

Abstract
High dietary protein imposes a metabolic acid load requiring excretion and buffering by the kidney. Impaired acid excretion in CKD, with potential metabolic acidosis, may contribute to the progression of CKD. Here, we investigated the renal adaptive response of acid excretory pathways in mice to high-protein diets containing normal or low amounts of acid-producing sulfur amino acids (SAA) and examined how this adaption requires the RhCG ammonia transporter. Diets rich in SAA stimulated expression of enzymes and transporters involved in mediating NH4+ reabsorption in the thick ascending limb of the loop of Henle. The SAA-rich diet increased diuresis paralleled by downregulation of aquaporin-2 (AQP2) water channels. The absence of Rhcg transiently reduced NH4+ excretion, stimulated the ammoniagenic pathway more strongly, and further enhanced diuresis by exacerbating the downregulation of the Na+/K+/2Cl− cotransporter (NKCC2) and AQP2, with less phosphorylation of AQP2 at serine 256. The high protein acid load affected bone turnover, as indicated by higher Ca2+ and deoxypyridinoline excretion, phenomena exaggerated in the absence of Rhcg. In animals receiving a high-protein diet with low SAA content, the kidney excreted alkaline urine, with low levels of NH4+ and no change in bone metabolism. Thus, the acid load associated with high-protein diets causes a concerted response of various nephron segments to excrete acid, mostly in the form of NH4+, that requires Rhcg. Furthermore, bone metabolism is altered by a high-protein acidogenic diet, presumably to buffer the acid load.
The kidney is the central organ that excretes acid and replenishes bicarbonate buffer used by metabolism.1,2 The importance of the kidney in acid-base balance is demonstrated by inherited and acquired renal diseases, reducing its ability to excrete acid and reabsorb and synthesize bicarbonate (HCO3−).3–5 Recent studies suggest that the progression of CKD is delayed by alkalinizing therapies aiming to reduce the metabolic acidosis occurring with the disease.6–10
In a Western diet, protein intake exceeds by up to 50% the recommended average daily consumption of 0.8 g of protein per kg per day; most protein is from animal sources, which are rich in sulfur-containing acidogenic amino acids.11,12 Diets high in animal protein have gained additional popularity in the context of obesity and its treatment.13,14 Adverse effects of high protein intake on many organs have been described. Whether high-protein diets negatively affect bone or kidney function has remained an open question.15–23 In kidney disease, high animal protein may accelerate decay of renal function; thus, current protocols strongly suggest that patients with CKD reduce animal protein intake.24–26
Dietary protein intake and its metabolism can provide a major acid load, but the metabolic acid load depends on the nature and composition of proteins. Proteins rich in sulfur-amino acids (SAA; i.e., cysteine and methionine) release protons (H+) and sulfate (SO42−) during metabolism and cause increased renal acid and NH4+ excretion paralleled by high urinary SO42− and urea removal.27,28 Plant proteins, such as soy protein, contain only small amounts of SAA and consequently cause a milder acid or even alkaline load.19,29,30 The acid content of a high-protein diet has been linked to the development of tubular-interstitial injury secondary to augmented intrinsic acid production provoked by endothelin-stimulated enhanced aldosterone activity.29,31,32
The renal ammonia (NH3) transporter RhCG is critical to eliminate NH4+ and maintain systemic acid-base balance.33–36 RhCG is localized in most cells along the collecting duct and mediates basolateral uptake of NH3 and final excretion into urine. Its expression is stimulated by acidosis in mice, and Rhcg becomes rate-limiting for urinary NH4+ excretion during strong acid loads (NH4Cl or HCl loading).33,36–38
Here we examined the effect of high-protein diets containing acidogenic SAA (casein diet) or those almost devoid of these amino acids (soy protein diet) on renal mechanisms mediating ammoniagenesis and excretion of the acid load. Our data demonstrate a concerted response of various nephron segments to eliminate the metabolic acid load, the requirement of Rhcg-mediated NH4+ excretion, and effects on bone. Together, these data provide a molecular explanation for the stimulation and requirement of renal acid excretion.
Results
Diet High in Casein Protein Induces a Transient Acid Load That Rhcg−/− Mice Can Excrete
Metabolic measures and acid-base status were assessed in Rhcg+/+, Rhcg+/−, and Rhcg−/− mice receiving a normal diet (basal status with 20% protein), acid-loading high casein (HC) protein diet, or non–acid-loading high soy control protein diet (HS) containing high or low SAA, respectively. Fifty percent of HC or HS diets were provided either as casein or soy protein, but the diets were otherwise isocaloric and identical in their composition. The HC but not the HS diet was associated with slightly reduced food intake (Table 1, Supplemental Table 1). However, no difference was found in food and water intakes among the three different genotypes at any time points measured and under all three types of diet (normal, HC, and HS) (Table 1, Supplemental Table 1). Baseline blood and urine variables (Table 1, Supplemental Tables 1–4) were similar among all three genotypes. A transient decrease in blood pH and HCO3− after 2 days of the HC diet (Figure 1, A and B), but not the HS diet (Supplemental Figure 1, A and B), was identically observed in Rhcg+/+, Rhcg+/−, and Rhcg−/−, confirming the acid-loading effect of the HC diet. The HC diet did not alter urinary pH but stimulated excretion of titratable acidity, most likely in the form of phosphate (Table 1, Supplemental Table 3) to a similar extent in all three genotypes (Figure 1, C and D). In contrast, the HS diet did not alter blood pH or HCO3− but led to a profound alkalinization of urine pH and a decrease in the urinary excretion of titratable acids (Supplemental Figure 1C, Supplemental Table 1). Urinary NH4+ excretion was markedly increased in the HC groups whereas it decreased in the HS groups (Table 1, Supplemental Table 1, Figure 1E, Supplemental Figure 1D). Rhcg deletion delayed the increase in urinary NH4+ excretion in response to the HC acid challenge (Figure 1E). At days 2 and 4 of the HC diet, respectively, Rhcg−/− exhibited a 38.5%±0.1% and 41.4%±0.1% decrease in excreted urinary NH4+ compared with Rhcg+/+ mice (Table 1). At day 9, Rhcg−/− adapted to the HC diet acid load and excreted similar amounts of NH4+, as did Rhcg+/+ mice. The HS diet did not induce any difference in urinary NH4+ excretion in all three genotypes (Supplemental Figure 1D). Taken together, these data confirm that a diet containing high amounts of SAA causes a metabolic acid load and demonstrate that its elimination depends partially on the presence of the ammonia transporter Rhcg.
Table 1.
Body weight, food intake, and urinary values of Rhcg+/+, Rhcg+/−, and Rhcg−/− mice during 9 days of HC protein loading
| Variable | Basal Status | 2 Days of HC Diet | 4 Days of HC Diet | 9 Days of HC Diet | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rhcg+/+ (n=8) | Rhcg+/− (n=8) | Rhcg−/− (n=8) | Rhcg+/+ (n=8) | Rhcg+/− (n=8) | Rhcg−/− (n=8) | Rhcg+/+ (n=8) | Rhcg+/− (n=8) | Rhcg−/− (n=8) | Rhcg+/+ (n=8) | Rhcg+/− (n=8) | Rhcg−/− (n=8) | |
| Body weight (g) | 28.2±0.6 | 29.7±0.4 | 27.0±0.8 | 28.0±0.5 | 29.5±0.7 | 26.0±0.9 | 27.6±0.6 | 29.0±0.7 | 25.7±0.9 | 27.8±0.4 | 27.3±0.9 | 25.9±0.9 |
| Food intake (g/24 h/body wt) | 0.14±0.02 | 0.14±0.03 | 0.15±0.02 | 0.10±0.02a | 0.08±0.04a | 0.10±0.04a | 0.10±0.02a | 0.10±0.03a | 0.10±0.02a | 0.12±0.02a | 0.10±0.05a | 0.10±0.02a |
| Water intake (g/24 h/body wt) | 0.16±0.01 | 0.14±0.01 | 0.15±0.01 | 0.24±0.03a | 0.21±0.02a | 0.21±0.02a | 0.22±0.03a | 0.23±0.02a | 0.26±0.03a | 0.25±0.03a | 0.24±0.03a | 0.26±0.03a |
| Urine values | ||||||||||||
| Volume (ml/24 h) | 1.89±0.32 | 1.91±0.18 | 1.74±0.19 | 4.51±0.44a | 4.34±0.29a | 5.20±0.37a | 4.24±0.50a | 4.38±0.38a | 5.523±0.55a | 4.52±0.17a | 4.36±0.24a | 5.65±0.30a,b |
| Urinary pH | 5.94±0.08 | 5.97±0.04 | 6.03±0.06 | 6.0±0.04 | 5.94±0.03 | 5.87±0.04 | 5.97±0.04 | 6.03±0.05 | 5.89±0.04 | 5.91±0.04 | 5.85±0.04 | 5.86±0.07 |
| NH4+/urine volume (mmol/24 h) | 0.028±0.01 | 0.028±0.01 | 0.027±0.01 | 0.26±0.08a | 0.24.0±0.07a | 0.16±0.04a,c | 0.29±0.08a | 0.24±0.07a | 0.17±0.05a,d | 0.28±0.08a | 0.28±0.05a | 0.29±0.08a |
| TA/urine volume (mmol/24h) | 0.08±0.06 | 0.09±0.03 | 0.07±0.03 | 0.22±0.09a | 0.25±0.05a | 0.22±0.06a | 0.20±0.08a | 0.23±0.06a | 0.23±0.08a | 0.24±0.07a | 0.28±0.06a | 0.27±0.05a |
| Ca2+/urine volume (μmol/24 h) | 1.5±0.1 | 1.6±0.1 | 1.2±0.1 | 3.3±0.1a | 3.2±0.7a | 6.5±0.2a,b | 5.4±0.3a | 5.5±0.2a | 8.2±0.4a,c | 4.1±0.4 | 6.8±0.2a | 5.1±0.4a |
| Dpd/urine volume (nmol/24 h) | 0.076±0.04 (n=6) | ND | 0.073±0.02 (n=6) | ND | ND | ND | 0.100±0.07 (n=6) | ND | 0.211±0.01a,c (n=6) | 0.093±0.02 (n=6) | ND | 0.149±0.05a,c (n=6) |
Body weight, food intake, and urinary values of Rhcg+/+, Rhcg+/−, and Rhcg−/− mice during 9 days HC protein loading (n=8 mice for each group). ND, not determined.
P≤0.05 significantly different from same genotype under control conditions.
P≤0.01 significantly different from Rhcg+/+ mice under same treatment conditions.
P≤0.05 significantly different from Rhcg+/+ mice under same treatment conditions.
P≤0.001 significantly different from Rhcg+/+ mice under same treatment conditions.
Figure 1.

Rhcg+/+, Rhcg+/−, and Rhcg−/− mice can adapt to HC diet. Blood and urine data were collected in Rhcg+/+, Rhcg+/−, and Rhcg −/− mice treated for 9 days on the HC diet. All animals showed a transient decrease of blood pH (A) and bicarbonate (B). (C) Titratable acids increased on the HC diet. (D) All mice rapidly increased urinary NH4+ excretion, but Rhcg−/− had lower NH4+ excretion than Rhcg+/+ during days 2 and 4 of the HC diet. Values are mean±SEM (n=8 mice) *P≤0.05 (Rhcg−/− versus Rhcg+/+), #P≤0.05 significantly different from same genotype under control conditions (day 0), ***P≤0.05 significantly different from Rhcg+/+ mice under same treatment conditions and for the same time point.
HC Diet Stimulates Ammoniagenesis in the Proximal Tubule
To assess the proximal tubule response to the HC diet, we studied the regulation of key molecules involved in NH4+ production and excretion (Figure 2, A–D, Supplemental Figures 2–4). The HC diet caused a transient increase in Rhcg mRNA, whereas Rhbg mRNA was not altered in Rhcg+/+ and Rhcg−/− kidneys (Supplemental Figure 2). Four days of the HC diet induced a transient increase in system N/A transporter 3 (SNAT3) and phosphate-dependent glutaminase (PDG) mRNA levels compared with normal diet (Figure 2, A and C). SNAT3 protein levels were also higher after 4 days of the HC diet, while PDG showed a higher protein expression at day 9 of the HC diet (Figure 2, B and D). Cytosolic phosphoenolpyruvate carboxykinase (PEPCK) and sodium-hydrogen exchanger 3 (NHE3) protein abundance remained unchanged in Rhcg+/+ during HC treatment (Supplemental Figure 3, A and B). To test whether the delayed adaption to HC diet acid load observed in Rhcg−/− mice could be partly explained by an adaptive enhanced NH4+ production, we compared SNAT3, NHE3, PDG, and PEPCK mRNA and protein abundances in all three groups of mice after 4 and/or 9 days of the HC diet. NHE3 and PEPCK mRNA and protein expression levels were similar among all three genotypes (Supplemental Figure 3, C–H). However, at day 4 of the HC diet, both Rhcg+/− and Rhcg−/− increased mRNA and protein expression of SNAT3 compared with Rhcg+/+ (Figure 2, A, B, E, and F). PDG mRNA and protein were higher in Rhcg−/− than Rhcg+/+ (Figure 2, C, D, G, and H). After 9 days of the HC diet, SNAT3 and PDG mRNA and protein were still higher in Rhcg−/− than in Rhcg+/+, while in Rhcg+/− mice only PDG protein levels were elevated. Thus, Rhcg−/− mice increased expression of some proteins critical for proximal tubular ammoniagenesis.
Figure 2.

The HC diet stimulated the ammoniagenic pathway. Kidneys collected from all three groups of mice after 4 or 9 days of the HC diet were analyzed by quantitative RT-PCR and immunoblotting. After 4 days of the HC diet, Rhcg+/+ highly increased mRNA (A) and protein (B) expression of SNAT3. Rhcg+/− and Rhcg −/− exhibited higher SNAT3 mRNA (A) and protein (E) levels than did Rhcg+/+. At day 9, only Rhcg−/− kept an enhanced level of SNAT3 mRNA (A) and protein (F). 4 or 9 days HC diet augmented Rhcg+/+ PDG mRNA (C) or protein (D) abundance. Rhcg−/− displayed higher mRNA (C) after 9 days HC and higher protein (G and H) expressions after both 4 and 9 days HC. Values are mean±SEM (n=4–8 mice) #P≤0.05 significantly different from same genotype under control conditions (day 0), *P≤0.05, **P≤0.01, ***P≤0.001 significantly different from Rhcg+/+ mice under the same treatment conditions and for the same time point.
Rhcg+/− and Rhcg−/− Mice Have Abnormal Accumulation of NH4+ in the Medullary Interstitium
Next we assessed mechanisms involved in the generation of the cortico-papillary NH4+ gradient. We hypothesized that Rhcg−/− mice could increase the NH4+ excretion by stimulating NH4+ reabsorption by the thick ascending limb Na+/K+/2Cl− cotransporter (NKCC2). NKCC2 protein expression in Rhcg+/+, Rhcg+/−, and Rhcg−/− mice after 4 and 9 days of the HC diet revealed two opposite regulations of the protein during HC treatment. NKCC2 levels were higher in Rhcg+/− and Rhcg−/− after 4 days (Figure 3A) but were lower in Rhcg−/− after 9 days (Figure 3B). NH4+ tissue content in the cortex, outer medulla, and inner medulla followed NKCC2 expression. At day 4 of the HC diet (Figure 3, C and D), both Rhcg+/− and Rhcg−/− accumulated more NH4+ in the inner medulla than did Rhcg+/+ (25.2%±0.1% for Rhcg+/− and 29.9%±0.1% for Rhcg−/−). In contrast, at day 9 of the HC diet (Figure 3D), Rhcg−/− had lower inner medullary NH4+ content than did Rhcg+/+ (−37.0%±0.1%). Thus, the adaption of Rhcg-deficient mice to a high-SAA diet involves regulation of the NKCC2 cotransporter and affects accumulation of NH4+ in the medullary interstitium.
Figure 3.

Altered medullary absorption and accumulation of ammonium in Rhcg-deficient mice. After 4 or 9 days of the HC diet, Rhcg+/+, Rhcg +/−, and Rhcg −/− mice kidneys were submitted to Western blot analysis and NH4+ concentration was measured in dissected cortex and inner and outer medulla. Expression of NKCC2 was higher in Rhcg+/− and Rhcg −/− than Rhcg+/+ at day 4 of the HC diet (A), while at day 9, Rhcg−/− showed a reduced level (B). (C) NH4+ concentration was similarly increased in the inner medulla of Rhcg+/− and Rhcg −/− mice at day 4 of the HC diet. (D) At day 9, Rhcg−/− had much lower inner medullary NH4+ content. Values are mean±SEM (n=6–7 mice). #P≤0.05 significantly different from same genotype under control conditions (day 0); *P≤0.05, ***P≤0.001 significantly different from Rhcg+/+ mice under same treatment conditions and for the same time point.
HC Diet Stimulates Diuresis and Downregulates NKCC2 and AQP2
Besides its role in NH4+ reabsorption, NKCC2 functions in the countercurrent mechanism establishing the cortico-papillary osmotic gradient required for water reabsorption along the collecting duct. The HC diet induced diuresis in all three genotypes (Figure 4E, Table 1), whereas diuresis was lower in animals receiving the HS diet despite quantitatively similar urea excretion (compare Table 1 with Supplemental Table 1). We measured NKCC2 and collecting-duct AQP2 protein expression levels in Rhcg+/+ (Figure 4, A and B) and Rhcg−/− animals (Figure 4, C and D) at 0, 4, and 9 days of the HC and HS diets. NKCC2 and AQP2 were downregulated by the HC diet in both genotypes, with reduced NKCC2 expression in Rhcg wild-type after 4 and 9 days of the HC die (Figure 4A) and after 9 days in Rhcg−/− mice (Figure 4C). Similarly, AQP2 levels were lower after 9 days of the HC diet in Rhcg+/+ and Rhcg−/− mice (Figure 4, B and D). Because Rhcg−/− excreted more urine than their littermates (5.7±0.5 versus 4.5±0.3 ml/24 hours after 9 days of the HC diet; P≤0.01) (Figure 4E), we tested whether the active phosphorylated form of AQP2 (pSer256-AQP2) was altered in Rhcg−/− mouse kidneys. The total abundance of mature pSer256-AQP2 (35-kDa band) was reduced, as well as the ratio of phosphorylated AQP2 over total AQP2 (Figure 4F). In animals receiving the HS diet, NKCC2 and AQP2 were not regulated (Supplemental Figure 4). In summary, a high-SAA diet stimulates diuresis by downregulating NKCC2 and AQP2, an effect amplified in Rhcg−/− mice.
Figure 4.

The HC diet stimulates diuresis and regulates NKCC2 and AQP2 expression. Protein expression levels were examined using immunoblotting, and urine production was measured at different time points of the HC diet in Rhcg+/+ and Rhcg −/− mice. NKCC2 protein levels were decreased after 4 and 9 days of the HC diet in Rhcg+/+ (A) and only after 9 days in Rhcg−/− (C), while AQP2 expression was reduced in Rhcg+/+ (B) and Rhcg −/− (D) mice at day 9 of the HC diet. Rhcg−/− showed higher urinary excretion than Rhcg+/+ (E). Mature pSer256-AQP2 showed decreased abundance at day 9 in Rhcg−/−, and the ratio of pSer256-AQP2 over total AQP2 was reduced in Rhcg−/− (F). Values are mean±SEM (n=4–8 mice). #P≤0.05 significantly different from same genotype under control conditions (day 0); *P≤0.05, **P≤0.01; ***P≤0.001 significantly different from Rhcg+/+ mice under same treatment conditions and for the same time point.
HC Diet Stimulates Bone Resorption Exaggerated by Absence of Rhcg
Two days of the HC diet caused a transient increase in ionized blood Ca2+ levels in both Rhcg+/+ and Rhcg−/− animals but significantly higher increases in Rhcg−/− mice. In contrast, Ca2+ levels remained higher in Rhcg−/− mice and returned to normal only on day 9 (Figure 5A). Urinary Ca2+ excretion was also transiently increased in both genotypes at days 2 and 4 of the HC diet, with significantly higher urinary Ca2+ levels in Rhcg−/− at day 4 (Figure 5B). To investigate bone remodelling, we measured urinary deoxypyridinoline (Dpd) excretion, a marker of bone resorption and plasma concentration of osteocalcin, a marker of bone formation (Figure 5, C and D). Dpd levels were elevated after 4 days of the HC diet in Rhcg+/+ and Rhcg−/− and remained higher in Rhcg−/− after 9 days of the HC diet. Plasma osteocalcin levels did not significantly differ during the treatment or between the two groups, suggesting that increased bone formation does not compensate for bone resorption in Rhcg+/+ and Rhcg−/− mice.
Figure 5.

Rhcg−/− mice develop hypercalcemia and hypercalciuria, and show signs of increased bone resorption during HC diet. (A) After 2 and 4 days of the HC diet, ionized Ca2+ concentration was higher in Rhcg−/− blood. (B) At day 4, urinary Ca2+ excretion was also increased in Rhcg −/−. (C) The release of the bone degradation marker Dpd was augmented in Rhcg−/− compared with Rhcg+/+ following 4 and 9 days the HC diet, whereas the plasma concentration of the bone formation marker osteocalcin (D) was not affected by diet and genotype. Values are mean±SEM (n=5–8 mice). #P≤0.05 significantly different from same genotype under control conditions (day 0); *P≤0.05, **P≤0.01 significantly different from Rhcg+/+ mice under same treatment conditions and for the same time point.
To further evaluate the direct effect of an acidogenic high-protein diet on bones, we measured tissue mineral density (TMD) of the midcortical region of femurs collected after 9 days of the HC diet (Figure 6, A and B). Micro–computed tomography (micro-CT) scans were analyzed by comparing TMD of a deep bone layer mineralized before the HC diet period and were therefore not influenced by the acidogenic protein load (layer 12) with TMD of a more superficial layer close to the periosteum, which contained bone formed during the HC diet (layer 2) (Figure 6A). Surprisingly, TMD of layer 12 in Rhcg−/− mice (diet independent) was higher than the corresponding layer in femurs from Rhcg+/+ mice (1267.5 mhHA/cm3 versus 1206.0 mhHA/cm3; P≤0.01). TMD of layer 2 was lower in both Rhcg+/+ and Rhcg−/−. However, the difference between genotypes was no longer detected in layer 2 (formed during the HC diet) (917.4 mhHA/cm3 versus 944.8 mhHA/cm3; P≥0.05) (Figure 6B). Cortical mineral content was not affected by the HS diet (data not shown). Moreover, no difference was detected in the standard morphometric measures, suggesting that all bones had similar size, shape, and internal microarchitecture (Supplemental Table 5).
Figure 6.

HC diet reduces bone mineral density in Rhcg−/− mice. (A) Micro-CT reconstruction of a femur with the midcortical region considered for TMD evaluation. The inset shows the different gray values (top), which carry information on TMD and a cross-section (bottom) with layer 2 close to the periosteum (light gray) and layer 12 (dark gray). (B) TMD of Rhcg−/− was higher than TMD of Rhcg+/+ in the diet-independent layer 12 but not in layer 2, which was formed during the diet. Data are represented by boxplots (i.e., the inner box contains 50% of all data, the whisker bars denote the full range, and the black line represents the median value [over all animals]). Statistical significance was obtained with two-way ANOVA with Bonferroni multiple comparisons test; n=5 per genotype. **P≤0.01 significantly different from wild-type mice under same treatment conditions and for the same time point.
Discussion
In the current study, we examined the effect of an acidogenic high-protein diet on the renal adaption through acid excretion. We compared two different diets with high protein content (50%) containing normal levels of SAA (casein protein) or low levels of these amino acids (soy protein). Intake of these diets was similar in all groups, as evident from total food intake and total urinary urea excretion.39 The dietary content of SAA is reflected by the much higher excretion of SO42− in urine among the animals ingesting the casein diet. On the basis of this animal model, we find that the kidney adapts to the high-SAA diet with a parallel response of various nephron segments: (1) stimulated NH4+ excretion and increased expression of key molecules of the ammoniagenic pathway in the proximal tubule; (2) reduced expression of the NKCC2 cotransporter; (3) increased diuresis and downregulation of the AQP2 water channel; (4) loss of bone TMD; and (5) all processes being dependent on the ammonia transporter Rhcg, as evident from reduced ammonium excretion, exaggerated induction of ammoniagenic molecules, enhanced diuresis, and downregulation of NKCC2 and AQP2, and more severe effects on bone remodeling.
Intake of high protein in the form of casein caused an increased urinary NH4+ and titratable acid excretion and a transient decrease in blood pH and HCO3−, indicating the metabolic acid load and the kidneys’ ability to adapt. In contrast, a diet high in soy protein reduced the dietary acid load as urinary NH4+ and titratable acid excretion decreased and urinary pH became more alkaline. Interestingly, titratable acid excretion increased strongly with the SAA diet and remained high. This is in contrast to findings in humans and rodents provided with an acid load in the form of NH4Cl or HCl, respectively, where most acid is excreted in the form of ammonium.36,40,41 These differences may be due in part to different types of acid loading.
The increased acid excretion found in animals fed a diet high in acidogenic protein was paralleled on the molecular level by stimulation of expression of the glutamine transporter SNAT3 and the phosphate-dependent glutaminase in the proximal tubule. The regulation of SNAT3 by high protein intake had been reported previously.42 In contrast, PEPCK, fueling α-keto-glutarate from ammoniagenesis into gluconeogenesis, was decreased during the HC diet in Rhcg+/+ kidneys, suggesting a decrease in renal gluconeogenesis. In rat liver, PEPCK is stimulated by high protein intake, which might indicate that high casein intake induces specifically renal ammoniagenesis and favors hepatic over renal gluconeogenesis.43
The response of the collecting duct system to high protein intake has been previously described, mostly on the basis of functional experiments demonstrating increased H+ and NH4+ secretion.31,32 Consistently, mice receiving an HC diet excreted high amounts of NH4+ into urine. Rhcg mRNA abundance increased transiently (day 4) at the time when Rhcg−/− mice showed decreased urinary ammonium excretion. This finding suggests that Rhcg may be directly regulated and mostly needed during the earlier phase of adaption. Similarly, during NH4Cl-induced acidosis, Rhcg protein abundance is increased and staining enhanced at the luminal and basolateral membrane, suggesting that Rhcg is regulated at different levels.44,45 Rhbg mRNA was not regulated, consistent with previous data from NH4Cl-loaded mice and a less important role of Rhbg in renal ammonium excretion.45–47 This later process along the collecting duct requires the formation of a NH4+ gradient from medullary interstitium into urine, which is generated at least in part by the reabsorption of NH4+ by the NKCC2 cotransporter in the thick ascending limb of the loop of Henle. This transporter is stimulated by acidosis induced by NH4Cl feeding.48–51 In contrast, the HC diet led to progressive decreases in expression of NKCC2 without disturbing medullary NH4+ accumulation.
The renal adaption to the HC diet was impaired in the absence of Rhcg. Rhcg−/− mice had a delayed increase in urinary NH4+ excretion and required a stronger and more sustained increase in PDG and SNAT3 expression, all indicating highly stimulated ammoniagenesis. Furthermore, NKCC2 expression was even more decreased than in wild-type animals and medullary NH4+ accumulation was impaired in the inner medulla.
High protein diets, such as casein and soy, stimulated diuresis in animals, but casein produced a stronger diuresis than did the soy diet. Increased excretion of urea from hepatic protein metabolism may be partly responsible for the diuresis causing an osmotic driving force. The acid content of the HC diet, however, probably provides an additional stimulus because mice receiving the acidogenic casein diet had higher diuresis despite almost identical urea excretion. Indeed, feeding mice or rats with NH4Cl causes similar diuresis.52,53 Here we found that diuresis was accompanied by a progressive reduction in AQP2 water channel expression. Rhcg−/− mice excreted even higher urine volumes than did Rhcg+/+ animals. NKCC2 and AQP2 were not regulated in mice receiving the HS diet, demonstrating that high protein per se does not regulate these proteins. Moreover, phosphorylation of AQP2 at serine 256, critical for the regulated insertion and activity of the channel in the membrane, was reduced in Rhcg−/− mice. This finding suggests that increased diuresis may be part of a compensatory mechanism. Thus, the more pronounced reduction in NKCC2 expression and reduced insertion of AQP2 at the plasma membrane would allow Rhcg−/− mice to excrete the NH4+ formed in the proximal tubule by shunting the medullary interstitium passage and diluting and excreting the NH4+ load in the thick ascending limb and collecting duct. The time course of achieving similar rates of urinary ammonium excretion and stronger downregulation of NKCC2, reduced medullary ammonium accumulation, and lower AQP2 expression is similar, possibly indicating a concerted compensatory mechanism. We speculate that several hormones might be involved in mediating the effect of the HC diet on NKCC2 and AQP2. Among them, aldosterone, endothelin 1, prostaglandin E2, and atrial natriuretic peptide are increased by high-protein diets.32,54,55 Atrial natriuretic peptide and prostaglandin E2 reduce AQP2 expression and NKCC2 function.56,57 In addition, endothelin 1 and aldosterone may reduce NKCC2 expression and function via a nitric oxide, cyclic guanosine monophosphate, and phosphodiesterase-2–dependent mechanism.
Whether high protein intake has a negative effect on bone is controversial, and the positive or negative impact may depend on the type of protein, the content of minerals, and the content of carbohydrates (e.g., whether diets are ketogenic).21–23 Acidosis has significant adverse effects on bone, stimulating osteoclast activity, increasing demineralization, and finally leading to loss of bone mineral density and stability.58,59 The HC diet stimulated bone degradation in Rhcg−/−, as evident from the increased urinary excretion of Dpd. Moreover, bone TMD was lower in a layer (layer 2) close to the bone surface containing newly formed bone, and no difference among genotypes could be detected. In wild-type animals, Dpd levels increased only transiently, whereas in Rhcg−/− mice, Dpd remained elevated, consistent with increased bone degradation; this possibly contributes to the compensation of reduced NH4+ excretion. Surprisingly, no evidence was found for higher osteoblast activity, reflected by constant osteocalcin levels. This finding suggests that the HC diet would eventually cause a small net loss of bone. Indeed, elevated urinary Ca2+ was paralleled with Dpd levels, indicating that this calcium load may at least in part originate from bone. However, effects of acidosis on calcium binding to albumin could also contribute to hypercalciuria and hypercalcemia, as well as stimulation of intestinal calcium absorption during acidosis.60 Consumption of an acidogenic HC diet was paralleled by a loss of difference between higher bone TMD in Rhcg−/− animals and lower TMD in wild-type animals. Whether the lower TMD in the younger layer is only an age-dependent effect or may be influenced by the acidogenic HC diet remains to be clarified. The higher TMD found in the deeper bone layer of Rhcg−/− suggests that Rhcg deletion might influence bone development toward a higher density. Expression of Rhcg has not been reported for bone, indicating that differences in TMD are rather the consequence of absent Rhcg function in other organs. Thus, the effects of HC diets and absence of Rhcg may be subtle in our data set but reflect only dietary changes over a very short period (9 days); longer exposure to HC-like diets may have more pronounced effects, which must be addressed in future studies.
In summary, we demonstrate that the kidney responds to acidogenic high-protein diets with increased ammoniagenesis and NH4+ excretion, and deciphers the transport pathways contributing to this adaptive response. The renal ammonium transporter Rhcg is critical for the adaption. Its absence or reduced activity in inherited or acquired kidney disease may contribute to metabolic acidosis and bone degradation, and eventually may be fed back on the kidney, contributing to the progression of kidney disease.
Concise Methods
Animals
Mice were genotyped by PCR directly on a 3-µl 25 mM NaOH ear biopsy digestion product. Genomic DNA was amplified using primer pairs specific for exon 1: forward (AGACCCCACAATGGAAAGCTATAA), Rhcg+/+ reverse (CAACCAGAACTCCCCAGTGTCAGA), and Rhcg−/− reverse (ATGGGCTGACCGCTTCCTCGTGCTTTAC).36 The products were separated by electrophoresis on 1% agarose gels (mutant product: 522 bp, wild-type product: 376 bp). Heterozygous mice were mated to generate mice of all genotypes. All animal experiments were conducted according to Swiss Laws of Animal Welfare and approved by the local Zurich Veterinary Authority (Kantonales Veterinäramt Zürich).
Metabolic Cage Studies
All experiments were performed using age-matched male Rhcg wild-type (Rhcg+/+), Rhcg knockout (Rhcg−/−), and Rhcg heterozygous (Rhcg+/−) littermate mice (3–4 months old), housed in standard or metabolic cages (Tecniplast, Buguggiate, Italy). Mice were given deionized water and were fed a standard powdered laboratory chow ad libitum (Kliba, Kaiseraugst, Switzerland). Mice were allowed to adapt to metabolic cages for 2 days; then, one 24-hour urine sample was collected under light mineral oil in urine collectors to determine daily urinary measures. Mice were then allowed to recover for 2 days in standard cages and were given a high-protein diet (50% casein or 50% soy) (Ssniff Spezialdiaeten GmbH, Soest, Germany) for 9 days. Four 24-hour urine samples were collected during the 9 days of the high-protein diet. Water and food intake and urine excretion were monitored at baseline and during 9 days of high-protein treatment. Blood was collected from the retroorbital plexus under isofluran anesthesia and analyzed in a Radiometer ABL 505 blood gas analyzer (Radiometer, Copenhagen, Denmark). Urinary pH was measured directly after collection using a pH microelectrode (691 pH meter; Metronohm). Urinary electrolytes concentrations were measured by flame photometry (IL943; Instruments Laboratory), and titratable acid was measured using a DL 50 titrator (Mettler Toledo36). Urinary NH3/NH4+ and creatinine were assessed using the Berthelot and Jaffe methods, respectively.61,62 Urinary SO42− was measured by ion exchange chromatography using an IonPac AS 11 analytical column on a Dionex DX-600 HPLC system (Dionex, Olten, Switzerland). Urinary deoxypyridinoline (DPD) was measured with a DPD Enzyme Immunoassay kit (Microvue DPD EIA; Quidel Corporation, San Diego, CA), and plasma osteocalcin was measured using a Mouse Osteocalcin Immunoradiometric Assay kit (Mouse Osteocalcin IRMA Kit; Immunotopics, San Clemente, CA). Mice were anesthetized with ketamine and xylazine and euthanized at different time points to collect blood, kidneys, and femurs. Kidneys were immediately flash-frozen in liquid nitrogen and placed at −80°C until further processing. Femurs were collected and stored in 70% ethanol at room temperature.
RNA Extraction and Reverse Transcription
Snap-frozen kidneys (eight or five half kidneys for each condition, 9 days of diet or 4 days of diet, respectively) were homogenized in RLT-Buffer (Qiagen, Basel, Switzerland) supplemented with β-mercaptoethanol to a final concentration of 1%. Total RNA was extracted from 200-μl aliquots of each homogenized sample using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Quality and concentration of the isolated RNA preparations were analyzed on the NanoDrop ND-1000 spectrophotometer (Wilmington, DE). Total RNA samples were stored at −80°C. Each RNA sample was diluted to a final concentration of 100 ng/μl, and cDNA was prepared using the TaqMan Reverse Transcription Reagent Kit containing 10× RT buffer, MgCl2, random hexamers, deoxyribonucleotide triphosphates, Rnase inhibitors, and Multiscribe reverse transcription enzyme (Applied Biosystems/Roche, Foster City, CA). Reverse transcription was performed with the Biometra TGradient thermocycler (Goettingen, Germany), with thermocycling conditions set at 25°C for 10 minutes, 48°C for 30 minutes, and 95°C for 5 minutes.
Real-Time Semi-Quantitative PCR
Relative mRNA expression was determined using semi-quantitative real-time RT-PCR using the Applied Biosystems 7500 Fast Real-Time PCR system. Thermocycling conditions consisted of denaturation (95°C; 10 minutes) followed by 40 cycles of denaturation at 95°C for 15 seconds and annealing/elongation (60°C; 60 seconds) with auto ramp time. All reactions were run in triplicate. Forward and reverse primers and probe concentrations were 25 μM and 5 μM, respectively. TaqMan Universal PCR master mix 2× (Applied Biosystems/Roche) was used as the Taq polymerase. Primers and probes for SNAT3, PDG, NHE3, PEPCK, NKCC2, and hypoxanthine-guanine phosphoribosyltransferase (HPRT) were generated using Primer Express software from Applied Biosystems and synthesized at Microsynth (Balgach, Switzerland) as described previously.36,63 Probes were generated with the reporter dye FAM at the 5′ end and carboxytetramethylrhodamine at the 3′ end. Reactions were run in triplicates, including a negative control (without Multiscribe reverse transcription enzyme). The cycle threshold (Ct) values obtained were ultimately compared with Ct values of the endogenous gene HPRT. Relative mRNA expression ratios were calculated as follows:
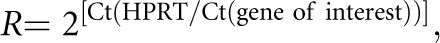 |
where Ct represents the cycle number at threshold 0.02.
Immunoblotting
Crude total membrane proteins or cytosolic fractions were obtained from kidneys homogenized in 250 mM sucrose, 10 mM Tris-HCl, pH 7.5, and in the presence of protease inhibitors (complete ULTRA tablets; Roche, Rotkreuz, Switzerland). Forty micrograms of crude membrane proteins or cytosolic proteins were solubilized in loading buffer containing dithiothreitol (2 M) and separated on 5%–10% polyacrylamide gels. For immunoblotting, proteins were transferred electrophoretically to polyvinylidene fluoride membranes (Immobilon-P; EMD Millipore, Bedford, MA). After blocking with 5% milk powder in Tris-buffered saline-0.1% Tween-20 for 60 minutes, membranes were incubated with rabbit polyclonal anti-SNAT3 (diluted 1:1000),63 anti-PDG (diluted 1:5000; kindly provided by Dr. N. Curthoys, University of Colorado), anti-PEPCK (diluted 1:5000; Cayman Chemicals, Ann Arbor, MI), rabbit polyclonal anti-NKCC2 and anti-AQP2 (diluted 1:5000; kindly provided by J. Loffing, Institute of Anatomy, University of Zurich), rabbit anti-pSer256-AQP2 1:3000 (kindly provided by Dr. S. Nielsen, University of Aarhus), rabbit polyclonal anti-NHE3 (StressMarq Biosciences Inc., Victoria, BC, Canada) and mouse monoclonal anti–β-actin antibody (1:20,000; Sigma-Aldrich, St. Louis, MO) overnight at 4°C. After washing and blocking with 5% milk powder for 60 minutes, membranes were then incubated for 2 hours at room temperature with secondary goat anti-rabbit antibodies 1:5000 or donkey anti-mouse antibodies 1:10,000 linked to alkaline phosphatase (Promega, Madison, WI) or horseradish peroxidase (Promega), respectively. The protein signal was detected with the appropriate substrate (CDP-Star; Roche, Rotkreuz, Switzerland for alkaline phosphatase; EMD Millipore for horseradish peroxidase) using the Las-4000 image analyzer system (Fujifilm; Life Science). All images were analyzed with Advanced Image Data Analyzer AIDA (Raytest, Straubenhardt, Germany) to calculate the protein of interest/β-actin ratio.
Measurement of Renal Ammonium Content
Renal tissue ammonium content was measured by an enzymatic technique (Sigma-Aldrich; Ammonia Assay Kit) as previously described.36 Mice were anesthetized and kidneys removed and immediately frozen in liquid nitrogen. Kidneys were then sliced frozen to yield a column of tissue, which extended from the cortex to the tip of the papilla. Sections were cut along the cortico-medullary axis to yield three slices: cortex, outer medulla, and inner medulla. Two kidneys were pooled for each sample. Tissue slices were then homogenized in 300 µl of ice-cold 7% trichloroacetic acid, and the solution was centrifuged. The supernatant was drawn off and the pH of a 250-µl sample was adjusted to near neutral by the addition of 12 µl of 10 mM Na2HPO4 in 9 N NaOH. A 200-µl sample of buffered supernatant was then analyzed for ammonium concentration. The pellet was resuspended in 1 N NaOH, shaken overnight, and analyzed for total protein concentration using the Bio-Rad protein assay (Bio-Rad, Hercules, CA).
Micro-CT Imaging and Quantitative Analysis
Whole femurs of five Rhcg+/+ and five Rhcg−/− were scanned with a desktop micro-CT (μCT40; Scanco Medical, Brüttisellen, Switzerland) operated at 55 kVp and 145 µA. The samples were scanned with the long axis perpendicular to the beam direction and using an integration time of 300 milliseconds and a frame averaging of 3, resulting in a total scanning time of approximately 5.1 hours per sample. Before image reconstruction, a voltage-specific third-order polynomial correction52 provided by the manufacturer was applied to minimize the influence of beam hardening. The reconstructed scans had a nominal isotropic resolution of 10 μm. A three-dimensional Gaussian filter (sigma 0.8, support 1) was applied to reduce the noise present in the images, and the gray levels of the scans were then transformed into TMD by using the manufacturer calibration record based on a phantom of 1200 mg HA/cm.3,64 The micro-CT scanner was calibrated weekly for mineral equivalent value and monthly for determining in-plane spatial resolution. All measurements and analyses were performed according to the guidelines for assessment of bone microstructure in rodents using micro-CT.65 Standard three-dimensional morphometric measures were computed for full, cortical, and trabecular bone as described elsewhere.66 TMD was evaluated in the cortical bone compartment having a size of 30% of the total femoral length by averaging the TMD values in different layers having the same distance to the bone surface, according to a recently developed layer analysis.67 Specifically, we analyzed the TMD in layer 2 (i.e., close to the bone periosteal surface and hence affected by diet) and in layer 12 (i.e., far from the bone surface and thus not affected by diet).
Statistical Analyses
Statistical comparisons were tested by ANOVA (one-way, Newman–Keuls multiple comparison test) and unpaired t test using GraphPad Prism (GraphPad Software). P values <0.05 were considered to represent statistically significant differences.
Disclosures
None.
Supplementary Material
Acknowledgments
We thank Julien Weber and Sébastien Druart for their help in the biochemical profiling of the mouse models. The use of the Zurich Integrative Rodent Physiology Core Facility is gratefully acknowledged.
This study was supported by a grant from the Swiss National Science Foundation to C.A.W. (31003A_138143). The studies were further supported in part by the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement number 305608 (EURenOmics) to O.D. and C.A.W., an Action de Recherche Concertée (ARC, Communauté Française de Belgique) to O.D., the FNRS and FRSM, the Inter-University Attraction Pole (IUAP, Belgium Federal Government), and the NCCR Kidney.CH program (Swiss National Science Foundation) to O.D.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2013050466/-/DCSupplemental.
References
- 1.Hamm LL, Alpern RJ, Preisig PA: Cellular mechanisms of renal tubular acidification. In: Seldin and Giebisch's The Kidney Physiology and Pathophysiology, edited by Alpern RJ, 4th Ed., Burlington, MA, Academic Press, 2008, pp 1539–1585 [Google Scholar]
- 2.Curthoys NP: Renal ammonium ion production and excretion. In: Seldin and Giebisch's The Kidney Physiology and Pathophysiology, edited by Alpern RJ, 4th Ed., Burlington, MA, Academic Press, 2008, pp 1601–1619 [Google Scholar]
- 3.Fry AC, Karet FE: Inherited renal acidoses. Physiology (Bethesda) 22: 202–211, 2007 [DOI] [PubMed] [Google Scholar]
- 4.Laing CM, Toye AM, Capasso G, Unwin RJ: Renal tubular acidosis: Developments in our understanding of the molecular basis. Int J Biochem Cell Biol 37: 1151–1161, 2005 [DOI] [PubMed] [Google Scholar]
- 5.Kraut JA, Kurtz I: Metabolic acidosis of CKD: Diagnosis, clinical characteristics, and treatment. Am J Kidney Dis 45: 978–993, 2005 [DOI] [PubMed] [Google Scholar]
- 6.de Brito-Ashurst I, Varagunam M, Raftery MJ, Yaqoob MM: Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol 20: 2075–2084, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahajan A, Simoni J, Sheather SJ, Broglio KR, Rajab MH, Wesson DE: Daily oral sodium bicarbonate preserves glomerular filtration rate by slowing its decline in early hypertensive nephropathy. Kidney Int 78: 303–309, 2010 [DOI] [PubMed] [Google Scholar]
- 8.Phisitkul S, Khanna A, Simoni J, Broglio K, Sheather S, Rajab MH, Wesson DE: Amelioration of metabolic acidosis in patients with low GFR reduced kidney endothelin production and kidney injury, and better preserved GFR. Kidney Int 77: 617–623, 2010 [DOI] [PubMed] [Google Scholar]
- 9.Wesson DE, Simoni J, Broglio K, Sheather S: Acid retention accompanies reduced GFR in humans and increases plasma levels of endothelin and aldosterone. Am J Physiol Renal Physiol 300: F830–F837, 2011 [DOI] [PubMed] [Google Scholar]
- 10.Goraya N, Simoni J, Jo C, Wesson DE: Dietary acid reduction with fruits and vegetables or bicarbonate attenuates kidney injury in patients with a moderately reduced glomerular filtration rate due to hypertensive nephropathy. Kidney Int 81: 86–93, 2012 [DOI] [PubMed] [Google Scholar]
- 11.Smit E, Nieto FJ, Crespo CJ, Mitchell P: Estimates of animal and plant protein intake in US adults: Results from the Third National Health and Nutrition Examination Survey, 1988-1991. J Am Diet Assoc 99: 813–820, 1999 [DOI] [PubMed] [Google Scholar]
- 12.European Food Safety Authority: Scientific opinion on dietary reference values for protein. EFSA Journal 10: 2257, 2012 [Google Scholar]
- 13.Westerterp-Plantenga MS, Lejeune MP, Nijs I, van Ooijen M, Kovacs EM: High protein intake sustains weight maintenance after body weight loss in humans. Int J Obes Relat Metab Disord 28: 57–64, 2004 [DOI] [PubMed] [Google Scholar]
- 14.Astrup A, Meinert Larsen T, Harper A: Atkins and other low-carbohydrate diets: Hoax or an effective tool for weight loss? Lancet 364: 897–899, 2004 [DOI] [PubMed] [Google Scholar]
- 15.Brändle E, Sieberth HG, Hautmann RE: Effect of chronic dietary protein intake on the renal function in healthy subjects. Eur J Clin Nutr 50: 734–740, 1996 [PubMed] [Google Scholar]
- 16.Brenner BM, Meyer TW, Hostetter TH: Dietary protein intake and the progressive nature of kidney disease: The role of hemodynamically mediated glomerular injury in the pathogenesis of progressive glomerular sclerosis in aging, renal ablation, and intrinsic renal disease. N Engl J Med 307: 652–659, 1982 [DOI] [PubMed] [Google Scholar]
- 17.Friedman AN: High-protein diets: potential effects on the kidney in renal health and disease. Am J Kidney Dis 44: 950–962, 2004 [DOI] [PubMed] [Google Scholar]
- 18.King AJ, Levey AS: Dietary protein and renal function. J Am Soc Nephrol 3: 1723–1737, 1993 [DOI] [PubMed] [Google Scholar]
- 19.Kontessis P, Jones S, Dodds R, Trevisan R, Nosadini R, Fioretto P, Borsato M, Sacerdoti D, Viberti G: Renal, metabolic and hormonal responses to ingestion of animal and vegetable proteins. Kidney Int 38: 136–144, 1990 [DOI] [PubMed] [Google Scholar]
- 20.Knight EL, Stampfer MJ, Hankinson SE, Spiegelman D, Curhan GC: The impact of protein intake on renal function decline in women with normal renal function or mild renal insufficiency. Ann Intern Med 138: 460–467, 2003 [DOI] [PubMed] [Google Scholar]
- 21.Eisenstein J, Roberts SB, Dallal G, Saltzman E: High-protein weight-loss diets: are they safe and do they work? A review of the experimental and epidemiologic data. Nutr Rev 60: 189–200, 2002 [DOI] [PubMed] [Google Scholar]
- 22.Darling AL, Millward DJ, Torgerson DJ, Hewitt CE, Lanham-New SA: Dietary protein and bone health: A systematic review and meta-analysis. Am J Clin Nutr 90: 1674–1692, 2009 [DOI] [PubMed] [Google Scholar]
- 23.Alexy U, Remer T, Manz F, Neu CM, Schoenau E: Long-term protein intake and dietary potential renal acid load are associated with bone modeling and remodeling at the proximal radius in healthy children. Am J Clin Nutr 82: 1107–1114, 2005 [DOI] [PubMed] [Google Scholar]
- 24.Odermatt A: The Western-style diet: A major risk factor for impaired kidney function and chronic kidney disease. Am J Physiol Renal Physiol 301: F919–F931, 2011 [DOI] [PubMed] [Google Scholar]
- 25.Pedrini MT, Levey AS, Lau J, Chalmers TC, Wang PH: The effect of dietary protein restriction on the progression of diabetic and nondiabetic renal diseases: a meta-analysis. Ann Intern Med 124: 627–632, 1996 [DOI] [PubMed] [Google Scholar]
- 26.National Kidney Foundation : 2012 Update. Am J Kidney Dis 60: 850–886, 2012 [DOI] [PubMed] [Google Scholar]
- 27.Remer T, Manz F: Estimation of the renal net acid excretion by adults consuming diets containing variable amounts of protein. Am J Clin Nutr 59: 1356–1361, 1994 [DOI] [PubMed] [Google Scholar]
- 28.Remer T: Influence of nutrition on acid-base balance—metabolic aspects. Eur J Nutr 40: 214–220, 2001 [DOI] [PubMed] [Google Scholar]
- 29.Wesson DE, Nathan T, Rose T, Simoni J, Tran RM: Dietary protein induces endothelin-mediated kidney injury through enhanced intrinsic acid production. Kidney Int 71: 210–217, 2007 [DOI] [PubMed] [Google Scholar]
- 30.Wesson DE, Simoni J: Increased tissue acid mediates a progressive decline in the glomerular filtration rate of animals with reduced nephron mass. Kidney Int 75: 929–935, 2009 [DOI] [PubMed] [Google Scholar]
- 31.Khanna A, Simoni J, Hacker C, Duran MJ, Wesson DE: Increased endothelin activity mediates augmented distal nephron acidification induced by dietary protein. J Am Soc Nephrol 15: 2266–2275, 2004 [DOI] [PubMed] [Google Scholar]
- 32.Khanna A, Simoni J, Wesson DE: Endothelin-induced increased aldosterone activity mediates augmented distal nephron acidification as a result of dietary protein. J Am Soc Nephrol 16: 1929–1935, 2005 [DOI] [PubMed] [Google Scholar]
- 33.Biver S, Belge H, Bourgeois S, Van Vooren P, Nowik M, Scohy S, Houillier P, Szpirer J, Szpirer C, Wagner CA, Devuyst O, Marini AM: A role for Rhesus factor Rhcg in renal ammonium excretion and male fertility. Nature 456: 339–343, 2008 [DOI] [PubMed] [Google Scholar]
- 34.Wagner CA, Devuyst O, Belge H, Bourgeois S, Houillier P: The rhesus protein RhCG: A new perspective in ammonium transport and distal urinary acidification. Kidney Int 79: 154–161, 2011 [DOI] [PubMed] [Google Scholar]
- 35.Weiner ID, Hamm LL: Molecular mechanisms of renal ammonia transport. Annu Rev Physiol 69: 317–340, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bourgeois S, Bounoure L, Christensen EI, Ramakrishnan SK, Houillier P, Devuyst O, Wagner CA: Haploinsufficiency of the ammonia transporter Rhcg predisposes to chronic acidosis: Rhcg is critical for apical and basolateral ammonia transport in the mouse collecting duct. J Biol Chem 288: 5518–5529, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee HW, Verlander JW, Bishop JM, Igarashi P, Handlogten ME, Weiner ID: Collecting duct-specific Rh C glycoprotein deletion alters basal and acidosis-stimulated renal ammonia excretion. Am J Physiol Renal Physiol 296: F1364–F1375, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee HW, Verlander JW, Bishop JM, Nelson RD, Handlogten ME, Weiner ID: Effect of intercalated cell-specific Rh C glycoprotein deletion on basal and metabolic acidosis-stimulated renal ammonia excretion. Am J Physiol Renal Physiol 299: F369–F379, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morris SM, Jr: Regulation of enzymes of urea and arginine synthesis. Annu Rev Nutr 12: 81–101, 1992 [DOI] [PubMed] [Google Scholar]
- 40.Sicuro A, Mahlbacher K, Hulter HN, Krapf R: Effect of growth hormone on renal and systemic acid-base homeostasis in humans. Am J Physiol 274: F650–F657, 1998 [DOI] [PubMed] [Google Scholar]
- 41.Wrong O, Davies HE: The excretion of acid in renal disease. Q J Med 28: 259–313, 1959 [PubMed] [Google Scholar]
- 42.Busque SM, Wagner CA: Potassium restriction, high protein intake, and metabolic acidosis increase expression of the glutamine transporter SNAT3 (Slc38a3) in mouse kidney. Am J Physiol Renal Physiol 297: F440–F450, 2009 [DOI] [PubMed] [Google Scholar]
- 43.Peret J, Chanez M: Influence of diet, cortisol and insulin on the activity of pyruvate carboxylase and phosphoenolpyruvate carboxykinase in the rat liver. J Nutr 106: 103–110, 1976 [DOI] [PubMed] [Google Scholar]
- 44.Seshadri RM, Klein JD, Smith T, Sands JM, Handlogten ME, Verlander JW, Weiner ID: Changes in subcellular distribution of the ammonia transporter, Rhcg, in response to chronic metabolic acidosis. Am J Physiol Renal Physiol 290: F1443–F1452, 2006 [DOI] [PubMed] [Google Scholar]
- 45.Seshadri RM, Klein JD, Kozlowski S, Sands JM, Kim YH, Han KH, Handlogten ME, Verlander JW, Weiner ID: Renal expression of the ammonia transporters, Rhbg and Rhcg, in response to chronic metabolic acidosis. Am J Physiol Renal Physiol 290: F397–F408, 2006 [DOI] [PubMed] [Google Scholar]
- 46.Chambrey R, Goossens D, Bourgeois S, Picard N, Bloch-Faure M, Leviel F, Geoffroy V, Cambillau M, Colin Y, Paillard M, Houillier P, Cartron JP, Eladari D: Genetic ablation of Rhbg in the mouse does not impair renal ammonium excretion. Am J Physiol Renal Physiol 289: F1281–F1290, 2005 [DOI] [PubMed] [Google Scholar]
- 47.Bishop JM, Lee HW, Handlogten ME, Han KH, Verlander JW, Weiner ID: Intercalated cell-specific Rh B glycoprotein deletion diminishes renal ammonia excretion response to hypokalemia. Am J Physiol Renal Physiol 304: F422–F431, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karim Z, Attmane-Elakeb A, Sibella V, Bichara M: Acid pH increases the stability of BSC1/NKCC2 mRNA in the medullary thick ascending limb. J Am Soc Nephrol 14: 2229–2236, 2003 [DOI] [PubMed] [Google Scholar]
- 49.Szutkowska M, Vernimmen C, Debaix H, Devuyst O, Friedlander G, Karim Z: Zeta-crystallin mediates the acid pH-induced increase of BSC1 cotransporter mRNA stability. Kidney Int 76: 730–738, 2009 [DOI] [PubMed] [Google Scholar]
- 50.Attmane-Elakeb A, Mount DB, Sibella V, Vernimmen C, Hebert SC, Bichara M: Stimulation by in vivo and in vitro metabolic acidosis of expression of rBSC-1, the Na+-K+(NH4+)-2Cl- cotransporter of the rat medullary thick ascending limb. J Biol Chem 273: 33681–33691, 1998 [DOI] [PubMed] [Google Scholar]
- 51.Laghmani K, Richer C, Borensztein P, Paillard M, Froissart M: Expression of rat thick limb Na/H exchangers in potassium depletion and chronic metabolic acidosis. Kidney Int 60: 1386–1396, 2001 [DOI] [PubMed] [Google Scholar]
- 52.Nowik M, Kampik NB, Mihailova M, Eladari D, Wagner CA: Induction of metabolic acidosis with ammonium chloride (NH4Cl) in mice and rats—species differences and technical considerations. Cell Physiol Biochem 26: 1059–1072, 2010 [DOI] [PubMed] [Google Scholar]
- 53.Mouri T, Inoue T, Nonoguchi H, Nakayama Y, Miyazaki H, Matsuzaki T, Saito H, Nakanishi T, Kohda Y, Tomita K: Acute and chronic metabolic acidosis interferes with aquaporin-2 translocation in the rat kidney collecting ducts. Hypertens Res 32: 358–363, 2009 [DOI] [PubMed] [Google Scholar]
- 54.Rodríguez-Iturbe B, Herrera J, Gutkowska J, Parra G, Coello J: Atrial natriuretic factor increases after a protein meal in man. Clin Sci (Lond) 75: 495–498, 1988 [DOI] [PubMed] [Google Scholar]
- 55.Yanagisawa H, Wada O: Effects of dietary protein on eicosanoid production in rat renal tubules. Nephron 78: 179–186, 1998 [DOI] [PubMed] [Google Scholar]
- 56.Bailly C: Effect of luminal atrial natriuretic peptide on chloride reabsorption in mouse cortical thick ascending limb: Inhibition by endothelin. J Am Soc Nephrol 11: 1791–1797, 2000 [DOI] [PubMed] [Google Scholar]
- 57.Klokkers J, Langehanenberg P, Kemper B, Kosmeier S, von Bally G, Riethmüller C, Wunder F, Sindic A, Pavenstädt H, Schlatter E, Edemir B: Atrial natriuretic peptide and nitric oxide signaling antagonizes vasopressin-mediated water permeability in inner medullary collecting duct cells. Am J Physiol Renal Physiol 297: F693–F703, 2009 [DOI] [PubMed] [Google Scholar]
- 58.Bushinsky DA, Smith SB, Gavrilov KL, Gavrilov LF, Li J, Levi-Setti R: Chronic acidosis-induced alteration in bone bicarbonate and phosphate. Am J Physiol Renal Physiol 285: F532–F539, 2003 [DOI] [PubMed] [Google Scholar]
- 59.Lemann J, Jr, Bushinsky DA, Hamm LL: Bone buffering of acid and base in humans. Am J Physiol Renal Physiol 285: F811–F832, 2003 [DOI] [PubMed] [Google Scholar]
- 60.Gafter U, Kraut JA, Lee DB, Silis V, Walling MW, Kurokawa K, Haussler MR, Coburn JW: Effect of metabolic acidosis in intestinal absorption of calcium and phosphorus. Am J Physiol 239: G480–G484, 1980 [DOI] [PubMed] [Google Scholar]
- 61.Slot C: Plasma creatinine determination. A new and specific Jaffe reaction method. Scand J Clin Lab Invest 17: 381–387, 1965 [DOI] [PubMed] [Google Scholar]
- 62.Berthelot M: Violet d'aniline. Rep Chim App 1: 284, 1859 [Google Scholar]
- 63.Moret C, Dave MH, Schulz N, Jiang JX, Verrey F, Wagner CA: Regulation of renal amino acid transporters during metabolic acidosis. Am J Physiol Renal Physiol 292: F555–F566, 2007 [DOI] [PubMed] [Google Scholar]
- 64.Burghardt AJ, Kazakia GJ, Laib A, Majumdar S: Quantitative assessment of bone tissue mineralization with polychromatic micro-computed tomography. Calcif Tissue Int 83: 129–138, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Müller R: Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res 25: 1468–1486, 2010 [DOI] [PubMed] [Google Scholar]
- 66.Kohler T, Stauber M, Donahue LR, Müller R: Automated compartmental analysis for high-throughput skeletal phenotyping in femora of genetic mouse models. Bone 41: 659–667, 2007 [DOI] [PubMed] [Google Scholar]
- 67.Lukas C, Ruffoni D, Lambers FM, Schulte FA, Kuhn G, Kollmannsberger P, Weinkamer R, Müller R: Mineralization kinetics in murine trabecular bone quantified by time-lapsed in vivo micro-computed tomography. Bone 56: 55–60, 2013 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.