

Abstract
Motivation: Several computational methods have been developed to identify cancer drivers genes—genes responsible for cancer development upon specific alterations. These alterations can cause the loss of function (LoF) of the gene product, for instance, in tumor suppressors, or increase or change its activity or function, if it is an oncogene. Distinguishing between these two classes is important to understand tumorigenesis in patients and has implications for therapy decision making. Here, we assess the capacity of multiple gene features related to the pattern of genomic alterations across tumors to distinguish between activating and LoF cancer genes, and we present an automated approach to aid the classification of novel cancer drivers according to their role.
Result: OncodriveROLE is a machine learning-based approach that classifies driver genes according to their role, using several properties related to the pattern of alterations across tumors. The method shows an accuracy of 0.93 and Matthew's correlation coefficient of 0.84 classifying genes in the Cancer Gene Census. The OncodriveROLE classifier, its results when applied to two lists of predicted cancer drivers and TCGA-derived mutation and copy number features used by the classifier are available at http://bg.upf.edu/oncodrive-role.
Availability and implementation: The R implementation of the OncodriveROLE classifier is available at http://bg.upf.edu/oncodrive-role.
Contact: abel.gonzalez@upf.edu or nuria.lopez@upf.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Research in cancer genomics has identified hundreds of genes involved in different stages of tumorigenesis due to specific somatic events. Single nucleotide variants, and large-scale amplifications and deletions of chromosomal regions have been identified as two of the main driver alterations in human tumors. The genes suffering these alterations are traditionally classified as oncogenes and tumor suppressors, depending on their role in cancer development. When the product of tumor suppressors lose their function, tumor cells tend to proliferate faster. Driver alterations in these genes frequently exhibit a recessive behavior. The loss of function (LoF) can be achieved through truncating or missense mutations, DNA deletions or hypermethylation of their promoters. Some known LoF genes, most notably BRCA1 and BRCA2, carry germline variants that increase the susceptibility to develop a tumor because only one hit is required to inactivate their function. Oncogenes, on the other hand, increase or change their function upon somatic variants in tumorigenesis. Therefore, their mode of action follow a dominant pattern, as one faulty copy of the gene is frequently enough to provide the required phenotype. A copy number gain may exponentiate the oncogenic function of the gene; a point mutation may achieve the same result by changing key amino acid residues, which results in constitutive activation of the protein, or produce a new biochemical function. These special cases are also regarded as activating driver mutations, as the new function is gained much like in the case of classic oncogenes. The Cancer Gene Census (CGC; Futreal et al., 2004) is a regularly updated compilation of well-studied cancer genes, which classifies their mode of action as dominant or recessive, following the oncogene/tumor suppressor paradigm, LoF and Act (activated), hereafter. The CGC contains some 500 genes implicated in cancer (November 2013). This is a rather small fraction of the 20 000 genomes in the human genome (International Human Genome Sequencing Consortium, 2004), but recent large-scale re-sequencing projects of tumor genomes (Hudson et al., 2010) suggest many additional genes may be involved in tumorigenesis. One important first step in the analysis of datasets of cancer genomics alterations is the identification of the genes that drive tumorigenesis. This is a non-trivial problem because tumor samples contain up to thousands of somatic alterations. The list of genes altered in tumors is heterogeneous, even within the same cancer type. Therefore, the difficult task is to distinguish between driver and passenger alterations.
The most intuitive way to identify driver genes is to detect signals of positive selection across tumor samples because cancer cell populations undergo a selection process during the progression of the disease. Different methods that aim to identify driver genes tackle different evidences to achieve their goal (Gonzalez-Perez et al., 2013a). Two recent efforts to comprehensively identify driver genes across large cohorts carried out by Lawrence et al. (2014) and Tamborero et al. (2013b), combining several signals of positive selection (Dees et al., 2012; Gonzalez-Perez and Lopez-Bigas, 2012; Lawrence et al., 2013; Reimand et al., 2013) detected, respectively, 291 and 260 likely driver genes.
Although years of experimental work have revealed the role of most well-known cancer genes, now our capability of detecting drivers has surpassed our capacity to probe their mode of action. Thus, revealing the mode of action of driver genes in tumorigenesis is becoming crucial to fully understand the mechanisms of tumorigenesis. This is essential for the development of new targeted cancer therapies because as a general rule only Act drivers are in principle susceptible to targeted drugs. Although exceptionally, some mutated tumor suppressors may be targeted (e.g. Lambert et al., 2009), other strategies, such as synthetic lethality, are needed to compensate for their LoF. This is the reason why we need to develop bioinformatics approaches to make this classification as accurately as possible. Vogelstein et al. recently described the so-called ‘20/20 rule’ to detect tumor suppressor genes and oncogenes based on their mutational pattern across tumor samples (Vogelstein et al., 2013). It states that genes with ≥20% truncating mutations are tumor suppressors, whereas genes with >20% of missense mutations in recurrent positions are oncogenes. While it correctly detects and classifies most of the well-known cancer genes, the rule fails to identify drivers included in newer catalogs (Tamborero et al., 2013b), mostly the lowly recurrent ones.
Building upon the same idea, Davoli et al. developed a machine learning approach to directly identify tumor suppressor genes and oncogenes from the somatic alterations observed across cohorts of tumor samples through their mutational and copy number patterns. Many cancer drivers are recognized correctly by carefully selected features (Davoli et al., 2013).
We recently proposed a strategy to obtain a comprehensive list of drivers minimizing the probability of detecting false-positive findings by combining complementary methods that detected different signals of positive selection (Tamborero et al., 2013b).
Once a list of high-confidence drivers (HCDs) is obtained, it is important to classify those in their mode of action. To this aim, we first carefully assessed the capability of 30 features to differentiate between these two groups of cancer genes. Then, we combined different sets of features with various classification algorithms to create several automated classifiers. We trained these classifiers with CGC genes, and after careful check of their performance, we selected a random forest algorithm that achieves an accuracy (ACC) of 93%, which we call OncodriveROLE. It is the first freely available automatic classifier that undertakes the task of assessing the mode of action of driver genes. Used in this setting, it may shed light upon the mechanisms of tumorigenesis in major cancer types. We have used it to classify the two previously mentioned lists of mutational drivers that have been recently published, namely, HCDs (Tamborero et al., 2013b) and Cancer5000 (Lawrence et al., 2014), and describe the results of this analysis.
2 METHODS
2.1 Mutation data, copy number alteration data and cancer driver lists
We retrieved data for the 17 TCGA (The Cancer Gene Census) projects currently available without restriction: BLCA, BRCA, COAD/READ, GBM, HNSC, KIRC, LAML, LGG, LUAD, LUSC, OV, PRAD, SKCM, STAD, THCA and UCEC. We designed and computed several features that we hypothesized might be useful to classify driver genes according to the role using mutation and copy number data. These features are based on the patterns of mutations and copy number alterations (CNAs) across tumor samples. Tumors with at least one mutation in the TCGA pan-cancer 17 dataset available at Synapse (syn1729383.2) were retrieved after excluding those considered as hypermutators (Kandoth, 2014; Kandoth et al., 2013). Hypermutators of a tumor type contained more than (Q3 + 4.5 × IQR) somatic mutations, where Q3 and IQR are the third quartile and the interquartile range of the distribution of mutations across all samples of the tumor type, respectively. After filtering, the pan-cancer 17 dataset was composed of 4327 samples. These mutations were mapped to protein positions, and their consequence types were assessed using the IntOGen-mutations pipeline (Gonzalez-Perez et al., 2013b), which makes use of the Ensembl Variant Effect Predictor (v70; Chen et al., 2010). The CNA status for all probed genes was downloaded from the January run of the TCGA FIREHOSE pipeline at the Broad Institute (http://gdac.broadinstitute.org/).
To apply the OncodriveROLE classifier, we gathered two lists of likely cancer drivers from the Supplementary Material of two independent papers (Lawrence et al., 2014; Tamborero et al., 2013b). From the Tamborero et al. (2013b), we selected the list of 291 genes annotated as HCDs, discarding one non-coding gene. From Lawrence et al. (2013), we obtained a list of 260 genes from the spreadsheet ‘Individual q-values’.
For comparison purposes, we retrieved the classifications of genes carried out by the previous work by Davoli et al. from the Supplementary Material of their paper, applying the same cutoffs described in the manuscript (Davoli et al., 2013). We also obtained the classification carried out by applying the 20/20 rule (Vogelstein et al., 2013) to the mutational dataset of 17 tumors types.
Whenever possible, data were obtained associated to Ensembl gene identifiers (Flicek et al., 2013). Other identifiers have been mapped to Ensembl gene identifiers with a dataset obtained from Ensembl v70.
2.2 Classifiers
We chose six different classifiers to test: cforest.party (cforest method in R), conditionalTree (ctree), logisticRegression (glm), naiveBayes (train), simpleTree (rpart) and randomForest (Breiman, 2001; Hothorn et al., 2006; Kuhn, 2008; Olshen et al., 1984; R Core Team, 2013). Some classifiers either do not accept missing values or perform variable imputation for those. Therefore, we opted to remove genes if they had missing values in one or more of the features and leave them unclassified. From each classifier we obtained a score of the certainty that each gene belongs to the Act class.
2.3 Training set
To use cancer genes with well-established roles as training set, we downloaded the material available at the CGC in November 2013 (Futreal et al., 2004). See below details on the curation of this dataset for training the classifier.
The CGC contains extensive and manually annotated information on well-known cancer genes and classifies the cancer genes into dominant (Dom) and recessive (Rec) influence on tumorigenesis. We have used the CGC classification into Rec and Dom classes as proxy for LoF and Act genes. Genes with ambiguous annotation, such as ‘Rec?’ or ‘Dom?’ or not citing observed somatic mutations were discarded, leaving 381 entries (see Supplementary Table S7 for their classification). To only include CGC driver genes, which are likely to act across the TCGA pan-cancer 17 cohort, we used a one-signal filter: we discarded genes not detected as significant by MutSigCV (recurrence signal), OncodriveFM (mutations impact signal) or OncodriveCLUST (mutations clustering signal). We also rejected genes with < 12 protein affecting mutations (PAMs; Gonzalez-Perez and Lopez-Bigas, 2012; Lawrence et al., 2013; Tamborero et al., 2013a). Only 115 CGC genes passed this filter. Equally, all CGC genes that were solely associated to translocation events—all labeled with Dom—were not allowed in the training set, finally leaving 76 entries in the training set.
2.4 Computing features
All features we computed are listed in Table 1 along with a brief explanation of their computation: some of them are similar to the ones used previously (Davoli et al., 2013; Vogelstein et al., 2013). Truncating mutations include mutations causing a frameshift, a gained or lost stop codon as well as mutations in splice donor or acceptor sites. PAMs include truncating mutations and missense mutations. Benign missense refers to missense mutations that are categorized as low or unknown functional impact by TransFIC (Gonzalez-Perez et al., 2012). OncodriveFM P-values (Gonzalez-Perez and Lopez-Bigas, 2012) and the location of OncodriveCLUST clusters of mutations (Tamborero et al., 2013a) for all driver genes were obtained by running the IntOGen-mutations pipeline on the TCGA pan-cancer 17 dataset.
Table 1.
List of mutational and CNA features for cancer driver genes
| Attribute name | Description |
|---|---|
| CNA_cbs_countGain | # samples in cohort with CBS value > 1.1 |
| CNA_cbs_countLoss | # samples in cohort with CBS value < 1.1 |
| CNA_cbs_logratio_GvL | Log10-ratio of countGain VS countLoss |
| CNA_gain_freq | # samples in cohort with CBS value > 1.1 / cohort size |
| CNA_loss_freq | # samples in cohort with CBS value < 1.1 / cohort size |
| MUTS_clusters_miss_VS_pam | Log10-ratio of missense VS PAM within OncodriveCLUST peaks |
| MUTS_freq_clustered | # of mutations in OncodriveCLUST peaks / # of samples with gene mutated |
| MUTS_freq_disruptive | # of samples with truncating mutations or high impact missense / # of samples having gene mutations |
| MUTS_freq_missH | # of high impact missense mutations not in OncodriveCLUST peaks / # samples with gene mutated |
| MUTS_freq_missHM | # of high and medium impact missense mutations not in OncodriveCLUST peaks / # samples with gene mutated |
| MUTS_freq_truncating | # of samples with truncating mutations / # of samples with at least one mutation |
| MUTS_missense_clustercov | # missense mutations in OncodriveCLUST peaks / # missense mutations / # amino acids covered by peaks |
| MUTS_missense_mutrec | # recurrent missense mutations / # high and medium impact missense mutations |
| MUTS_missense_rec_freq | # recurrent missense mutations / # mutations (as in Vogelstein et al.) |
| MUTS_missense_recHM | # samples with high and medium impact recurrent missense mutations / # samples with missense mutations |
| MUTS_OncoFM_pvalue | OncodriveFM P-value |
| MUTS_pams_count | # samples with PAM |
| MUTS_pams_freq | # samples with PAM / # samples with gene mutations |
| MUTS_pams_ratio | # samples with PAM VS # samples with no PAM |
| MUTS_pamsrec_freq | # samples with PAM VS # of samples with gene mutation |
| MUTS_trunc_count | # samples with truncating mutations |
| MUTS_trunc_freq_cohort | # of truncating mutations / # of samples with gene mutations |
| MUTS_trunc_mutfreq | # truncating mutations / # mutations (as in Vogelstein et al.) |
| MUTS_trunc_vs_missbenign_ratio | # samples with truncating mutations VS # samples with benign missense mutations |
| MUTS_trunc_vs_missense_ratio | # samples with truncating mutations VS # samples with missense mutations |
| MUTS_trunc_vs_notrunc_ratio | # samples with truncating mutations VS # samples without truncating mutations |
| MUTS_tuson_missHM_missbenign_ratio | # samples with high and medium impact mutations VS # samples with benign missense mutations (as described in Davoli et al.) |
| MUTS_tuson_splicing_missbenign_ratio | # samples splicing variants mutations VS # samples with benign missense mutations (as described in Davoli et al.) |
| MUTS_tuson_trunc_missbenign_ratio | # samples with truncating (excluding splicing variants) mutations VS # samples with benign missense mutations (as described in Davoli et al.) |
Note: List of features initially created for characterizing LoF and Act genes. The description reflects the formula applied for the calculation of the features. All features elaborated describe either mutation or CNA characteristics. Abbreviations used in the descriptions are: # (number sign): Count/number of, / (slash): divided by, CBS : circular binary segmentation, truncating mutations: frameshift, stop gained and lost, splice donor and acceptor, missense: all missense mutations and insertions and deletions not altering the reading frame, high and medium impact mutations: all missense mutations with and TransFIC impact of 1 and 2 , benign missense: all missense with low or unknown TransFIC impact, PAM : protein affecting: frameshift, stop gained and lost, splice donor and acceptor, missense, (gene) mutations: all mutations-affecting coding sequence, VS : versus—a ratio has been obtained.
The R implementation of Wilcoxon's signed rank (R Core Team, 2013) was used to compare the distribution of each feature between the CGC Rec and CGC Dom genes. We also used the variable importance function from the party library (Hothorn et al., 2006; Strobl et al., 2008) to rank features for their selection to be taken into account by the classifiers.
2.5 Training and prediction
The selected CGC genes were therefore used as training set of the classifiers. With all different classification settings, we performed a leave-one-out cross-validation: each item in the training set is classified with a model built with the rest of the training set items. We found three genes whose initial classification extremely contradicted their CGC category: NOTCH1, NPM1 and CEBPA genes, which have evidence in the literature for a dual role (Halmos et al., 2002; Sportoletti et al., 2008; Vogelstein et al., 2013). Therefore, we decided to discard them from the training set. Thus, the final, trimmed CGC training set included 28 Dom and 45 Rec genes.
For the classification of HCD and Cancer5000 genes, we considered that values between 0.7 and 1 as Act and those with values between 0 and 0.3 as LoF. We computed the ACC and MCC (Matthew's correlation coefficient) of each classifier at the leave-one-out cross-validation of the training set. Furthermore, we calculated the coverage (COV) of the classifier, which reflects the percentage of the entire training set for which a prediction could be made.
3 RESULTS
3.1 Identifying features that differentiate Act from LoF driver genes
We tested 30 features that we initially hypothesized could be used to characterize and discriminate between LoF and Act drivers (see Table 1 for detailed description of each). All features elaborate on somatic mutation and CNA patterns across data from the pan-cancer 17 cohort. We expected LoF genes to be affected more frequently by deleterious events such as CNA loss and truncating mutations. Act genes should be more frequently amplified and receive protein-affecting non-truncating mutations, which may increase and/or alter the protein function.
To select the most informative features for the task of distinguishing between Act and LoF genes, we compared the distribution of the features in both categories of CGC genes (Fig. 1). The features we considered can be divided into four broad categories (Fig. 1A): (i) features that measure the relative abundance of truncating mutations, (ii) features that reflect the CNA status of the gene across tumors, (iii) features that account for the relative abundance of PAMs and (iv) features that measure the degree of clustering of missense mutations along the protein sequence.
Fig. 1.
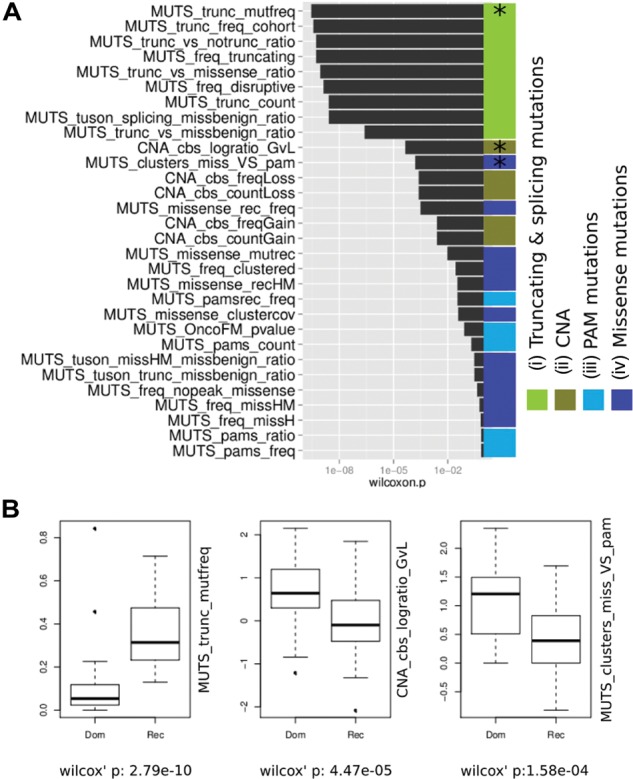
A) The list of features ordered by Mann–Whitney–Wilcoxon rank sum test P-value significance. Features dependant on truncating mutations are the best discriminators for LoF and Act genes. Features described in (B) are marked with asterisk. A detailed explanation of each feature can be found in Table 1. (B) Box plots comparing the distribution of the three non-redundant top-ranking features that have been selected for the OncodriveROLE classifier in CGC genes annotated as Dom and Rec
Features in Group iii show the poorest performance to discriminate between CGC Dom and CGC Rec genes (light blue in Fig. 1A). On the other hand, all the features in Group i (green in Fig. 1A) rank at the top of performance of all features analyzed. As expected, this reflects that Act genes (or proto-oncogenes) are intolerant to truncating mutations because an active protein product is required for tumorigenesis. In LoF (or tumor suppressor) genes the truncation of the protein product gene is positively selected, which facilitates the identification of LoF candidates. The best performing feature in this group was the ratio of truncating mutations to the total number of coding mutations in the protein (Fig. 1B).
The distribution of mutations within the gene (Group iv, dark blue in Fig. 1A) differs significantly between CGC Dom and CGC Rec genes. The CGC Dom genes have fewer mutational hotspots, detected as clusters by OncodriveCLUST, than CGC Rec genes, whose mutations tend to be more evenly distributed (Supplementary Fig. S1) along the protein sequence. This is probably because Act driver genes receive mutations that potentiate their function, e.g. by constitutively activating a regulatory site, or cause a switch of the protein function. To achieve such behavior through mutations, these must occur at specific places in the sequence, which results in fewer numbers of recurrent sites (clusters) than in CGC Rec genes (Supplementary Fig. S1). We elaborated a series of features based on impact, frequency and clustering of missense mutations. Many did not show any power of discrimination of CGC Rec and Dom. The features that perform reasonably well are based on the recurrence of missense mutations. The best-performing feature in this group compares the ratio of missense mutations with total number of PAMs within OncodriveCLUST peaks (MUTS_clusters_miss_VS_PAM; Fig. 1). Another feature in this group that performs relatively well is the ratio recurrent missense mutations (MUTS_missense_rec_freq).
All features in Group ii are designed to capture the known fact that LoF genes have a tendency to be deleted, whereas Act genes are more frequently affected by amplifications (Davoli et al., 2013). In this case, we found that the ratio of amplifications to deletions across all tumors in the cohort achieved the best separation of the two groups of genes.
3.2 Developing a classifier to differentiate between LoF drivers and Act drivers
Thereafter, we created a feature set that contained non-redundant best-performing features from Groups i, ii and iv, disregarding those of Group iii because of their poor performance resulting in three features: MUTS_trunc_mutfreq, MUTS_clusters_miss_VS_PAM and CNA_cbs_logratio_GvL. We tested six machine learning approaches trained with the trimmed version of the CGC (see Section 2). For each gene, the classifiers produced a score of the likelihood that it belonged to the CGC Dom class. A score of value 0 means that the classifier regards the gene as an LoF beyond all doubt, whereas a score of value 1 means it exactly resembles the model of an Act gene. We assessed the performance of each classifier through the ACC, the MCC and the COV of the driver set (all listed in Supplementary Table S1). ACC and MCC validate the performance of the classifiers on the 76 CGC driver genes by means of a leave-one-out cross-validation approach. We computed these values for different classification probabilities thresholds to select the cutoff that maximize the ACC and MCC, even at the cost of reducing the COV. Then, we used these sets of values to choose the classifier with the best performance and a reasonable COV. Overall, randomForest produced the best results (Supplementary Table S1). We also trained classifiers with different combinations of the three selected features and included MUTS_missense_rec_freq feature for testing purposes. We found that multiple combinations of these features perform similarly (Supplementary Table S2 and Supplemental Text). We decided to use the randomForest classifier trained with the three non-redundant features shown in Figure 1B to create OncodriveROLE, under the rationale that features representing the three independent groups could provide more information to classify novel drivers. The method shows an ACC of 0.94, MCC of 0.84 and COV of 88% in the leave-one-out cross-validation. We further tested OncodriveROLE in an independent set of tumor suppressor genes (Zhao et al., 2013) that are not present in the CGC. OncodriveROLE accurately classified 91.7% of those genes as LoF drivers (Supplemental Text).
3.3 Applying OncodriveROLE to lists of cancer driver genes
We identified two recent studies in which identified novel cancer driver genes could be classified with OncodriveROLE. The first study detected cancer drivers by integrating four methods that assess different signals of positive selection across samples of the pan-cancer 12 dataset. This analysis resulted in 291 high-confidence cancer drivers (Tamborero et al., 2013b). In the second study, MutsigCV was applied in a cohort of about 5000 tumor samples to obtain a cancer driver list composed of 260 genes (Lawrence et al., 2013, 2014). The two lists will be referred to as HCD and Cancer5000 further on. Even though both lists have similar sizes, their overlap is only 50%, making the two gene sets different as can be seen in Figure 2. As for the training set, we applied the one-signal filter to only predict the role of genes possibly acting as drivers in the dataset under evaluation resulting in 200 HCD and 144 Cancer5000 genes.
Fig. 2.
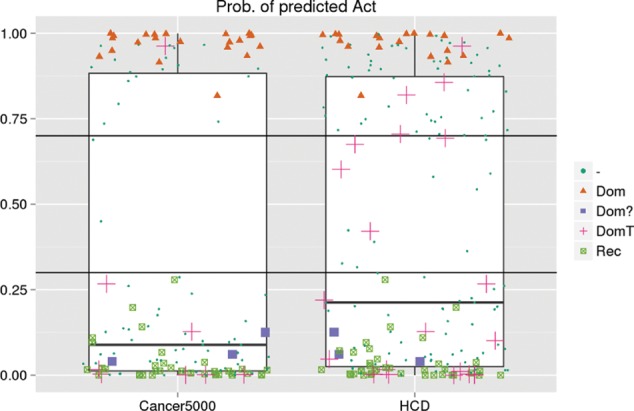
Classification of 200 (HCD list) and 144 (Cancer5000 list) cancer driver genes into the classes Act and LoF. The training set of OncodriveROLE constitutes of all ‘Dom’ and ‘Rec’ labeled data points. ‘Dom?’ are CGC-annotated dominant genes excluded from the training set because of strong resemblance to the ‘Rec’ genes and previous literature evidence of this role. ‘DomT’ genes are CGC-annotated dominant genes only citing translocation events as prove and therefore not included in the training set. All ‘-’ labeled data points are driver genes not annotated in CGC, and whose prediction was the main goal of the study. The thresholds are drawn at 0.3 (as top limit of the LoF class) and 0.7 (as bottom limit of the Act class). Working with classification score thresholds of 0.3 (as top limit of the LoF class) and 0.7 (as bottom limit of the Act class), we classified 109 genes as LoF, 76 as Activating and left 15 genes as unclassified in the HCD list; meanwhile, we classified 97 genes as LoF, 43 as Activating and left 4 genes as unclassified (Fig. 2) in the Cancer5000 list. Genes for which we have observed <12 mutations were directly classified as ‘No class’ and assigned NA values in the classifications results (see Supplementary Tables S4 and S6)
The overall distribution of probabilities of these two groups of genes is roughly bimodal in both driver lists, which allowed us to choose these symmetric cutoff values (Fig. 2 and Supplementary Fig. S2) such as 0.3 and 0.7 for LoF and Act genes, respectively. Other cutoffs may be used for the datasets under analysis depending on how strict a classification the user wants for their list of cancer drivers. Interestingly, we classified three CGC Dom genes as LoF (‘Dom?’ in Fig. 2). The genes in question are NOTCH1, NPM1 and CEBPA. All three have been implicated in leukemia (Cancer Genome Atlas Research Network, 2013; Liu et al., 2013; Ohlsson et al., 2014) and both NOTCH1 and NPM1 are annotated in the CGC as partners of translocation events in leukemia. NOTCH1 has been described as an oncogene as well as a tumor suppressor. Its actual role may depend on the tumor type (Licciulli et al., 2013; Liu et al., 2013; Vogelstein et al., 2013). Equally, CEBPA and NPM1 have been characterized as tumor suppressors in the literature (Halmos et al., 2002; Sportoletti et al., 2008). We cannot be certain of the functional impact of the translocation on the function of the product of the fused gene. It may associate to a new promoter and change its expression accordingly, or it may be truncated as a result of the fusion and thus function as an LoF. For this reason, we had previously excluded all CGC Dom genes that are solely associated to translocation events in the Census. The plot in Figure 2 shows those genes labeled as DomT, and their classification shows no clear resemblance to LoF or Act, which supports our decision to remove them from the training set.
3.4 Comparison of OncodriveROLE with other bioinformatics approaches
The 20-20 rule was created to identify mutational driver genes, both oncogenes and tumor suppressor genes (Vogelstein et al., 2013). Therefore, it differs from OncodriveROLE, designed to classify previously identified driver genes into their most probable roles. The simple 20-20 rule reaches a high ACC (Table 2) when applied to the trimmed CGC list. However, it is unable to reach a decision on many drivers where none of its two estimators (see Section 2) surpasses the threshold of 20% (Tables 2 and 3).
Table 2.
List of approaches and their performance on trimmed CGC dataset
| Method | ACC | MCC | COV (%) |
|---|---|---|---|
| OncodriveROLEa | 0.925 | 0.848 | 83 |
| 20-20 rule | 0.895 | 0.769 | 75 |
| Tuson | 0.914 | 0.817 | 92 |
aResults of leave-one-out cross-validation.
Table 3.
List of approaches and their performance on the 290 drivers from the HCD list and 260 drivers from the Cancer5000 list
| Method | Act/Oncogene | LoF/Tumour suppressor | Unclassified | Coverage (%) |
|---|---|---|---|---|
| HCD | ||||
| Oncodrive ROLE 0.3/0.7 | 76 | 109 | 15 | 92 |
| Oncodrive ROLE 0.2/0.8 | 58 | 96 | 46 | 77 |
| 20-20 rule | 23 | 96 | 81 | 60 |
| Tuson | 44 | 92 | 64 | 68 |
| Cancer5000 | ||||
| Oncodrive ROLE 0.3/0.7 | 43 | 97 | 4 | 97 |
| Oncodrive ROLE 0.2/0.8 | 40 | 91 | 13 | 91 |
| 20-20 rule | 18 | 90 | 36 | 75 |
| Tuson | 32 | 90 | 22 | 85 |
We also compared the results obtained by the approach designed by Davoli et al. (2013), implemented in a classifier named Tuson. As with the 20-20 rule, Tuson was created to distinguish oncogenes and tumor suppressor genes from genes with passenger mutations, instead of classifying previously identified cancer drivers as is the case of OncodriveROLE. We found OncodriveROLE slightly outperforms Tuson in ACC and MCC on the trimmed CGC dataset. Note that Tuson method was trained with CGC genes, and the performance reported in Table 2 does not remove genes in the training set, as it is done in the leave-one-out cross-validation of OncodriveROLE. We can conclude that well-known cancer genes are classified with a high ACC with all approaches. The main difference between the three approaches lies in the COV that can be reached when predicting the role of novel cancer drivers in tumorigenesis.
4 DISCUSSION
Two main rationales to detect LoF and Act driver genes acting across tumor samples exist. The first approach consists in directly detecting genes that exhibit known alterations patterns corresponding to these two classes from mutations and CNA data. This strategy was first conceptualized by Vogelstein et al. (2013) to be implemented later on as a machine learning algorithm by Davoli et al. (2013). In the second approach, first driver genes acting in tumor samples are detected by combining the signals of positive selection they exhibit (Lawrence et al., 2014; Tamborero et al., 2013b). Then, in a second step, these drivers are classified into the two aforementioned classes exploiting similar alteration patterns as in the first approach. This second two-step approach has two main advantages. First, genes that do not exhibit clear alterations pattern that define them as LoF or Act can still be detected as drivers if they show clear signals of positive selection. Second, the combination of several signals controls the ratio of false-positive drivers identified (Tamborero et al., 2013b), which is unattainable to the direct classification of genes.
This is the reason why we have decided to develop OncodriveROLE, a machine learning classifier, which takes a list of pre-selected driver genes and sorts them according to their mode of action. We first carefully compared and selected a set of features that best captures the differences of alterations patterns of these two groups of drivers. We then used those features to train the classifier, on a carefully trimmed subset of the CGC genes. When applied to two recent lists of drivers, we found that, even under strict classification conditions, OncodriveROLE was able to classify more drivers than the 20-20 rule and the Tuson machine learning algorithm.
The OncodriveROLE validation procedure identified several likely misclassified drivers in the CGC. The most salient examples of these are probably some genes that drive hematopoietic malignancies upon translocation and fusion with other genomic regions, all classified as Dom in the GCG. However, when analyzed using mutational and CNAs data from the pan-cancer 17 dataset, some of them appear as clear LoF drivers. For instance, OncodriveROLE assigns MLL, RUNX1 and SUZ12 classification probabilities under 0.003 (see Supplementary Tables S3–S6 for feature and classification values). These genes could be Act drivers upon fusion to other genes, but LoF upon mutations.
Even though OncodriveROLE is able to classify most of the genes in the two drivers lists as LoF or Act, it still leaves few of them unclassified. Some of these correspond to lowly recurrent drivers whose mutational features are not correctly computed because of the scarcity of their alterations. Sequencing more tumors will certainly improve their classification. Others may not have a clear enough pattern to be classified in one of the two classes, as they could be exhibiting different roles in different contexts. In some rare cases, the method misclassifies known cancer genes. For example, KEAP1 is classified as an Act driver, although it is reported to lose its function upon mutation (Hayes and McMahon, 2009; Shibata et al., 2008). A close look at its mutational pattern reveals missense mutations dominate and accumulate in certain regions of the protein. As member of a ubiquitin-mediated proteolysis complex, the function of KEAP1 is probably essential to the cell, and its impairment is likely lethal. Therefore, few truncating mutations may appear in KEAP1, and it is ultimately misclassified by OncodriveROLE. Future finer measurements of the impact of missense mutations may help correcting this problem.
Summing up, in this article, we have described the development and validation of OncodriveROLE, an approach to differentiate between LoF and Act driver genes. The OncodriveROLE classifier is freely available at http://bg.upf.edu/oncodrive-role as an R object that researchers may use to classify the drivers they have detected across a cohort of tumor samples. At the same URL, the pre-computed TCGA pan-cancer 17 mutational and copy number features used for the classification are available for download.
Funding: We acknowledge funding from the Spanish Ministry of Economy and Competitivity (grant number SAF2012-36199) and the Spanish National Institute of Bioinformatics (INB). M.P.S. and C.R.-P. are supported by FPI fellowships.
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Breiman L. Random forests. Mach. Learn. 2001;45:5–32. [Google Scholar]
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, et al. Ensembl variation resources. BMC Genomics. 2010;11:293. doi: 10.1186/1471-2164-11-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, et al. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013;155:948–962. doi: 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dees ND, et al. MuSiC: identifying mutational significance in cancer genomes. Genome Res. 2012;22:1589–1598. doi: 10.1101/gr.134635.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, et al. Ensembl 2013. Nucleic Acids Res. 2013;41:D48–D55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal PA, et al. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perez A, Lopez-Bigas N. Functional impact bias reveals cancer drivers. Nucleic Acids Res. 2012;40:e169. doi: 10.1093/nar/gks743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perez A, et al. Improving the prediction of the functional impact of cancer mutations by baseline tolerance transformation. Genome Med. 2012;4:89. doi: 10.1186/gm390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perez A, et al. Computational approaches to identify functional genetic variants in cancer genomes. Nat. Methods. 2013a;10:723–729. doi: 10.1038/nmeth.2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perez A, et al. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods. 2013b;10:1081–1082. doi: 10.1038/nmeth.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halmos B, et al. Down-regulation and antiproliferative role of C/EBPα in lung cancer. Cancer Res. 2002;62:528–534. [PubMed] [Google Scholar]
- Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009;34:176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Hothorn T, et al. Unbiased recursive partitioning: a conditional inference framework. J. Comput. Graph. Stat. 2006;15:651–674. [Google Scholar]
- Hudson TJ, et al. International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- Kandoth C. 2014 MAF files - strictly filtered. http://dx.doi.org/10.7303/syn1729383.2. [Google Scholar]
- Kandoth C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M. Building predictive models in R using the caret package. J. Stat. Softw. 2008;28:1–26. [Google Scholar]
- Lambert JMR, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009;15:376–388. doi: 10.1016/j.ccr.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licciulli S, et al. Notch1 is required for Kras-induced lung adenocarcinoma and controls tumor cell survival via p53. Cancer Res. 2013;73:5974–5984. doi: 10.1158/0008-5472.CAN-13-1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, et al. The emerging roles of Notch signaling in leukemia and stem cells. Biomark. Res. 2013;1:23. doi: 10.1186/2050-7771-1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlsson E, et al. Initiation of MLL-rearranged AML is dependent on C/EBPα. J. Exp. Med. 2014;211:5–13. doi: 10.1084/jem.20130932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshen LB, et al. Classification and Regression Trees. 1984. Wadsworth Int. Group. CHAPMAN & HALL/CRC. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. 2013 R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Reimand J, et al. The mutational landscape of phosphorylation signaling in cancer. Sci. Rep. 2013;3:2651. doi: 10.1038/srep02651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata T, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc. Natl Acad. Sci. USA. 2008;105:13568–13573. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sportoletti P, et al. Npm1 is a haploinsufficient suppressor of myeloid and lymphoid malignancies in the mouse. Blood. 2008;111:3859–3862. doi: 10.1182/blood-2007-06-098251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobl C, et al. Conditional variable importance for random forests. BMC Bioinformatics. 2008;9:307. doi: 10.1186/1471-2105-9-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamborero D, et al. OncodriveCLUST: exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics. 2013a;29:2238–2244. doi: 10.1093/bioinformatics/btt395. [DOI] [PubMed] [Google Scholar]
- Tamborero D, et al. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci. Rep. 2013b;3 doi: 10.1038/srep02650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, et al. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, et al. TSGene: a web resource for tumor suppressor genes. Nucleic Acids Res. 2013;41:D970–D976. doi: 10.1093/nar/gks937. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.