initiation and exacerbation of asthma and chronic obstructive pulmonary disease, and is directly linked to various extrapulmonary complications (1–7). M. pneumoniae colonizes the respiratory epithelium by means of adhesins and cytadherence accessory proteins and elicits tissue damage and other cytopathic effects (8–10). Historically, M. pneumoniae-mediated host cell injury has been correlated with mycoplasma production of hydrogen peroxide and superoxide anions (11–13) and immune-mediated induction of proinflammatory cytokines and chemokines (14, 15).
Recently, we identified a 68-kDa surfactant protein A (SP-A)-binding, ADP-ribosylating, and vacuolating toxin of M. pneumoniae designated community-acquired respiratory distress syndrome (CARDS) toxin (16). The N-terminal ADP-ribosyltransferase domain of CARDS toxin shares amino acid sequence similarity with Bordetella pertussis pertussis toxin S1 subunit (17). CARDS toxin, which is substantially upregulated during the early stages of infection and readily detectable in infected lung epithelial cells and bronchoalveolar lavage fluid specimens (18, 19) induces inflammatory responses and airway dysfunction in intoxicated animals, similar to that observed during M. pneumoniae infection (20, 21). Exposure of mice to recombinant CARDS toxin alone recapitulates the spectrum of pathologies observed during mycoplasma infection (20). Furthermore, the extent of pulmonary damage caused by M. pneumoniae infection appears to be dependent on the biological properties of individual mycoplasma strains and CARDS toxin concentrations (22).
Bacterial toxins act either at the level of the host cell surface or intracellularly. ADP-ribosylating toxins target cytosolic proteins, achieved through receptor-mediated binding and internalization. Host cell susceptibility to toxins is usually determined by the presence and abundance of appropriate receptors, which provide a molecular basis for toxin target cell specificities. CARDS toxin binds to mammalian cells at 4°C and is internalized by clathrin-mediated pathways (23), which requires a temperature shift to 37°C, reinforcing active receptor-mediated uptake. Although we initially identified CARDS toxin as an SP-A-binding protein (17), we noted that CARDS toxin carries out ADP-ribosylating and vacuolating activities in a wide range of mammalian cell lines, including some that lack SP-A, suggesting the utilization of alternative receptors (24). As a result, in order to understand the range of CARDS toxin activities and tissue distribution in susceptible hosts, we searched for additional receptor families that mediate CARDS toxin binding and internalization.
Here, we show that the C-terminal domain of CARDS toxin interacts with the host protein annexin A2 (also called annexin II, calpactin 1, and AnxA2) (referred to as AnxA2 here), a member of the annexin family of proteins, which are Ca2+- and phospholipid-binding proteins that exhibit many signaling functions. The interaction between CARDS toxin and AnxA2 likely plays an important role in the observed localized and disseminated inflammation and tissue pathologies associated with M. pneumoniae infections.
RESULTS
The CARDS toxin binds to AnxA2.
To identify an A549 cell membrane target(s) that binds CARDS toxin, we immobilized histidine (His)-tagged CARDS toxin onto nickel-nitrilotriacetic acid (Ni-NTA) resin and added solubilized A549 cell membrane extracts. Membrane proteins that bound to CARDS toxin were eluted by boiling with SDS lysis buffer, resolved on 4 to 12% NuPAGE gel, and visualized by Coomassie blue staining. Although some background proteins were associated with uncoupled Ni-NTA resin, several protein bands were selectively bound to the Ni-NTA–CARDS toxin resin (Fig. 1A, lane 2). These bands were excised, digested with trypsin, and identified using matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS). The mass profiles of the trypsin-generated peptides of ~70-, ~40-, and ~34-kDa proteins (Fig. 1A, short dashed arrows) matched CARDS toxin, and the ~36-kDa protein (Fig. 1A, long solid arrow) was identified as annexin A2 (AnxA2).
FIG 1 .

CARDS toxin binds to A549 cell membrane-associated AnxA2. (A) Identification of AnxA2 bound to CARDS toxin. Membrane-enriched fractions of A549 cells were incubated with Ni-NTA alone or CARDS toxin coupled to Ni-NTA. Ni-NTA-bound membrane proteins (lane 1) or CARDS toxin-coupled Ni-NTA-bound membrane proteins (lane 2) were separated on NuPAGE (4 to 12% gradient) gels and stained with Coomassie brilliant blue G-250. Mass spectrometry analysis was performed on eluted proteins. The short dashed arrows point to protein bands that were identified as FL or processed/degraded CARDS toxin, and the long solid arrow points to AnxA2. The capital boldface letters in the AnxA2 sequence are AnxA2-specific amino acids identified by mass spectrometry. The molecular masses (in kilodaltons) of molecular mass markers are indicated to the left of the gel. (B) Immunoblot confirmation of AnxA2 bound to CARDS toxin during pulldown assay. Eluted proteins from panel A were resolved on 4 to 12% NuPAGE gels, transferred to nitrocellulose membranes, and probed with anti-AnxA2 monoclonal antibody. Eluted proteins from control uncoupled Ni-NTA beads (lane 1) show no immunoreactivity, whereas eluted proteins from CARDS toxin-coupled Ni-NTA beads (lane 2) demonstrate clear immunoreactivity at ~36-kDa range.
To further confirm the identity of AnxA2, A549 cell membrane proteins enriched by the receptor pulldown assay (Materials and Methods) were transferred to nitrocellulose membranes and probed with monoclonal antibody specific to AnxA2 protein. An intense immunoreactive band was observed at ~36 kDa, and the band was absent in the negative-control lane (Fig. 1B).
Binding of CARDS toxin to AnxA2 is specific and concentration dependent.
To further characterize the CARDS toxin-AnxA2 interaction, we performed a ligand overlay binding assay (Materials and Methods) using recombinant glutathione S-transferase (GST)-tagged AnxA2. The CARDS toxin-AnxA2 complex was readily detected using anti-CARDS toxin antibody reagent; no toxin was detected in the bovine serum albumin (BSA) control lane (Fig. 2A). To determine the specificity of binding of CARDS toxin to AnxA2, purified AnxA2 or BSA was coated onto individual wells of microtiter plates and incubated with various concentrations of CARDS toxin followed by rabbit anti-CARDS toxin polyclonal antibody and horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody. CARDS toxin bound to AnxA2 in a concentration-dependent, saturable manner (Fig. 2B).
FIG 2 .
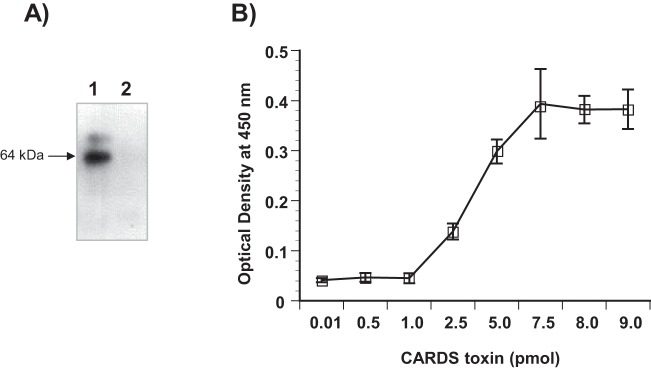
Binding of CARDS toxin to recombinant AnxA2. (A) Binding of CARDS toxin to AnxA2 by ligand blotting. GST-AnxA2 or BSA (2 µg each) was separated on SDS-polyacrylamide gels, transferred to nitrocellulose membranes, and incubated with CARDS toxin (7 µg/ml) for 2 h. CARDS toxin binding was detected by incubation with rabbit polyclonal anti-CARDS toxin antibody followed by incubation with goat anti-rabbit IgG and visualization with ECL. Lane 1, AnxA2; lane 2, BSA. (B) Dose-dependent binding of CARDS toxin to AnxA2. Microtiter wells were coated with 100 ng AnxA2, and increasing concentrations of CARDS toxin or BSA were added to individual wells for 1 h at room temperature. Bound protein was detected with rabbit polyclonal anti-CARDS toxin antibody and goat anti-rabbit HRP-conjugated polyclonal antibody, followed by development with TMB substrate. Wells with BSA alone served as negative controls, and the nonspecific bound values were subtracted from individual test scores. Values are means ± standard errors of the means (error bars) for triplicate wells from three separate experiments. No immunological cross-reactivity was observed between anti-CARDS toxin antibody and AnxA2.
The C terminus of CARDS toxin mediates binding to AnxA2.
In order to delineate the region of CARDS toxin responsible for binding to AnxA2, we added equimolar concentrations of purified His-tagged full-length (FL) and truncated N-terminal (amino acids 1 to 249; CARDS249) and C-terminal (amino acids 266 to 591; 266CARDS) CARDS toxin derivatives (30) to wells coated with AnxA2. The results show that only FL CARDS toxin and the C terminus of CARDS toxin bind to AnxA2 (Fig. 3A)
FIG 3 .
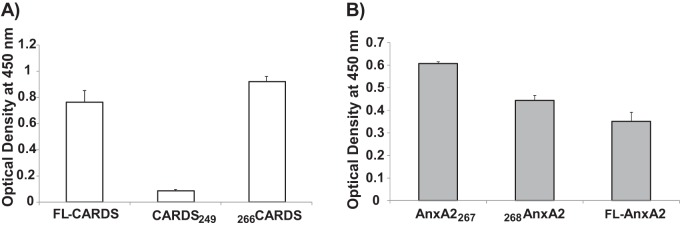
The C terminus of CARDS toxin mediates binding to full-length (FL) and truncated AnxA2. (A) Interaction of CARDS toxin C terminus with FL AnxA2. AnxA2 (100 ng/well) was bound to individual wells on ELISA plates and incubated with equimolar concentrations of FL CARDS toxin and to N-terminal (CARDS249; amino acids [aa] 1 to 249) and C-terminal (266CARDS; aa 266 to 591) proteins. Binding was detected using anti-His tag monoclonal antibody. (B) Interaction of FL and truncated AnxA2 derivatives with FL CARDS toxin. Equimolar concentrations of FL AnxA2, truncated AnxA2267, or truncated 268AnxA2 were bound to individual wells of ELISA plates and incubated with CARDS toxin. Toxin binding was detected using rabbit polyclonal anti-CARDS toxin antibody.
AnxA2 consists of an N terminus of 30 amino acids followed by a C terminus containing four 61-amino-acid repeats that include amino acids 42 to 102 (repeat I), 114 to 174 (repeat II), 199 to 259 (repeat III), and 274 to 334 (repeat IV). To identify AnxA2 domain(s) responsible for binding to CARDS toxin, we generated and purified GST-tagged AnxA2267 (amino acids 1 to 267 containing the N terminus and repeats I to III) and 268AnxA2 (amino acids 268 to 339; repeat IV alone). We coated individual wells with FL AnxA2 or truncated derivatives of AnxA2 and analyzed their ability to facilitate CARDS toxin binding. Like FL AnxA2, both truncated AnxA2 proteins bound toxin, although AnxA2267 demonstrated maximal binding (Fig. 3B).
AnxA2 monoclonal antibody inhibits CARDS toxin binding to A549 cells.
To further characterize the role that AnxA2 plays in CARDS toxin binding, we preincubated A549 cells with AnxA2 monoclonal antibody or negative-control monoclonal antibody (10 µg/ml) for 10 min before adding CARDS-mCherry fusion protein (10 µg/ml). CARDS toxin binding was substantially reduced by AnxA2 monoclonal antibody (~40%) in contrast to negative-control antibody (Fig. 4A).
FIG 4 .

AnxA2 surface accessibility promotes CARDS toxin binding to A549 cells. (A) CARDS toxin binding to A549 cells was reduced by anti-AnxA2 monoclonal antibody (mAb). A549 cells were incubated with or without anti-AnxA2 monoclonal antibody or negative-control isotype-matched monoclonal antibody for 10 min followed by incubation with CARDS toxin-mCherry protein for 30 min at 4°C. Binding of CARDS toxin-mCherry protein to A549 cells was analyzed by fluorometry as described in Materials and Methods. (B) Reduced CARDS toxin binding following suppressed AnxA2 expression. CARDS toxin (1 µg) was added to A549 cells transfected with siRNAs specific to AnxA2 or control random siRNAs and incubated for 1 h at 37°C. AnxA2 expression and CARDS toxin association with A549 cells were analyzed by immunoblotting with anti-AnxA2 monoclonal antibody and anti-CARDS polyclonal antibody. Comparative GAPDH intensities were used as loading controls. (C) Quantification of CARDS toxin associated with A549 cells upon suppression of AnxA2. Immunostained bands from panel B were quantified as detailed in Materials and Methods and normalized with GAPDH. Experiments were repeated two times (experiments 1 and 2), and the relative signal differences in binding levels of CARDS toxin and expression levels of AnxA2 are presented. (D) Decreased CARDS toxin-mediated vacuolization in A549 cells following suppressed AnxA2 expression. CARDS toxin was added to A549 cells transfected with control random siRNAs (a) or siRNAs specific to AnxA2 (b) and incubated for 24 h at 37°C. Vacuole formation was analyzed microscopically. (E) Quantification of CARDS toxin-induced vacuoles in A549-AnxA2-siRNA cells at 24 h. As described above for panel C, the cells were incubated with CARDS toxin, and the numbers of vacuoles per cell were counted and compared.
Knockdown of AnxA2 expression decreases total cell-associated CARDS toxin and subsequent vacuolization.
Transfection of A549 cells with small interfering RNAs (siRNAs) specific to AnxA2 (siRNA1 and siRNA2) decreased the levels of AnxA2 expression compared to control nontargeting siRNA as determined by immunoreactivity using anti-AnxA2 monoclonal antibody (Fig. 4B). Further, the amount of cell-associated CARDS toxin was examined in parallel with AnxA2 expression by probing test samples with anti-CARDS toxin antibody (Fig. 4B). In A549 cells transfected with AnxA2-specific siRNA, reduced amounts of CARDS toxin were detected in the total cell lysate (consistent with AnxA2 levels) compared to cells treated with control nontargeting siRNA (Fig. 4B and C). In addition, CARDS toxin induced smaller and reduced numbers of vacuoles in A549 cells transfected with AnxA2-specific siRNA than in cells transfected with control random siRNA (Fig. 4D and E).
AnxA2 colocalizes with the CARDS toxin.
To examine the earliest interaction between CARDS toxin and AnxA2, we treated A549 cells with CARDS toxin at 4°C for 1 h, washed cells to remove excess CARDS toxin, and analyzed colocalization of CARDS and AnxA2 by confocal microscopy. At 4°C, CARDS toxin concentrated at the cell surface (green; Fig. 5A, panel b), whereas AnxA2 was detected both on the surface and in the cytoplasm (red; Fig. 5A, panel c). When the images of both fluorescently labeled immunological probes were superimposed, colocalization of CARDS toxin and AnxA2 at the cell surface was evident as structures that appear yellow due to the combined contributions of green and red fluorescence (Fig. 5A, panel d). Under these experimental conditions, z sections clearly indicated the colocalization of CARDS toxin with only surface-associated AnxA2 (Fig. 5B). When the temperature was raised to 37°C for 1 h, we observed green (internalized CARDS toxin), red (cytoplasmic AnxA2), and yellow (colocalized AnxA2 and toxin) puncta (Fig. 5C), clearly indicating that a subpopulation of internalized toxin remains associated with AnxA2.
FIG 5 .

CARDS toxin colocalizes with cell surface and intracellular AnxA2. (A) Colocalization of CARDS toxin with cell surface-associated AnxA2. (a to c) A549 cells were incubated with 10 µg of CARDS toxin at 4°C for 1 h, fixed in the presence of DAPI to stain nuclei (blue) (a) and probed with rabbit polyclonal anti-CARDS toxin antibody followed by secondary goat anti-rabbit IgG conjugated with Alexa Fluor 488 (green) (b) and anti-AnxA2 monoclonal antibody (1:500) followed by secondary goat anti-mouse IgG antibody conjugated with Alexa Fluor 555 (red) (c). (d) The merged image shows colocalization of CARDS toxin and AnxA2 at the membrane surface as yellow. (B) Interaction of CARDS toxin with A549 cell surface-associated AnxA2 using confocal laser-scanning microscopy. White arrows indicate the surface colocalization of CARDS toxin with AnxA2 (yellow) based upon serial z sections (0.44 μm; z series) obtained by analyzing x-y scans. (C) Interaction of internalized CARDS toxin with cytoplasmic AnxA2. (a to c) A549 cells were incubated with 10 µg of CARDS toxin at 4°C for 1 h and shifted to 37°C for 1 h, fixed in the presence of DAPI to stain nuclei (blue) (a) and probed with specific antibodies against CARDS toxin (green) (b) and AnxA2 (red) (1:1,000) (c) as described above for panel A. White arrows indicate colocalized intracellular CARDS toxin and AnxA2. (d) Images were collected sequentially from different channels with a confocal laser-scanning microscope and merged to show colocalization (yellow). To demonstrate the colocalization of intracellular AnxA2 and CARDS toxin, a section of the merged image shown by the white square is enlarged (top right panel).
Expression of AnxA2 increases CARDS toxin binding and subsequent vacuolization in HepG2 cells.
HepG2 cells, which are deficient in AnxA2 expression, were stably transfected with pCDNA-AnxA2 (HepG2-AnxA2) and screened for AnxA2 expression (Fig. 6A, lanes 2 to 4). The clone with the highest AnxA2 expression (Fig. 6A, lane 4) was selected for further studies. As expected, nontransfected control HepG2 cells possess very little AnxA2 (Fig. 6A, lane 1). Upon treatment with CARDS-mCherry (1 h at 4°C; 1 µg/ml), HepG2-AnxA2 cells exhibited ~50% higher CARDS toxin binding than nontransfected, control HepG2 cells (Fig. 6B). Consistent with these observations, we incubated HepG2-AnxA2 or nontransfected HepG2 cells with CARDS toxin at 37°C for 4 h and observed greater numbers (~98%) of HepG2-AnxA2 cells with vacuoles compared to HepG2 cells (~70%). Furthermore, individual vacuoles in HepG2-AnxA2 cells were larger (Fig. 6C, panel d) and more abundant (Fig. 6D) than nontransfected HepG2 cells.
FIG 6 .

AnxA2 expression enhances CARDS toxin binding and vacuolization in transfected HepG2 cells. (A) Expression of AnxA2 in stably transfected HepG2 cells. HepG2 cells were transfected with pCDNA plasmid carrying the AnxA2 gene, and G418-resistant stable clones were isolated and screened for expression of AnxA2 (lanes 2 to 4) and compared to normal HepG2 cells (lane 1). The clone expressing the highest level of HepG2-AnxA2 (lane 4) was selected for further studies. (B) Increased binding of CARDS toxin-mCherry to HepG2-AnxA2 cells. HepG2 and HepG2-AnxA2 cells were incubated with CARDS toxin-mCherry protein for 30 min at 4°C, and binding of CARDS toxin-mCherry protein to cells was analyzed by fluorometry as described in Materials and Methods. (C) AnxA2 expression enhances CARDS toxin-mediated vacuolization. HepG2 and HepG2-AnxA2 cells were incubated with CARDS toxin (50 µg/ml) for 4 h at 37°C, and vacuole formation was analyzed microscopically. (a to d) HepG2 cells alone (a) and with CARDS toxin (b) and HepG2-AnxA2 cells alone (c) and with CARDS toxin (d) are shown. (D) Increased numbers of CARDS toxin-induced vacuoles in HepG2-AnxA2 cells at 4 h. As described above for panel C, the cells were incubated with CARDS toxin, and the numbers of vacuoles per cell were counted and compared.
Suppression of AnxA2 and SP-A reduces CARDS toxin binding and subsequent vacuolization in SP-A-expressing H441 cells.
Compared to the pulmonary adenocarcinoma A549 and NCI-H358 (H358) cell lines, NCI-H441 (H441) cells, a pericardial fluid-derived pulmonary adenocarcinoma cell line, expresses both SP-A and AnxA2 (Fig. 7A). Transfection of H441 cells with siRNAs specific to AnxA2 or SP-A1 decreased the levels of AnxA2 and SP-A expression compared to nontransfected controls, as confirmed by immunoreactivity using anti-AnxA2 or anti-SP-A monoclonal antibodies (Fig. 7B). However, when AnxA2 and SP-A were silenced simultaneously in the same cells, we observed less reduction in the amounts of both AnxA2 and SP-A, possibly due to competition and inefficiencies using both siRNAs. Nonetheless, the amount of CARDS toxin bound was proportional to AnxA2 and SP-A levels (Fig. 7B and C). Interestingly, SP-A appears to be preferred to AnxA2 as a binding target, based upon the relative abundance of SP-A versus AnxA2 (Fig. 7B and C).
FIG 7 .

CARDS toxin binding and subsequent vacuolization in siRNA-transfected H441 cells. (A) Screening of different human cell lines for the expression of AnxA2 and SP-A. Total cell lysates (5 µg for analysis of AnxA2 and 30 µg for analysis of SP-A) were separated using 4 to 12% gels, transferred to nitrocellulose membranes, and probed with anti-AnxA2 monoclonal antibody (MAb) and anti-SP-A MAb. H441 cells that expressed both AnxA2 and SP-A were used for further studies. (B) Association of CARDS toxin with the cell surface upon suppression of AnxA2 and SP-A expression. H441 cells were transfected with siRNAs specific to AnxA2, SP-A, AnxA2 and SP-A, and control random siRNAs and incubated individually with CARDS toxin (5 µg/ml) for 1 h at 4°C. Five-microgram amounts of total cell lysates were analyzed by immunoblotting with anti-AnxA2 MAb or anti-SP-A MAb and anti-CARDS toxin polyclonal antibody. Comparative β-actin intensities were used as loading controls. (C) Quantification of CARDS toxin relative to AnxA2 and SP-A in siRNA-transfected H441 cells. Using β-actin (loading control), CARDS toxin, AnxA2, and SP-A immunoblot intensities, we calculated the relative signal difference of receptor-mediated association of CARDS toxin, along with expression levels of AnxA2 and SP-A. Data are from two independent experiments (experiments 1 and 2). (D) Suppression of AnxA2 or SP-A or AnxA2 and SP-A expression decreases CARDS toxin-mediated vacuolization in H441 cells. Untreated and siRNA-transfected H441cells were incubated with CARDS toxin (50 µg/ml) for 24 h at 37°C, and vacuole formation was analyzed microscopically. (a) Control random siRNA, (b) AnxA2-siRNA, (c) SP-A1-siRNA and (d) AnxA2 and SP-A1 siRNAs. (E) Quantification of numbers of CARDS toxin-induced vacuoles in H441 and siRNA-transfected H441 cells at 24 h. As described above for panel C, the cells were incubated with CARDS toxin, and the numbers of vacuoles per cell were counted and compared as described in Materials and Methods.
Similarly, upon CARDS toxin treatment for 24 h at 37°C, AnxA2- and/or SP-A-silenced H441 cells developed smaller vacuoles than control cells did (Fig. 7D). Furthermore, the number and distribution of medium to large vacuoles per cell in these receptor-specific silenced cells were reduced (Fig. 7D and E) compared to nontransfected H441 cells.
DISCUSSION
Bacterial ADP-ribosylating toxins manipulate host factors to reach their target proteins. Such factors include host cell membrane receptors that mediate toxin binding and components of the cellular endocytic machinery that bacterial toxins usurp for internalization and intracellular trafficking. Previously, our laboratory showed that M. pneumoniae CARDS toxin binds to human SP-A (17). Interestingly, CARDS toxin interacts with a wide range of mammalian cells, including cell lines that lack SP-A, indicating that additional receptor recognition events precede clathrin-facilitated endocytosis of CARDS toxin (23). Therefore, identification of CARDS toxin receptors is important in order to understand how the toxin distributes to airway and extrapulmonary sites, performs ADP-ribosylating and vacuolating activities (16), and elicits inflammatory pathways and tissue injury (20, 21). We used detergent-solubilized human cell membrane fractions combined with cell-free toxin affinity binding and immunoreactive assays to monitor the interaction between CARDS toxin and AnxA2 (Fig. 1). Similar methodologies have been utilized in the identification of the diphtheria toxin receptor from Vero cells, botulinum toxin type B receptor from synaptosomal membranes (25, 26), fibronectin-binding proteins from M. pneumoniae and aerolysin receptor from Aeromonas hydrophila (27–29). Using Escherichia coli-expressed and purified recombinant full-length CARDS toxin and full-length AnxA2 proteins, we confirmed that the interaction between CARDS toxin and AnxA2 is specific and concentration dependent (Fig. 2). We also showed that the C terminus of CARDS toxin (266CARDS) and not the N terminus (CARDS249) binds to AnxA2 (Fig. 3A), which is consistent with the functional role of the C-terminal region of CARDS toxin in receptor binding (30). Concerning the structural properties of AnxA2, the N terminus of AnxA2 is composed of amino acids 1 to 30 and the C terminus is composed of amino acids 31 to 339. The C-terminal domain has four annexin repeats and confers Ca2+-, phospholipid-, and actin-binding properties (31, 32). We compared CARDS toxin binding to 268AnxA2, which retains only one annexin repeat, with AnxA2267, which possesses three annexin repeats. We observed a statistically significant increase in CARDS toxin binding to AnxA2267, implicating the repeats as mediators of CARDS toxin interactions with AnxA2 (Fig. 3). Furthermore, AnxA2267 demonstrated increased CARDS toxin binding compared to FL AnxA2, possibly caused by conformational changes in AnxA2267 that favorably expose more CARDS toxin-interactive sites. The CARDS toxin-AnxA2 association was further reinforced by evidence that a single treatment of A549 cells with anti-AnxA2 monoclonal antibody, and not irrelevant monoclonal antibody, resulted in a significant suppression of toxin binding (Fig. 4A). In addition, decreased expression of AnxA2 using siRNA-mediated interference (Fig. 4B and C) markedly diminished the amount of CARDS toxin present in A549 total cell lysates. Using confocal microscopy and immunofluorescence, we demonstrated that CARDS toxin colocalizes with AnxA2 at the plasma membrane prior to cell entry and that a subpopulation of internalized CARDS toxin remains associated with intracellular AnxA2. These observations indicate that AnxA2 is functionally involved in early surface-associated CARDS toxin-mediated events and intracellular trafficking of CARDS toxin (Fig. 5). Data with HepG2-AnxA2-transfected cells further implicated AnxA2 as a functional receptor for CARDS toxin (Fig. 6). Interestingly, we still observed measurable toxin binding and internalization capabilities when AnxA2 was reduced or eliminated (Fig. 4 and 6) in A549 and HepG2 cell lines, suggesting the following possibilities: a subpopulation of cells was not effectively silenced for AnxA2; small amounts of AnxA2 are enough to promote CARDS toxin interactions; intracellular AnxA2 protein is relatively stable; or alternative receptor molecules continue to facilitate CARDS toxin binding and internalization.
To further analyze the importance of AnxA2 as a receptor of CARDS toxin, we compared CARDS toxin binding to AnxA2 in the presence of its other known receptor, SP-A (17). We observed reduced CARDS toxin binding and subsequent diminished vacuolization in targeted siRNA-transfected H441 cells, indicating that AnxA2 is a major target for CARDS toxin. Still, CARDS toxin utilizes both receptors (Fig. 7), suggesting that receptor composition and abundance regulate the ability of CARDS toxin to target specific anatomical sites.
AnxA2 has recently drawn attention for its ability to regulate multiple key processes in both cells and pathogens, including viruses and bacteria. AnxA2 is a member of the annexin family of proteins and exists either in monomeric form or as a heterotetramer containing two light chains of S100A10/p11 and two chains of AnxA2. Apart from their intracellular distribution, AnxA2 has also been detected on the membrane surface of diverse mammalian cells. Although the mechanisms by which surface expression of AnxA2 occurs are still unclear, cell surface AnxA2 acts as a receptor and/or adhesin (33, 34) and also participates in cell-cell interactions. When AnxA2 is accessible on the cell surfaces of macrophages or epithelial cells, it provides a physiological signal that facilitates phagocytosis of apoptotic cells (35–37). Furthermore, the AnxA2-S100A10 heterotetramer has been implicated in tight junction maintenance in epithelial MDCK cells (38). Recently, AnxA2 has been reported to regulate multiple key processes in both mammalian cells and pathogens, including viruses and bacteria. For example, AnxA2 assists in the invasion of epithelial cells by Pseudomonas aeruginosa (39) and Salmonella enterica serotype Typhimurium (40) and is recruited to bacterial attachment sites during enteropathogenic and enterohemorrhagic E. coli infections (41). AnxA2 also plays a role in the cellular entry of cytomegalovirus (42) and influenza viruses (43), assembly of human immunodeficiency virus type 1 (44, 45), release of blue tongue virus, replication of hepatitis C virus (46), and infectivity of enterovirus 71 (47).
Since AnxA2 performs multiple cellular functions, including regulation of vesicular trafficking, phagocytosis, and budding of clathrin-coated vesicles, we propose that AnxA2 not only mediates CARDS toxin binding but also acts as a key mediator of CARDS toxin ADP-ribosylating and vacuolating events. Consistent with these scenarios, AnxA2 assists in clathrin- (48) and caveola-mediated internalization (49) and is involved in macropinocytic rocketing (50), which correlates with our previous report that CARDS toxin uses clathrin-mediated pathways to enter cells (23). The correlation of vacuole size with the presence of receptors (increased numbers and larger sizes of vacuoles in CARDS toxin-treated/AnxA2-transfected HepG2 cells [Fig. 6C and D] and decreased numbers and smaller sizes of vacuoles in AnxA2-silenced A549 cells [Fig. 4D and E] and in AnxA2- and/or SP-A-silenced H441 cells [Fig. 7D and E]) suggests that CARDS toxin-receptor binding and internalization events lead to subsequent pathogenic activities mediated by intracellular toxin. Understanding how AnxA2 or SP-A transports CARDS toxin through distinct endocytic pathways is likely to delineate the complexity of CARDS toxin entry, trafficking, and pathogenic behaviors.
The relationship between CARDS toxin and AnxA2 is especially appealing in terms of intimate and successful pathogen-host interactions. To perform its ADP-ribosylating and vacuolating activities, CARDS toxin must bind to host cell surfaces via receptor-mediated events and be internalized and trafficked effectively. AnxA2, which exists both on the cell surface and intracellularly and performs many signaling functions, appears to be a “perfect partner” for CARDS toxin purposes. We will continue to examine receptor-mediated CARDS toxin interactions with AnxA2, SP-A, and other host molecules that facilitate CARDS toxin action with the intent to define therapeutic interventions that limit or eliminate the ability of CARDS toxin to usurp normal cell functions during M. pneumoniae acute and chronic infections.
MATERIALS AND METHODS
Bacteria and plasmids.
E. coli Top10 (Invitrogen), E. coli BL21(DE3) [F' ompT hsdS(rB mB) gal dcm λ(DE3)/pLysS] (Stratagene) were grown in Luria-Bertani (LB) broth and used to clone and express recombinant community-acquired respiratory distress syndrome (CARDS) toxin, annexin A2 (AnxA2), and their derivatives using the primers given in Table 1. For DNA manipulations, pCR2.1 (Apr Kmr TA cloning vector; Invitrogen), pET19b (Apr, N-terminal His10 tag, expression vector; Novagen), pGEX-6p-1 (GE Healthcare), and pCDNA3.1(+) (Invitrogen) were utilized. Glutathione S-transferase (GST)-AnxA2 plasmid was kindly provided by Marta Miaczynska (International Institute of Molecular and Cell Biology [IIMCB], Poland). Using GST-AnxA2 plasmid as the template, we amplified and cloned truncations of the C terminus and N terminus of AnxA2 in pGEX-6P-1 vector. pCDNA3.1(+) plasmid containing the full-length (FL) AnxA2 (pCDNA-AnxA2) was constructed with primers given in Table 1 which were inserted between the BamHI and XbaI restriction sites of pCDNA3.1(+). For construction of CARDS-mCherry fusion plasmid, UGA-corrected CARDS toxin (16) was PCR amplified using specific primers (Table 1) and fused with the N terminus of mCherry (51) by overlap extension PCR. This construct was cloned in PCR 2.1 and subcloned in pET19b vector between NdeI and BamHI to yield pET-CARDS-mCherry. All plasmid construct inserts were verified by sequencing at the Department of Microbiology and Immunology Nucleic Acids Facility (University of Texas Health Science Center at San Antonio [UTHSCSA])
TABLE 1 .
Primers used for amplification of CARDS toxin and AnxA2 derivatives
| Plasmid and primera | Primer sequence (5′ to 3′)b | Positionsc |
|---|---|---|
| pET-CARDS-mCherry | ||
| CARDSF | CATATGCCAAATCCTGTTAGATTTGTTTACCGTGTT | 1 → 33 |
| CARDSR | GCTGCCGCCGCCGCCGCTGCCAAAGCGATCAAAACCATCTTT | 1753 ← 1773 |
| mCherryF | GGCAGCGGCGGCGGCGGCAGCATGGTGAGCAAGGGCGAGGAGGATAAC | 1 → 27 |
| mCherryR | GGATCCTTACTTGTACAGCTCGTCCATGCCGCC | 685 ← 711 |
| pGEX-AnxA2267 | ||
| ANX2F | TACGGATCCTCTACTGTTCACGAAATCCTG | 4 → 24 |
| ANX2NR | TACCTGGAGTCAGGGCTTGTTCTGAATGCACTG | 781 ← 801 |
| pGEX-268AnxA2 | ||
| ANX2CF | TACGGATCCCTGTATTTTGCTGATCGGCTG | 802 → 822 |
| ANX2R | TACCTCGAGTCAGTCATCTCCACCACACAG | 999 ← 1020 |
| pCDNA-AnxA2 | ||
| ANXA2F | TCTGCGGGATCCATGTCTACTGTT | 1 → 12 |
| ANXA2R | CATGCGTCTAGATCAGTCATCTCC | 1008 ← 1020 |
The plasmid and the primers used to construct the plasmid are shown. The forward (F) and reverse (R) primers are indicated by the appropriate letter at the end of the primer designation.
In the primer sequences, the spacer sequences are shown in italic type, and introduced restriction endonuclease sites are shown in boldface type.
Positions of nucleotides within the coding open reading frame of CARDS toxin, AnxA2, and mCherry DNA sequence.
Cell lines and culture conditions.
Human alveolar adenocarcinoma A549 cells (CCL-185; ATCC), pulmonary adenocarcinoma NCI-H441 (H441) cells (HTB-174; ATCC), hepatocellular carcinoma HepG2 cells (HB-8065; ATCC), and cervical adenocarcinoma HeLa cells (CCL-2; ATCC) were cultured at 37°C in 95% air−5% CO2 in Kaighn’s modification of Ham’s F-12 medium (F12K), Roswell Park Memorial Institute medium (RPMI 1640), and in minimal essential medium (MEM) supplemented with 10% fetal bovine serum (HyClone), 2 mM l-glutamine, 1,000 U/ml penicillin G, and 50 µg/ml streptomycin (Invitrogen). AnxA2-expressing HepG2 cells were generated by transfection (X-tremeGENE HP DNA transfection reagent; Roche) using pCDNA-AnxA2, and cultures were maintained in selection medium containing 400 µg/ml G418 (Cellgro, VA). An individual transfected HepG2 clone that expressed high levels of recombinant AnxA2 (HepG-AnxA2) was used for further studies.
Expression and purification of recombinant proteins.
Plasmids encoding FL GST-AnxA2 and the C- and N-terminus truncations of AnxA2 were transformed into E. coli BL21, induced with 0.5 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) at 20°C for 16 h and purified according to the manufacturer’s instructions (GE Healthcare). For generation of CARDS-mCherry fusion protein, pET-CARDS toxin-mCherry plasmid was transformed in E. coli BL21 and overexpressed by induction with 1 mM IPTG at 18°C for 16 h. Soluble recombinant fusion protein was purified by nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography under native conditions (Qiagen) and eluted with lysis buffer containing 500 mM imidazole. Each collected fraction was desalted in 50 mM Tris-HCl buffer (pH 7.4) and 5% glycerol by using PD-10 columns and stored at −80°C. CARDS toxin and various truncated CARDS proteins that we had already generated, were used in the present study.
Receptor pulldown assay.
A549 cells were grown to 80% confluence in T75 flasks, and membranes were isolated using a subcellular fractionation kit (Pierce). Solubilized membrane fractions were incubated with control Ni-NTA beads or Ni-NTA beads coupled with CARDS toxin at 4°C overnight. After the beads were washed four times with phosphate-buffered saline (PBS), SDS lysis buffer was added, and samples were boiled. Protein profiles were resolved using 4 to 12% NuPAGE gels and analyzed by mass spectrometric analyses (UTHSCSA Institutional Mass Spectrometry Laboratory) or transferred to nitrocellulose membranes. For immunoblotting, nitrocellulose membranes were blocked with 2% skim milk in TBST (Tris-buffered saline with 0.1% Tween 20) for 1 h, washed three times with TBST, and incubated with AnxA2 antibody (Santa Cruz; 1 µg/ml dilution in TBST plus 1% skim milk [TBST-skim milk]) at 4°C overnight, followed by washing and incubation with anti-mouse IgG conjugated to horseradish peroxidase (anti-mouse IgG–HRP) (1:2,000 dilution in TBST-skim milk) for 45 min at room temperature. Reactivity was visualized using enhanced chemiluminescence (ECL) detection system (PerkinElmer).
CARDS toxin-AnxA2 overlay gel assay.
GST-AnxA2 or bovine serum albumin (BSA) (2 µg) was mixed with SDS lysis buffer, boiled at 100°C for 10 min, separated on 12% SDS-polyacrylamide gels, and transferred to nitrocellulose membranes by semidry transfer prior to blocking with PBS containing 2% (wt/vol) skim milk for 1 h. Then, CARDS toxin (7 µg/ml) in PBS-skim milk (1%) was added, and incubation continued for 2 h. After the membranes were washed three times with PBS-Tween 20 (0.05%), CARDS toxin binding to AnxA2 was detected with polyclonal rabbit antibody specific to CARDS toxin (diluted 1:4,000) followed by anti-rabbit secondary antibody conjugated with HRP (1:2,000; Invitrogen). Immunocomplex formation was detected using ECL reagent (PerkinElmer).
CARDS toxin-AnxA2 ELISA binding assay.
One hundred nanograms of GST-AnxA2 or BSA was coated overnight onto individual wells of 96-well plates at 4°C. After the wells were washed with PBS, they were blocked with 2% skim milk in PBS for 1 h and washed with PBS. Then, various concentrations of CARDS toxin (0.7 to 700 ng/well; diluted in PBS containing 1% skim milk [1% skim milk−PBS]) or equimolar concentrations of truncated CARDS toxin derivatives were added in triplicate and incubated for 1 h at room temperature, followed by 1-h incubation with anti-His tag monoclonal antibody (Clontech; diluted 1:4,000 in 1% skim milk−PBS) or polyclonal rabbit antibody specific to CARDS toxin (diluted 1:4,000). Wells were washed with PBS-Tween 20 (0.05%) and HRP-conjugated goat anti-mouse or goat anti-rabbit secondary antibody (Invitrogen; diluted 1:2,000 in 1% skim milk−PBS) was added for 45 min before the addition of TMB (3,3′,5,5′-tetramethylbenzidine) substrate and measurement at 450 nm.
Inhibition of CARDS toxin binding to A549 cells by AnxA2 antibody.
A549 cells were seeded at a density of 4 × 104 cells per well (96-well optical bottom black plates; Nunc) and grown for 24 h at 37°C. After we removed medium and washed the cells with PBS, we added AnxA2 monoclonal antibody (1 µg/well; Novus Biologicals) or negative-control monoclonal antibody (1 µg/well) in F12K medium to A549 cells at 4°C for 10 min. Then, CARDS-mCherry protein (1 µg) was added at 4°C for 30 min. The medium was removed, the wells were washed with PBS three times to remove unbound toxin, and mCherry fluorescence was measured using a Modulus microplate reader (Promega).
siRNA knockdown of AnxA2 and SP-A expression.
A549 cells were transfected with small interfering RNAs (siRNAs) (ORIGENE) against AnxA2 (siRNA1 and siRNA2) or AnxA2 Silencer Select siRNA (catalog no. 4390824; Life Technologies) or negative control (control siRNA) using Lipofectamine 2000 (Invitrogen) or Lipofectamine RNAiMAX transfection reagent (Life Technologies) according to the manufacturer’s instructions. H441 cells were treated with 30 pmol of AnxA2 Silencer Select siRNA (catalog no. 4390824; Life Technologies), SFTPA1 (SP-A1) or SFTPA2 (SP-A2) 27-mer siRNA duplexes (catalog no. SR318909 and SR319048; ORIGENE) complexed with Lipofectamine RNAiMAX reagent (Life Technologies) suspended in fetal bovine serum (FBS)-free Opti-MEM medium (Life Technologies). After 72 h of transfection, fresh medium containing CARDS toxin (1 to 10 µg/ml) was added to individual wells, and incubation continued at 4°C for 1 h. At the end of incubation, the cells were washed with PBS and lysed with SDS lysis buffer. Equal amounts of protein samples were resolved on SDS-polyacrylamide gels, transferred to nitrocellulose membranes, and probed with mouse monoclonal antibodies specific to AnxA2 (1:500 or 1:1,000), human surfactant protein A (hSP-A) (Abcam; 1:500), β-actin (Sigma-Aldrich; 1:1,000), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Cell Signaling; 1:1,000) along with in-house polyclonal antibodies against CARDS toxin (1:4,000) followed by incubation with respective HRP-conjugated goat anti-mouse or anti-rabbit IgG (Invitrogen) (1:5,000). Quantification of band intensities was performed by scanning immunostained bands from X-ray films and analyzing images with KODAK Image software. Relative levels of cell-associated CARDS toxin in receptor-suppressed cells were determined by comparing with negative-control siRNA-treated cells. Immunostained GAPDH (A549) or β-actin (H441) proteins were used as normalizers. Human SP-A is encoded by two highly similar genes, SP-A1 and SP-A2. Since SP-A1 siRNA itself is enough to suppress the expression of total SP-A we used SP-A1 alone.
Immunofluorescence microscopy.
A549 cells grown on coverslips in 24-well plates to 50% to 60% confluence were incubated at 4°C for 30 min, followed by the addition of CARDS toxin (10 µg/ml in cold serum-free F12K medium) for 1 h at 4°C. Unbound toxin was removed, and cells were either fixed in 2% paraformaldehyde to detect surface-associated CARDS toxin colocalization with AnxA2 or incubated in fresh prewarmed medium at 37°C for 1 h and then fixed in 2% paraformaldehyde to monitor intracellular colocalization of CARDS toxin and AnxA2. The cells were permeabilized with 0.2% Triton X-100 in PBS for 5 min and blocked for 30 min with 1% normal goat serum in PBS. Then, the cells were incubated with rabbit polyclonal anti-CARDS toxin antibody (1:500 dilution) in 0.2% normal goat serum in PBS for 1 h, washed three times with 0.2% normal goat serum in PBS, and further incubated with secondary goat polyclonal anti-rabbit antibody (1:400 dilution) labeled with Alexa Fluor 488 (Invitrogen) in 0.2% normal goat serum in PBS for 1 h. The samples were washed three times with 0.2% normal goat serum in PBS and incubated with anti-human AnxA2 mouse monoclonal primary antibody 1G7 (Novus Biologicals; 1:500) in 0.2% normal goat serum in PBS for 1 h. After the standard washes, the cells were incubated with secondary goat anti-mouse antibody labeled with Alexa Fluor 555 (Invitrogen) (1:400) with 0.2% normal goat serum in PBS for 1 h. Samples were washed with PBS and mounted on glass slides using Vectashield hard fix mounting medium containing DAPI (4',6-diamidino-2-phenylindole dihydrochloride) stain (Vector Laboratories Inc.). Confocal image acquisition was done using an Olympus XI-81 confocal laser-scanning microscope with Flow view 1000 imaging software (UTHSCSA Core Optical Imaging Facility) and 60× oil immersion lens. z series at 0.44-μm sections were obtained by combining x-y scans taken along the z axis. Cells without CARDS toxin were processed similarly to serve as negative controls.
Mammalian cell vacuolization.
HepG2 and HepG2-AnxA2 cells were grown in 6-well plates at 50% confluence, and similarly, normal and siRNA-treated A549 and H441 cells were grown at 50% confluence. Then, CARDS toxin at various concentrations (0.5 to 50 µg/ml) was added as described before (24). The timing of vacuolization, number of vacuoles per cell, size of the vacuoles, and number of vacuolated cells were observed at different time points. All experiments were repeated in triplicate, and 20 fields of 20 to 25 cells per sample were examined to determine the vacuolization patterns. Statistical analysis was performed using Microsoft Excel.
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award U19 AI070412 and the Kleberg Foundation.
The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
We thank Rose Garza for assembling the manuscript.
Footnotes
Citation Somarajan SR, Al-Asadi F, Ramasamy K, Pandranki L, Baseman JB, Kannan TR. 2014. Annexin A2 mediates Mycoplasma pneumoniae community-acquired respiratory distress syndrome toxin binding to eukaryotic cells. mBio 5(4):e01497-14. doi:10.1128/mBio.01497-14.
REFERENCES
- 1. Baseman JB, Tully JG. 1997. Mycoplasmas: sophisticated, reemerging, and burdened by their notoriety. Emerg. Infect. Dis. 3:21–32. 10.3201/eid0301.970103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kraft M, Cassell GH, Pak J, Martin RJ. 2002. Mycoplasma pneumoniae and Chlamydia pneumoniae in asthma: effect of clarithromycin. Chest 121:1782–1788. 10.1378/chest.121.6.1782 [DOI] [PubMed] [Google Scholar]
- 3. Biscardi S, Lorrot M, Marc E, Moulin F, Boutonnat-Faucher B, Heilbronner C, Iniguez JL, Chaussain M, Nicand E, Raymond J, Gendrel D. 2004. Mycoplasma pneumoniae and asthma in children. Clin. Infect. Dis. 38:1341–1346. 10.1086/392498 [DOI] [PubMed] [Google Scholar]
- 4. Waites KB, Talkington DF. 2004. Mycoplasma pneumoniae and its role as a human pathogen. Clin. Microbiol. Rev. 17:697–728. 10.1128/CMR.17.4.697-728.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nisar N, Guleria R, Kumar S, Chand Chawla T, Ranjan Biswas N. 2007. Mycoplasma pneumoniae and its role in asthma. Postgrad. Med. J. 83:100–104. 10.1136/pgmj.2006.049023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Atkinson TP, Balish MF, Waites KB. 2008. Epidemiology, clinical manifestations, pathogenesis and laboratory detection of Mycoplasma pneumoniae infections. FEMS Microbiol. Rev. 32:956–973. 10.1111/j.1574-6976.2008.00129.x [DOI] [PubMed] [Google Scholar]
- 7. Peters J, Singh H, Brooks EG, Diaz J, Kannan TR, Coalson JJ, Baseman JG, Cagle M, Baseman JB. 2011. Persistence of community-acquired respiratory distress syndrome toxin-producing Mycoplasma pneumoniae in refractory asthma. Chest 140:401–407. 10.1378/chest.11-0221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baseman JB. 1993. The cytadhesins of Mycoplasma pneumoniae and M. genitalium. Subcell. Biochem. 20:243–259. 10.1007/978-1-4615-2924-8_9 [DOI] [PubMed] [Google Scholar]
- 9. Krause DC, Balish MF. 2001. Structure, function, and assembly of the terminal organelle of Mycoplasma pneumoniae. FEMS Microbiol. Lett. 198:1–7. 10.1111/j.1574-6968.2001.tb10610.x [DOI] [PubMed] [Google Scholar]
- 10. Tyron VV, Baseman JB. 1992. Pathogenic determinants and mechanisms, p 457–471 In Maniloff J, McElhaney RN, Finch LR, Baseman JB. (ed), Mycoplasmas: molecular biology and pathogenesis. American Society for Microbiology, Washington, DC [Google Scholar]
- 11. Somerson NL, Walls BE, Chanock RM. 1965. Hemolysin of Mycoplasma pneumoniae: tentative identification as a peroxide. Science 150:226–228. 10.1126/science.150.3693.226 [DOI] [PubMed] [Google Scholar]
- 12. Cohen G, Somerson NL. 1967. Mycoplasma pneumoniae: hydrogen peroxide secretion and its possible role in virulence. Ann. N. Y. Acad. Sci. 143:85–87. 10.1111/j.1749-6632.1967.tb27648.x [DOI] [PubMed] [Google Scholar]
- 13. Lynch RE, Cole BC. 1980. Mycoplasma pneumoniae: a prokaryote which consumes oxygen and generates superoxide but which lacks superoxide dismutase. Biochem. Biophys. Res. Commun. 96:98–105. 10.1016/0006-291X(80)91186-9 [DOI] [PubMed] [Google Scholar]
- 14. Tanaka H, Narita M, Teramoto S, Saikai T, Oashi K, Igarashi T, Abe S. 2002. Role of interleukin-18 and T-helper type 1 cytokines in the development of Mycoplasma pneumoniae pneumonia in adults. Chest 121:1493–1497. 10.1378/chest.121.5.1493 [DOI] [PubMed] [Google Scholar]
- 15. Yang J, Hooper WC, Phillips DJ, Talkington DF. 2004. Cytokines in Mycoplasma pneumoniae infections. Cytokine Growth Factor Rev. 15:157–168. 10.1016/j.cytogfr.2004.01.001 [DOI] [PubMed] [Google Scholar]
- 16. Kannan TR, Baseman JB. 2006. ADP-ribosylating and vacuolating cytotoxin of Mycoplasma pneumoniae represents unique virulence determinant among bacterial pathogens. Proc. Natl. Acad. Sci. U. S. A. 103:6724–6729. 10.1073/pnas.0510644103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kannan TR, Provenzano D, Wright JR, Baseman JB. 2005. Identification and characterization of human surfactant protein A binding protein of Mycoplasma pneumoniae. Infect. Immun. 73:2828–2834. 10.1128/IAI.73.5.2828-2834.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kannan TR, Coalson JJ, Cagle M, Musatovova O, Hardy RD, Baseman JB. 2011. Synthesis and distribution of CARDS toxin during Mycoplasma pneumoniae infection in a murine model. J. Infect. Dis. 204:1596–1604. 10.1093/infdis/jir557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kannan TR, Musatovova O, Balasubramanian S, Cagle M, Jordan JL, Krunkosky TM, Davis A, Hardy RD, Baseman JB. 2010. Mycoplasma pneumoniae community acquired respiratory distress syndrome toxin expression reveals growth phase and infection-dependent regulation. Mol. Microbiol. 76:1127–1141. 10.1111/j.1365-2958.2010.07092.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hardy RD, Coalson JJ, Peters J, Chaparro A, Techasaensiri C, Cantwell AM, Kannan TR, Baseman JB, Dube PH. 2009. Analysis of pulmonary inflammation and function in the mouse and baboon after exposure to Mycoplasma pneumoniae CARDS toxin. PLoS One 4:e7562. 10.1371/journal.pone.0007562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Medina JL, Coalson JJ, Brooks EG, Winter VT, Chaparro A, Principe MF, Kannan TR, Baseman JB, Dube PH. 2012. Mycoplasma pneumoniae CARDS toxin induces pulmonary eosinophilic and lymphocytic inflammation. Am. J. Respir. Cell Mol. Biol. 46:815–822. 10.1165/rcmb.2011-0135OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Techasaensiri C, Tagliabue C, Cagle M, Iranpour P, Katz K, Kannan TR, Coalson JJ, Baseman JB, Hardy RD. 2010. Variation in colonization, ADP-ribosylating and vacuolating cytotoxin, and pulmonary disease severity among Mycoplasma pneumoniae strains. Am. J. Respir. Crit. Care Med. 182:797–804. 10.1164/rccm.201001-0080OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krishnan M, Kannan TR, Baseman JB. 2013. Mycoplasma pneumoniae CARDS toxin is internalized via clathrin-mediated endocytosis. PLoS One 8:e62706. 10.1371/journal.pone.0062706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnson C, Kannan TR, Baseman JB. 2011. Cellular vacuoles induced by Mycoplasma pneumoniae CARDS toxin originate from Rab9-associated compartments. PLoS One 6:e22877. 10.1371/journal.pone.0022877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mekada E, Okada Y, Uchida T. 1988. Identification of diphtheria toxin receptor and a nonproteinous diphtheria toxin-binding molecule in Vero cell membrane. J. Cell Biol. 107:511–519. 10.1083/jcb.107.2.511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nishiki T, Kamata Y, Nemoto Y, Omori A, Ito T, Takahashi M, Kozaki S. 1994. Identification of protein receptor for Clostridium botulinum type B neurotoxin in rat brain synaptosomes. J. Biol. Chem. 269:10498–10503 [PubMed] [Google Scholar]
- 27. Cowell S, Aschauer W, Gruber HJ, Nelson KL, Buckley JT. 1997. The erythrocyte receptor for the channel-forming toxin aerolysin is a novel glycosylphosphatidylinositol-anchored protein. Mol. Microbiol. 25:343–350. 10.1046/j.1365-2958.1997.4691828.x [DOI] [PubMed] [Google Scholar]
- 28. Dallo SF, Kannan TR, Blaylock MW, Baseman JB. 2002. Elongation factor Tu and E1 beta subunit of pyruvate dehydrogenase complex act as fibronectin binding proteins in Mycoplasma pneumoniae. Mol. Microbiol. 46:1041–1051. 10.1046/j.1365-2958.2002.03207.x [DOI] [PubMed] [Google Scholar]
- 29. Nelson KL, Raja SM, Buckley JT. 1997. The glycosylphosphatidylinositol-anchored surface glycoprotein Thy-1 is a receptor for the channel-forming toxin aerolysin. J. Biol. Chem. 272:12170–12174. 10.1074/jbc.272.18.12170 [DOI] [PubMed] [Google Scholar]
- 30. Kannan TR, Krishnan M, Ramasamy K, Becker A, Pakhomova ON, Hart PJ, Baseman JB. 20 June 2014. Functional mapping of community acquired respiratory distress syndrome (CARDS) toxin of Mycoplasma pneumoniae defines regions with ADP-ribosyltransferase, vacuolating, and receptor-binding activities. Mol. Microbiol. 10.1111/mmi.12680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gerke V, Moss SE. 2002. Annexins: from structure to function. Physiol. Rev. 82:331–371 [DOI] [PubMed] [Google Scholar]
- 32. Filipenko NR, Waisman DM. 2001. The C terminus of annexin II mediates binding to F-actin. J. Biol. Chem. 276:5310–5315. 10.1074/jbc.M009710200 [DOI] [PubMed] [Google Scholar]
- 33. Hajjar KA, Guevara CA, Lev E, Dowling K, Chacko J. 1996. Interaction of the fibrinolytic receptor, annexin II, with the endothelial cell surface. Essential role of endonexin repeat 2. J. Biol. Chem. 271:21652–21659. 10.1074/jbc.271.35.21652 [DOI] [PubMed] [Google Scholar]
- 34. Peterson EA, Sutherland MR, Nesheim ME, Pryzdial EL. 2003. Thrombin induces endothelial cell-surface exposure of the plasminogen receptor annexin 2. J. Cell Sci. 116:2399–2408. 10.1242/jcs.00434 [DOI] [PubMed] [Google Scholar]
- 35. Fan X, Krahling S, Smith D, Williamson P, Schlegel RA. 2004. Macrophage surface expression of annexins I and II in the phagocytosis of apoptotic lymphocytes. Mol. Biol. Cell 15:2863–2872. 10.1091/mbc.E03-09-0670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Law AL, Ling Q, Hajjar KA, Futter CE, Greenwood J, Adamson P, Wavre-Shapton ST, Moss SE, Hayes MJ. 2009. Annexin A2 regulates phagocytosis of photoreceptor outer segments in the mouse retina. Mol. Biol. Cell 20:3896–3904. 10.1091/mbc.E08-12-1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fang YT, Lin CF, Wang CY, Anderson R, Lin YS. 2012. Interferon-gamma stimulates p11-dependent surface expression of annexin A2 in lung epithelial cells to enhance phagocytosis. J. Cell. Physiol. 227:2775–2787. 10.1002/jcp.23026 [DOI] [PubMed] [Google Scholar]
- 38. Lee DB, Jamgotchian N, Allen SG, Kan FW, Hale IL. 2004. Annexin A2 heterotetramer: role in tight junction assembly. Am. J. Physiol. Renal Physiol. 287:F481–F491. 10.1152/ajprenal.00175.2003 [DOI] [PubMed] [Google Scholar]
- 39. Kirschnek S, Adams C, Gulbins E. 2005. Annexin II is a novel receptor for Pseudomonas aeruginosa. Biochem. Biophys. Res. Commun. 327:900–906. 10.1016/j.bbrc.2004.12.089 [DOI] [PubMed] [Google Scholar]
- 40. Jolly C, Winfree S, Hansen B, Steele-Mortimer O. 2014. The annexin A2/p11 complex is required for efficient invasion of Salmonella typhimurium in epithelial cells. Cell. Microbiol. 16:64–77. 10.1111/cmi.12180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Munera D, Martinez E, Varyukhina S, Mahajan A, Ayala-Sanmartin J, Frankel G. 2012. Recruitment and membrane interactions of host cell proteins during attachment of enteropathogenic and enterohaemorrhagic Escherichia coli. Biochem. J. 445:383–392. 10.1042/BJ20120533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wright JF, Kurosky A, Wasi S. 1994. An endothelial cell-surface form of annexin II binds human cytomegalovirus. Biochem. Biophys. Res. Commun. 198:983–989. 10.1006/bbrc.1994.1140 [DOI] [PubMed] [Google Scholar]
- 43. LeBouder F, Morello E, Rimmelzwaan GF, Bosse F, Péchoux C, Delmas B, Riteau B. 2008. Annexin II incorporated into influenza virus particles supports virus replication by converting plasminogen into plasmin. J. Virol. 82:6820–6828. 10.1128/JVI.00246-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ryzhova EV, Vos RM, Albright AV, Harrist AV, Harvey T, González-Scarano F. 2006. Annexin 2: a novel human immunodeficiency virus type 1 Gag binding protein involved in replication in monocyte-derived macrophages. J. Virol. 80:2694–2704. 10.1128/JVI.80.6.2694-2704.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Harrist AV, Ryzhova EV, Harvey T, González-Scarano F. 2009. Anx2 interacts with HIV-1 Gag at phosphatidylinositol (4,5) bisphosphate-containing lipid rafts and increases viral production in 293T cells. PLoS One 4:e5020. 10.1371/journal.pone.0005020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Backes P, Quinkert D, Reiss S, Binder M, Zayas M, Rescher U, Gerke V, Bartenschlager R, Lohmann V. 2010. Role of annexin A2 in the production of infectious hepatitis C virus particles. J. Virol. 84:5775–5789. 10.1128/JVI.02343-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang SL, Chou YT, Wu CN, Ho MS. 2011. Annexin II binds to capsid protein VP1 of enterovirus 71 and enhances viral infectivity. J. Virol. 85:11809–11820. 10.1128/JVI.00297-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Turpin E, Russo-Marie F, Dubois T, de Paillerets C, Alfsen A, Bomsel M. 1998. In adrenocortical tissue, annexins II and VI are attached to clathrin coated vesicles in a calcium-independent manner. Biochim. Biophys. Acta 1402:115–130. 10.1016/S0167-4889(97)00151-1 [DOI] [PubMed] [Google Scholar]
- 49. Schnitzer JE, Liu J, Oh P. 1995. Endothelial caveolae have the molecular transport machinery for vesicle budding, docking, and fusion including VAMP, NSF, SNAP, annexins, and GTPases. J. Biol. Chem. 270:14399–14404 [DOI] [PubMed] [Google Scholar]
- 50. Merrifield CJ, Rescher U, Almers W, Proust J, Gerke V, Sechi AS, Moss SE. 2001. Annexin 2 has an essential role in actin-based macropinocytic rocketing. Curr. Biol. 11:1136–1141. 10.1016/S0960-9822(01)00321-9 [DOI] [PubMed] [Google Scholar]
- 51. Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. 2004. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22:1567–1572. 10.1038/nbt1037 [DOI] [PubMed] [Google Scholar]