

Abstract
Motivation: Ancestral character state reconstruction describes a set of techniques for estimating phenotypic or genetic features of species or related individuals that are the predecessors of those present today. Such reconstructions can reach into the distant past and can provide insights into the history of a population or a set of species when fossil data are not available, or they can be used to test evolutionary hypotheses, e.g. on the co-evolution of traits. Typical methods for ancestral character state reconstruction of continuous characters consider the phylogeny of the underlying data and estimate the ancestral process along the branches of the tree. They usually assume a Brownian motion model of character evolution or extensions thereof, requiring specific assumptions on the rate of phenotypic evolution.
Results: We suggest using ridge regression to infer rates for each branch of the tree and the ancestral values at each inner node. We performed extensive simulations to evaluate the performance of this method and have shown that the accuracy of its reconstructed ancestral values is competitive to reconstructions using other state-of-the-art software. Using a hierarchical clustering of gene mutation profiles from an ovarian cancer dataset, we demonstrate the use of the method as a feature selection tool.
Availability and implementation: The algorithm described here is implemented in C++ as a stand-alone program, and the source code is freely available at http://algbio.cs.uni-duesseldorf.de/software/RidgeRace.tar.gz.
Contact: mchardy@hhu.de
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Many biological studies investigate the ancestral states of one or several discrete and continuous characters on a phylogenetic tree (Felsenstein, 2004). Comparative methods correlate the evolution of alleles at different loci with each other or with a trait and thus often require the reconstruction of ancestral values. Typical examples for discrete characters are the reconstruction of the absence, presence or state of genes or traits for the internal nodes of the tree (i.e. for the ancestral organisms), while typical examples for continuous characters are environmental preferences of different species, measures of morphology or physiology or behavioral or metabolic properties (Nunn, 2011). Such reconstructions are also of interest when fossil records cannot be retrieved, when the phenotype of interest cannot be determined from the fossil tissue or when studying the evolution of a gene family across different environmental conditions.
Statistical approaches such as regression or correlation can fail to estimate correlations between traits correctly when they assume that closely related species are statistically independent (Felsenstein, 1985; Grafen, 1989; Harvey and Pagel, 1991). Comparative methods account for such dependencies by including estimates of the phylogeny underlying the data into their predictions. In the case of continuous characters, most of these techniques are based on a simple model assuming neutral evolution of the respective character. The Brownian motion (BM) model (Felsenstein, 1985) assumes that the trait of a leaf node in a phylogeny develops as a random walk starting from the ancestral root. The duration of that walk—and therefore the change and variance in the trait—is assumed to be proportional to the change in branch length covered between those two nodes. At each inner node, the random walk bifurcates, creating two dependent processes and thus defining a stochastic distribution for all leaves. Several methods that apply the BM model are available to reconstruct continuous ancestral characters, and implemented in widely used software packages, such as APE (Paradis et al., 2004), Geiger (Harmon et al., 2008), Phytools (Revell, 2012), Mesquite (Maddison and Maddison, 2011), BayesTrait/Continuous (Pagel, 1999), PAUP* (Swofford, 2003) and Contml (Felsenstein, 1993).
One of the simplest ways to reconstruct a continuous ancestral character state was established with Felsenstein’s algorithm for ‘Phylogenetic Independent Contrasts’ (Felsenstein, 1985). In the Phylogenetic Independent Contrasts algorithm, ancestral values are computed recursively as the weighted average of their child values, with the weights set according to the distance (i.e. the branch length) of these children. In addition, branch lengths leading to reconstructed internal nodes are rescaled to account for the uncertainty of the reconstruction. This leads to a maximum likelihood estimation of the ancestral value for the root node alone. Other algorithms estimate the values for the whole tree by re-rooting or by a squared change maximum parsimony approach (Felsenstein, 2004; Schluter et al., 1997). Linear regression allows another framework to formulate the reconstruction of ancestral character states, and generalized least squares has been suggested as a technique to reconstruct ancestral values as a weighted average of the values of all extant species, while taking the correlation structure described by the phylogenetic tree into account (Martins and Hansen, 1997). This approach is particularly flexible, as it allows detailed assumptions of the evolutionary process by inclusion of an appropriate covariance matrix. Several possible extensions and modifications of the BM process have been suggested and can be considered for ancestral character state reconstruction. For example, the Ornstein–Uhlenbeck (OU) process models adaptation explicitly by defining a single global optimum (Hansen, 1997) or several local optima (Butler and King, 2004) of directional selection. A major restriction of the BM and the OU models, noted in, for example, Harmon et al. (2010), is the assumption of a constant rate of trait variation throughout the underlying phylogeny. The early burst model (Blomberg et al., 2003; Freckleton and Harvey, 2006) offers an alternative that decreases the rate of evolution exponentially through time, and describes a process of adaptive radiation [Harmon et al. (2010) provide a detailed comparison]. The ACDC model (Blomberg et al., 2003) describes a process of accelerating versus decelerating rates of character evolution toward an optimum, i.e. a combination of OU and early burst. Several methods allow us to estimate BM rates (Garland, 1992; Lynch, 1990; Martins, 1994), to test for variations in that rate or use different rates in different parts of the tree (McPeek, 1995; O’Meara et al., 2006; Revell, 2008) or to suggest global branch length transformations in the phylogeny to account for variable rates (Blomberg et al., 2003; Pagel, 1999). However, most of the phylogenetic methods aforementioned do not explicitly aim to reconstruct ancestral character states. They are originally concerned with correlations between two or more traits [‘phylogenetic regression’, Grafen (1989); Felsenstein (2004)] or with tests for deviation from the assumption of a globally constant rate, and they do not suggest how to infer ancestral character states for more complex scenarios. In addition, critical studies note that deciding on the correct model might be difficult and warn of over-interpreting the phylogenetic patterns (Blomberg et al., 2003; Boettiger et al., 2012; Losos, 2011; Price, 1997).
2 APPROACH
We here describe RidgeRace (Ridge Regression for Ancestral Character Estimation), a new and simple method inspired by the least-squares optimization technique of Cavalli-Sforza and Edwards (1967) for the inference of branch weights in a phylogeny via pairwise distances. RidgeRace does not assume certain rates at certain regions of the phylogeny or a particular model of rate change over time. It treats phenotypic measurements at the terminal nodes of a phylogeny as sample observations and relies on a linear regression with L2-Norm regularization, allowing phenotypic rates to vary at each branch. It estimates branch-wise rates and ancestral characters simultaneously, in a way that best describes the phenotypes observed at the terminal nodes.
In an extensive simulation study, we evaluated different variations of BM on randomly created trees and show that our method performs equally well as or better than established implementations of state-of-the-art reconstruction algorithms. We suggest using RidgeRace in studies aiming to reconstruct ancestral character states of continuous characters when no definite assumptions can be made about the type of evolutionary process, or when the assumption of a model for phenotypic evolution is not appropriate at all. The latter might, for example, be the case in studies that rely only on a hierarchical clustering of samples instead of phylogenies.
Branch weights inferred by the ridge regression based on phenotype measurements can be interpreted as rates of phenotypic change (i.e. phenotypic rates) and provide insights into particularly interesting areas of the phylogeny. They can also be used to judge the phenotypic impact of genetic changes or other types of events associated with branches within the phylogeny [see, for example, Steinbrück and McHardy (2012)]. To demonstrate a possible application of RidgeRace integrating phenotypic and genotypic data, we studied an ovarian cancer dataset, created by the Cancer Genome Atlas research network and recently analyzed with network-based stratification (Hofree et al., 2013).
3 METHODS
RidgeRace estimates ancestral character states on a phylogenetic tree. As in the original BM model, we consider the leaf values to be the result of a weighted sum of intermediate contributions gi created along the tree, beginning at the root (Fig. 1). The contributions represent the gain or loss in character value on each branch of the tree so that, for example, the character value of sample y4 can be described as follows:
where a, b and c represent the branches in the tree, and g0 holds a bias term representing the original contribution of the root node. The contribution gj of a single branch j can be seen as being analogous to the formulation of BM: the gain or loss in the phenotype is dependent on the length lj of branch j and the speed of the process, in analogy to the variance term in the BM model:
One can then write the solution for the vector of leaf phenotypes y in matrix form:
| (1) |
where
 |
and β is a vector with a length equal to the number of branches in the phylogeny, including a single virtual branch above the root to account for its original contribution g0. This scheme is overparameterized, as it adds a parameter for each inner branch, and only considers one sample observation for each terminal node. However, it also allows the inclusion of measurements at inner nodes (e.g. from fossil records), and it is suitable for accounting for multiple measurements at single leaf nodes. Such samples can be added by appending rows to y and L.
Fig. 1.

Model of phenotype evolution on a phylogenetic tree. The observed continuous character values at the nodes yi are the result of a sum of contributions on ancestral branches. A virtual branch ‘above’ the root node x1 contributes the global phylogenetic mean, i.e. the ancestral state of x1
Ridge regression is a simple extension of ordinary least squares regression. As ordinary least squares, ridge regression also aims to minimize the squared error term, but adds a quadratic regularization penalty on large values of the weight vector β. A tuning parameter λ controls the relative impact of both terms. The regularization does not only help to reduce the variance of the model, it also acts as an integrated parameter selection method for overparameterized models [see Gareth et al. (2013) for details]. We here use ridge regression to estimate a vector that explains the known observations y best:
| (2) |
The textbook solution (Gareth et al., 2013; Hastie et al., 2009) to this optimization problem is as follows:
| (3) |
Equation (2) shows how the optimization balances the leaf reconstruction error versus the quadratic term that penalizes large variance in the phenotypic rates. A trivial but undesirable solution to the optimization would set the gain at each terminal branch equal to the according terminal node value, leaving all other gains empty and making ancestral reconstruction impossible.
We here use quadratic regularization instead of L1 regularization. The latter penalizes the absolute value of the weight vector β, driving single weights toward zero. This would correspond to a phylogeny with many phenotypic rates at zero and only few branches with rates of high absolute values, describing a rather implausible model for phenotypic evolution.
For a given estimate of as defined in Equation (3), the vector containing the phenotypic reconstruction of all inner nodes can then be computed in analogy to Equation (1):
| (4) |
where
 |
This formulation is similar to the generalized least squares method proposed by Martins and Hansen (1997). Similarly they suggest inferring ancestral character states as the weighted average of leaf contributions, with weights assigned according to the covariance between an ancestor and a leaf [Equation (10) in Cunningham et al. (1998); Martins and Hansen (1997)]:
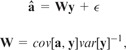 |
where the covariance between an inner node a and a leaf node y is defined as , with being the distance between the root of the tree and the most recent common ancestor of a and y. RidgeRace differs in the sense that it allows us to estimate a weight for each branch instead of assuming a constant rate or, more generally, the predefined covariances between nodes. Extensions of the generalized least squares approach under the BM model use more complex matrices W. However, the design of W has to be defined in advance based on specific model assumptions, whereas RidgeRace is able to estimate rates independently.
An important assumption of linear regression is that the standard deviations of the error terms are constant and do not depend on the covariates (here: the branch lengths). This assumption is violated under the BM model, as leaf nodes with a long distance to the root will have a high variance in their trait value and phenotypic measurements will produce larger errors at these nodes compared with the predicted value. The estimation of β might thus be biased if the depth of single leaf nodes is large compared with the rest of the tree. We therefore recommend RidgeRace for approximately balanced trees.
3.1 Estimation of the regularization weight
The regularization weight parameter λ in Equation (2) balances the impact of accuracy at the leaves versus the complexity of the model and variance of β. To find the optimal value of λ, we performed a leave-one-out iteration over all leaves of the tree. To estimate the goodness of fit of a particular λ0, we iteratively removed a single leaf x from the tree, estimated β on the remaining tree and used the rate of the branch leading to the parent of x as an approximation of the branch rate of the missing node. The leave-one-out error for x is defined as the squared difference between the inferred phenotypic value for x and the actual value according to the input data. The leave-one-out error for a particular λ0 is the sum over all leave-one-out errors for all leaves. Iterating , we selected the final λ to be the one that minimized the leave-one-out error.
3.2 Simulation study
We created random trees with an increasing number N of leaves using the function rtree in the R-package APE (Paradis et al., 2004; R Core Team, 2012). We simulated BM with variation along the branches of the tree, resulting in a character assignment for each inner or leaf node. The process was repeated several times for different trees and different values for the parameters and N. Supplementary Text S1 provides details on the simulation algorithm and the parameter settings. The random tree and the simulated values obtained at the leaf nodes were provided as input to RidgeRace and to implementations of the maximum likelihood and generalized least squares algorithms (Felsenstein, 1985; Hansen, 1997) in the APE package for ancestral character state estimation (Paradis et al., 2004). The reconstructed values thus obtained were mapped back to the inner nodes of the tree and compared with the simulated ones (leaf nodes were excluded from the comparison), and the mean squared error was computed for evaluation.
3.3 Cancer study
A binary matrix describing the absence or presence of non-synonymous mutations in 9850 genes for 325 patients was taken from the supplementary data of an ovarian cancer dataset provided by Hofree et al. (2013). Analogous to the description of the authors in their article and Supplementary Material, we used their network-based stratification software (NBS, version 0.2, available at http://idekerlab.ucsd.edu) with four clusters, the HM network and default parameters, creating 1000 bootstrap samples. We then inferred a hierarchical clustering (average linkage) on the bootstrap similarity matrix using the methods provided in the scripts of the authors. We used this inferred topology as the input tree for RidgeRace. We then downloaded information on each patient’s survival time from the TCGA database (Cancer Genome Atlas Research Network, 2011). Treating a patient’s survival time as a ‘trait’ of each patient, phenotypic rates were inferred with RidgeRace as described above. The binary genetic profile of each patient was then mapped to the leaf nodes and reconstructed to inner nodes with the Sankoff algorithm implemented in RidgeRace, using a simple 0/1 cost matrix and the ACCTRAN principle in case of ambiguities (Felsenstein, 1985). Changes in the genetic profiles of neighboring nodes were then reconstructed on the branches of the tree. Finally, the tree was visualized using FigTree (Rambaut, 2013).
4 RESULTS
4.1 Simulation study
Our evaluation of RidgeRace on data consisting of randomly drawn phylogenetic trees and continuous ancestral characters states created by a simulated BM process showed that the method performs similarly or better than other state-of-the-art techniques. We compared the performance of RidgeRace with generalized least squares (Hansen, 1997) and REML (Felsenstein, 1985), and simulated ancestral character evolution in a BM setting. Figure 2 shows that all three methods were able to reconstruct ancestral states well, achieving low mean squared errors, even for small trees or high variation values. The variation of the mean squared error of all three methods is large to observe statistically significant differences in the methods; however, on average, RidgeRace performed similar or better in our simulation than the two alternative methods.
Fig. 2.
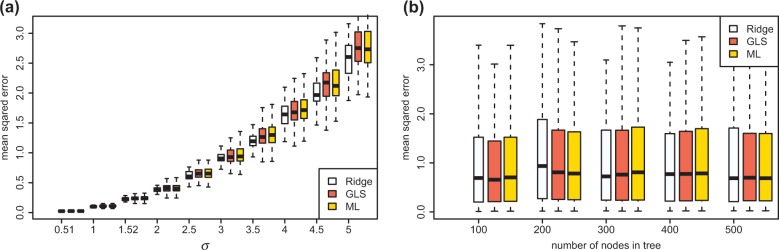
Mean squared error between the inferred ancestral characters and the true simulated values, when using maximum likelihood reconstruction (yellow), generalized least squares (red) and RidgeRace (light blue). The plot shows (a) the dependence of performance on the standard deviation σ of the BM process or (b) performance when increasing the number of leaf nodes in the tree
To show another practical application of the RidgeRace method, we mapped the inferred rates β to their associated branches using an arbitrary random tree from the simulation. Figure 3 shows such a tree with 25 leaves and BM simulated in three different regimes that have the internal rate parameters of 5.3, 1.3 and 2.3. Simulated phenotypic values are shown as node labels. The correlation coefficient between the simulated ancestral states and those inferred by RidgeRace was r = 0.988. The inferred phenotypic rates vector β was plotted at the branches, and the branches were colored according to the relative size of these rates, with blue branches indicating strongly negative weights, red branches indicating strongly positive weights and gray branches indicating weights close to zero. Large changes in the phenotype value mainly occurred in Regime I, which features the largest σ parameter. One can observe that the inferred phenotypic rate is large when the absolute change in phenotypic value is large compared with the length of the associated branch. Therefore, plotting the inferred phenotypic rates to the phylogeny can be useful when studying the evolution of a population or a set of species. It will visualize regions in the tree that are associated with rapid phenotypic evolution.
Fig. 3.

Reconstruction of the phenotypic rates β along the branches of a random tree with 25 leaves, simulated with three regimes and a hypothetical phenotypic trait that resulted from a BM process with original mean zero and standard deviations , and in regimes I, II and III. The inferred rates visualize the speed of phenotypic evolution from strongly decreasing (red) to strongly increasing (blue). Absolute phenotypic rates are clearly largest in the regime with the highest σ parameter
4.2 Application to ovarian cancer data
According to the World Health Organization, cancer is a leading cause of disease-related deaths worldwide and was responsible for 7.6 million deaths in the year 2008 (WHO, 2013). The disease is the result of a complex interplay of genetic preconditions, external influences and interactions with the immune system (Hanahan and Weinberg, 2000, 2011). For a wide variety of cancer types, recent studies have identified genes that are significantly associated with cancer risk, onset and progression (e.g. Cancer Genome Atlas Research Network, 2008, 2012a, b, 2013; Kandoth et al., 2013).
Hofree et al. (2013) argue that somatic mutations are likely to contain the causal driver events of tumor progression, and that this type of data provides a promising source of information to identify clinically relevant subclusters. Such subclusters are identified with methods that find groups of samples with significant differences in their allele frequency profile, a process described as stratification. Network-based stratification is a new clustering method that smooths the sparse and diverse genetic profiles with the help of gene interaction networks (Hofree et al., 2013), and the authors show that it produces clinically meaningful clusterings. We used a dataset and the software provided by the authors to reconstruct a hierarchical clustering on somatic mutation data of ovarian cancer samples. We thus created a tree structure showing similarities in the genetic profiles of the tumors of ovarian cancer patients (Fig. 4). The tree structure may be error-prone because of the high diversity of genetic aberrations in tumors, but their main branches closest to the root are likely to represent biologically meaningful subclusters (see also argumentation in Hofree et al., 2013).
Fig. 4.

Application of RidgeRace to a hierarchical clustering on somatic mutations inferred for an ovarian cancer dataset. Colors on the side of the tree indicate the subtypes inferred with network-based stratification (Hofree et al., 2013). Branches are colored according to the phenotypic rate parameter β; the thickness of branches is proportional to the number of nodes below them. Branches leading directly to leaf nodes were colored gray for improved visibility. Labels m1 to m5 indicate branches with strong changes in patient survival time. Changes in the absence or presence of mutations in the selected genes are indicated on all branches with four or more children
It was not possible to determine whether our inferred clustering was completely identical to that of Hofree et al. (2013), as the exact tree was not provided by the authors. However, we observed similar cluster sizes and distributions of survival time in the clusters. We found that patients assigned to the smallest of the four subtypes showed an increased survival time (Fig. 4, green cluster). A RidgeRace analysis of patient survival time as a phenotype consistently showed a strong positive rate increase in the branch leading to that cluster (Fig. 4, marker m1). Similarly, RidgeRace inferred a decrease in survival time for the branch leading to the yellow cluster (branch m2). Branch m3 was associated with a rather small decrease in survival time because the red cluster splits in distinct two subtypes with a successive second increase (branch m5) or a decrease (branch m4) in survival time, with branch m4 leading to the majority of the red cluster, which had the lowest survival time of all four clusters.
RidgeRace reconstruction can be combined with the reconstruction of discrete genetic events. We mapped the binary data encoding the absence or presence of non-synonymous mutations in a selection of genes to the tree. The mapping confirmed the diverse nature of the somatic mutations. Only P53 was found to be altered in almost all patients and was reconstructed to have mutated at the root of the tree. Beside P53, only TTN was reconstructed to change on a higher level node: it was ‘gained’ (mutated) at branch m3 and was present in 83 of 85 patients of the red cluster. RYR2 was gained on branch m5 and present in 9 of 85 patients in the red cluster. Besides these changes, no change appeared on a branch higher than five levels below the root.
4.3 System requirements
RidgeRace requires only minimal system resources (<100 MB RAM). The C++ implementation relies on the boost ublas library (BOOST, 2014) to solve the ridge optimization [Equation(3)]. The running time of a full RidgeRace inference is larger than the time required by comparable methods (Table 1, measured using an Intel Xeon X5660 with 2.8 GHz) but still within the range of a few minutes. The majority of the running time for RidgeRace is consumed by estimating the λ parameter, performing a leave-one-out iteration over all leaf nodes of the tree and testing . Decreasing the evaluation range for λ or performing the leave-one-out iteration only on a subset of the leaf nodes can considerably decrease the running time for larger trees.
Table 1.
Comparison of average running times in seconds for RidgeRace and the APE implementations of GLS and REML, shown for trees of different size, ranging from 100 to 500 leaf nodes
| Method | 100 | 200 | 300 | 400 | 500 |
|---|---|---|---|---|---|
| RidgeRace | 4 + 0 | 29 + 1 | 135 + 1 | 1074 + 7 | 3372 + 24 |
| GLS | 1 | 2 | 3 | 7 | 10 |
| REML | 2 | 6 | 15 | 38 | 123 |
Note: RidgeRace running time is provided as the running time required for full λ inference plus time required for ACR.
5 CONCLUSION
We here describe a new method for the inference of ancestral character states for continuous characters by performing a ridge regression between the total branch length reaching from the root to a leaf node and the phenotypic value of that leaf. The inference is made by minimizing the prediction error at extant nodes as well as an additional L2-norm regularization factor. The regularization forces the phenotypic rate to be distributed more equally across the whole tree and to branches shared by several nodes, circumventing the trivial case that explains the phenotype by a gain at the terminal branches owing to the overparameterization.
Our evaluation showed that RidgeRace achieved a good congruence between inferred and simulated ancestral character states, and that it performed similarly to or better than two other state-of-the-art methods in terms of the mean squared error. RidgeRace does not assume any underlying model of evolution, and thus, the method allows large flexibility when no definite assumptions can be made about the type of evolutionary process. The formulation of the optimization problem [Equation(2)] allows the straightforward inclusion of measurements at ancestral nodes (e.g. from fossil records). In a similar fashion, multiple measurements at a node can be easily included to lend further support to the inference. Such measurements might originate from multiple observations of the same trait for the same species, or they might represent several traits fitted together. Finally, visualization of the inferred branch weights β along the tree allows a detailed interpretation of the specific phenotypic rates and can indicate short periods of strong directional selection or of increasingly fast evolution, or regimes of the phylogeny that feature larger variation overall.
In our application to the genetic profiles of ovarian cancer data, we demonstrated that RidgeRace was able to reconstruct the main clusters of the phenotype distribution. We also reconstructed changes in the genetic profiles on the branches of the tree. However, no associations between known genetic aberrations and change in survival rate were found for these data. Nevertheless, this study demonstrated the general functionality of the method and suggests future extensions. Patient survival time as a phenotype is a biased measurement, as it is based on the time of diagnosis and the (potential) death of the patient. It may also be dependent on many other factors, such as the patient’s age and the type of therapy received. As RidgeRace can perform a regression on the patient data, such information could easily be included as additional covariates (features) in the regression, if available. This would allow us to control for the influence of such factors and provide insights into their relevance relative to the genetic factors.
Funding: This work was supported by Heinrich Heine University.
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Blomberg SP, et al. Testing for phylogenetic signal in comparative data: behavioral traits are more labile. Evolution. 2003;57:717–745. doi: 10.1111/j.0014-3820.2003.tb00285.x. [DOI] [PubMed] [Google Scholar]
- Boettiger C, et al. Is your phylogeny informative? Measuring the power of comparative methods. Evolution. 2012;66:2240–2251. doi: 10.1111/j.1558-5646.2011.01574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOOST (2014). ublas library. http://www.boost.org.
- Butler MA, King AA. Phylogenetic comparative analysis: a modeling approach for adaptive evolution. Am. Nat. 2004;164:683–695. doi: 10.1086/426002. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012a;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012b;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli-Sforza LL, Edwards AW. Phylogenetic analysis. Models and estimation procedures. Am. J. Hum. Genet. 1967;19(3 Pt 1):233–257. [PMC free article] [PubMed] [Google Scholar]
- Cunningham CW, et al. Reconstructing ancestral character states: a critical reappraisal. Trends Ecol. Evol. 1998;13:361–366. doi: 10.1016/s0169-5347(98)01382-2. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Phylogenies and the comparative method. Am. Nat. 1985;125:1–15. [Google Scholar]
- Felsenstein J. 1993. PHYLIP (Phylogeny Inference Package) http://evolution.gs.washington.edu/phylip/software.html. [Google Scholar]
- Felsenstein J. Inferring Phylogenies. Sunderland, MA, USA: Sinauer Associates Inc; 2004. [Google Scholar]
- Freckleton RP, Harvey PH. Detecting non-Brownian trait evolution in adaptive radiations. PLoS Biol. 2006;4:e373. doi: 10.1371/journal.pbio.0040373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gareth J, et al. An Introduction to Statistical Learning. Springer Series in Statistics, New York; 2013. [Google Scholar]
- Garland T. Rate tests for phenotypic evolution using phylogenetically independent contrasts. Am. Nat. 1992;140:509–519. doi: 10.1086/285424. [DOI] [PubMed] [Google Scholar]
- Grafen A. The phylogenetic regression. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1989;326:1–39. doi: 10.1098/rstb.1989.0106. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hansen TF. Stabilizing selection and the comparative analysis of adaptation. Evolution. 1997;51:1341–1351. doi: 10.1111/j.1558-5646.1997.tb01457.x. [DOI] [PubMed] [Google Scholar]
- Harmon LJ, et al. GEIGER: investigating evolutionary radiations. Bioinformatics. 2008;24:129–131. doi: 10.1093/bioinformatics/btm538. [DOI] [PubMed] [Google Scholar]
- Harmon LJ, et al. Early bursts of body size and shape evolution are rare in comparative data. Evolution. 2010;64:2385–2396. doi: 10.1111/j.1558-5646.2010.01025.x. [DOI] [PubMed] [Google Scholar]
- Harvey PH, Pagel MD. The Comparative Method in Evolutionary Biology. Oxford, UK: Oxford University Press; 1991. [Google Scholar]
- Hastie T, et al. Elements of Statistical Learning. 2nd edn. Berlin: Springer; 2009. [Google Scholar]
- Hofree M, et al. Network-based stratification of tumor mutations. Nat. Methods. 2013;10:1108–1115. doi: 10.1038/nmeth.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losos JB. Seeing the forest for the trees: the limitations of phylogenies in comparative biology. (American Society of Naturalists Address) Am. Nat. 2011;177:709–727. doi: 10.1086/660020. [DOI] [PubMed] [Google Scholar]
- Lynch M. The rate of morphological evolution in mammals from the standpoint of the neutral expectation. Am. Nat. 1990;136:727–741. [Google Scholar]
- Maddison WP, Maddison DR. 2011. Mesquite: a modular system for evolutionary analysis. http://mesquiteproject.org. [Google Scholar]
- Martins EP. Estimating the rate of phenotypic evolution from comparative data. Am. Nat. 1994;144:193–209. [Google Scholar]
- Martins EP, Hansen TF. Phylogenies and the comparative method: a general approach to incorporating phylogenetic information into the analysis of interspecific data. Am. Nat. 1997;149:646–667. [Google Scholar]
- McPeek MA. Testing hypotheses about evolutionary change on single branches of a phylogeny using evolutionary contrasts. Am. Nat. 1995;145:686–703. [Google Scholar]
- Nunn CL. The Comparative Approach in Evolutionary Anthropology and Biology. Chicago: The University of Chicago Press; 2011. [Google Scholar]
- O’Meara BC, et al. Testing for different rates of continuous trait evolution using likelihood. Evolution. 2006;60:922–933. [PubMed] [Google Scholar]
- Pagel MD. Inferring the historical patterns of biological evolution. Nature. 1999;401:877–884. doi: 10.1038/44766. [DOI] [PubMed] [Google Scholar]
- Paradis E, et al. APE: analyses of phylogenetics and evolution in R language, v. 3.0-8. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- Price T. Correlated evolution and independent contrasts. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1997;352:519529. doi: 10.1098/rstb.1997.0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2012. [Google Scholar]
- Rambaut A. 2013. FigTree v1.4. http://tree.bio.ed.ac.uk/software/figtree/ [Google Scholar]
- Revell LJ. On the analysis of evolutionary change along single branches in a phylogeny. Am. Nat. 2008;172:140–147. doi: 10.1086/588078. [DOI] [PubMed] [Google Scholar]
- Revell LJ. Phytools: an R package for phylogenetic comparative biology (and other things) Methods Ecol. Evol. 2012;3:217–223. [Google Scholar]
- Schluter D, et al. Likelihood of ancestral states in adaptive radiation. Evolution. 1997;51:1699–1711. doi: 10.1111/j.1558-5646.1997.tb05095.x. [DOI] [PubMed] [Google Scholar]
- Steinbrück L, McHardy AC. Inference of genotype-phenotype relationships in the antigenic evolution of human influenza A (H3N2) viruses. PLoS Comput. Biol. 2012;8:e1002492. doi: 10.1371/journal.pcbi.1002492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford DL. 2003. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods) Version 4. paup.csit.fsu.edu/ [Google Scholar]
- WHO. (2013). Cancer Fact Sheet No 297. http://www.who.int/mediacentre/factsheets/fs297/en/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.