

Abstract
The endoplasmic reticulum (ER) stress response is activated in the diabetic kidney and functions to reduce ER protein accumulation and improve cellular function. We previously showed that tribbles homolog 3 (TRB3), an ER stress-associated protein, is upregulated in the diabetic kidney. Here, we investigated whether absence of TRB3 alters outcomes in diabetic nephropathy. Type 1 diabetes was induced in TRB3 wild-type and knockout (−/−) mice by low-dose streptozotocin, and the mice were followed for 12 weeks. Diabetic TRB3−/− mice developed higher levels of albuminuria and increased expression of inflammatory cytokine and chemokine mRNA in renal cortices relative to wild-type littermates, despite similar hyperglycemia. Diabetic TRB3−/− mice also expressed higher levels of ER stress-associated molecules in both the renal cortices and glomeruli. This change was associated with higher renal cortical phosphorylation of AKT at serine 473 (Ser473), which is the AKT site phosphorylated by mammalian target of rapamycin complex-2 (mTORC2). We show in renal tubular cells that TRB3 binds to mTOR and the rapamycin-insensitive companion of mTOR (Rictor), a protein specific to mTORC2. Finally, we demonstrate in murine tubular cells that TRB3 can inhibit secretion of IL-6. Thus, TRB3 reduces albuminuria and inflammatory gene expression in diabetic kidney disease by a mechanism that may involve inhibition of the mTORC2/AKT pathway and may prove to be a novel therapeutic target.
Diabetic nephropathy is the most common cause of CKD and a growing public health challenge.1 Clinical treatment is limited, emphasizing the importance of developing new therapeutic strategies that target novel signaling pathways. The diabetic milieu promotes the accumulation of misfolded proteins in the endoplasmic reticulum (ER), which alters optimal cellular function. In the diabetic kidney, we and others have shown that the ER stress response is activated2 to repress protein synthesis and increase ER chaperone content to alleviate ER protein accumulation.2
Tribbles homolog 3 (TRB3) is a pseudokinase that is upregulated during the ER stress response.3,4 TRB3 lacks kinase activity, and in the liver it binds and inhibits the activation of AKT/protein kinase.3 We have shown that TRB3 suppresses inflammatory pathways by inhibiting monocyte chemokine protein-1 (MCP-1) expression in podocytes.4 TRB3 is expressed ubiquitously, functions as a scaffold protein to inhibit mitogen-activated protein kinase signaling cascades,5 and reduces the activation of downstream inflammatory transcription factors, including NF-κB and activator protein-1.5–7 In humans, a polymorphism of TRB3 is associated with insulin resistance and cardiovascular risk.8 Not surprisingly, TRB3 may be an attractive target for modulating insulin resistance and the complications of diabetes.9
Manipulation of ER stress-associated molecules, including chemical chaperones that improve ER folding, can reduce ER stress and improve manifestations of diabetic nephropathy.2,10 Here we demonstrate that in diabetic mice constitutive knockout of TRB3 increases albuminuria and renal cortical mRNA expression of proinflammatory cytokines and chemokines relative to wild-type (WT) littermates in the streptozotocin (STZ)-induced model of diabetes. Expression of ER stress-associated molecules was higher in the renal cortices and glomeruli of the diabetic TRB3−/− mice. Absence of TRB3 is associated with augmented phosphorylation of AKT at residue serine 473 (Ser473). To our knowledge, this is the first study to show that TRB3 may inhibit the phosphorylation of AKT by binding to mammalian target of rapamycin (mTOR) and rapamycin-insensitive companion of mTOR (Rictor), components of the mTOR complex 2 (mTORC2). Thus, TRB3 may provide a new molecular target for the treatment of diabetic kidney disease through its actions on mTORC2.
Results
In Diabetic Mice TRB3 Knockout Increases Albuminuria
TRB3 knockout (−/−) mice were healthy and born in normal Mendelian ratios. Body weight was similar in the two treatment groups at the time of diabetes induction (mean weight±SD, 25.3±0.5 g in control mice versus 24.6±0.4 in TRB3−/− mice; P=NS; n=37 total mice). We induced diabetes by low-dose STZ injection in WT and TRB3−/− mice (control mice were injected with citrate), and mice were followed for 12 weeks after STZ injection. STZ-injected mice developed similar levels of hyperglycemia (Figure 1A). Twenty-four-hour urine collections were performed and the diabetic TRB3−/− mice had significantly higher urine albumin excretion and urinary albumin-to-creatinine ratios (Figure 1, B and C) than the diabetic WT mice.
Figure 1.
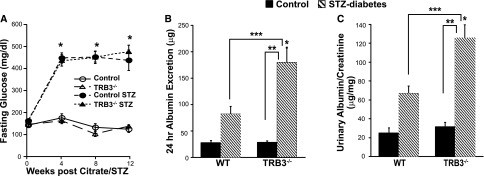
TRB3 knockout does not affect hyperglycemia but increases albuminuria in diabetic mice. (A) Diabetes was induced in WT and TRB3−/− mice by low-dose STZ. Blood glucose levels were significantly elevated in both STZ-treated groups. *P<0.05 versus control and TRB3−/− mice (no STZ). At 12 weeks after STZ administration, the diabetic TRB3−/− mice had significantly higher (B) 24-hour albuminuria and (C) urinary albumin-to-creatinine ratios. *P<0.05 versus WT control; **P<0.05 versus TRB3−/− control; ***P<0.05 versus STZ WT. Bars represent SEM; n=5 WT control, n=6 TRB3−/− control, n=9 WT STZ, n=17 TRB3−/− STZ.
The mice were euthanized 12 weeks after STZ injection. Body weight was similarly reduced in both diabetic groups compared with nondiabetic controls (Table 1). Kidney weights were normalized to body weight, and both the diabetic WT and TRB3−/− mice had heavier kidneys than the nondiabetic mice. There was a modest trend for higher serum creatinine levels in the diabetic TRB3−/− mice (Table 1).
Table 1.
Characterization of experimental mice 12 weeks after STZ administration
| Variable | WT Control | WT STZ | TRB3−/− Control | TRB3−/− STZ |
|---|---|---|---|---|
| Body weight (g) | 29.3±0.6 | 26.2±0.6a | 29.1±1.2 | 25.8±0.5a,b |
| Kidney weight (mg) | 379±4.0 | 405±10.4 | 372±12.5 | 406±15.7 |
| Kidney weight/body weight (mg/g) | 12.9±0.3 | 15.5±0.5a | 12.8±0.3 | 15.9±0.6a,b |
| Serum creatinine (mg/dl) | 0.087 +/− 0.015 | 0.091±0.008 | 0.093±0.007 | 0.103±0.005 |
Values are expressed as mean±SEM.
P<0.05 versus WT control.
P<0.05 versus TRB3−/− control.
In Diabetic Mice TRB3 Knockout Does Not Markedly Alter Glomerular Histologic Features
Morphometry studies revealed no significant differences in the periodic acid-Schiff (PAS)–positive matrix in the mesangium (Figure 2, A–E), glomerular nuclear number (Figure 2F), or glomerular area (Figure 2G) in response to diabetes or due to the absence of TRB3.
Figure 2.
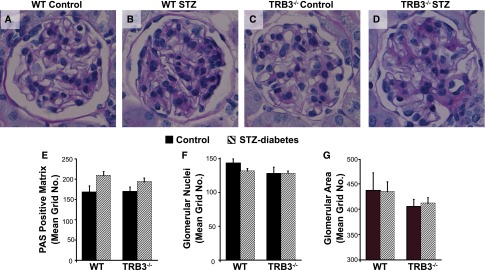
In diabetic mice, absence of TRB3 does not markedly alter glomerular histologic features. Morphometry was performed on PAS-stained kidney sections. (A) WT control (B) WT STZ, (C) TRB3−/− control, and (D) TRB3−/− STZ-treated mice. (E) PAS-positive matrix, (F) glomerular nuclei, and (G) glomerular area were assessed as described in Concise Methods. n=4 WT control, n=5 TRB3−/− control, n=5 WT STZ, n=6 TRB3−/− STZ. Bars represent SEM.
Effect of TRB3 Knockout on Renal Gene Expression
Similar to our previous studies,4 TRB3 expression in the renal cortices of the WT diabetic mice increased significantly compared with nondiabetic controls. As expected, expression of TRB3 in the TRB3−/− mice was very low (0.009 versus 3.0 relative units; diabetic TRB3−/− versus diabetic WT mice; Figure 3A). To evaluate the effect of absence of TRB3 on candidate genes involved in renal inflammation and fibrosis, the mRNA expression of proinflammatory and fibrotic markers was measured by real-time PCR and diabetic mice were compared with their nondiabetic controls. We observed higher MCP-1 (Figure 3B), IL-6 (Figure 3C), Lcn2/Ngal (Figure 3D), and fibronectin mRNA (Figure 3E) in the diabetic TRB3−/− mice than in diabetic WT mice. TGF-β expression did not significantly differ (Figure 3F). In parallel we assessed IL-6 (at 4 weeks) and MCP-1 and Lcn2/Ngal (at 8 weeks) in 24-hour urine and observed higher IL-6 in both diabetic WT and TRB3−/− mice compared with their nondiabetic controls. There was a trend for higher IL-6, MCP-1, and Lcn2/Ngal expression in the urine of the diabetic TRB3−/− mice compared with the diabetic WT mice, but this did not achieve statistical significance (Figure 3, J–L).
Figure 3.

Diabetic TRB3 knockout mice have higher renal inflammatory gene expression and evidence of ER stress than WT controls. Real-time PCR on kidney cortices comparing diabetic with nondiabetic controls revealed the following: (A) increased TRB3 expression in the diabetic WT mice compared with nondiabetic WT mice and higher (B) MCP-1, (C) IL-6, (D) Lcn2/Ngal, and (E) fibronectin expression in diabetic TRB3−/− mice than in WT diabetic mice. (F) There was no difference in TGF-β mRNA expression in the diabetic WT and TRB3−/− mice. (G–I) There was higher renal cortical expression of the ER stress markers (GRP78/BiP and ATF4) and a trend for higher CHOP in the diabetic TRB3−/− mice. In both genotypes, there was higher excretion of (J) the early-response cytokine IL-6 in 24-hour urine collected 4 weeks after STZ administration and (K) MCP-1 and (L) Lcn2/Ngal in urine collected at 8 weeks after diabetes induction. *P<0.05 versus WT control mice, **P<0.05 versus TRB3−/− control mice; n=5 WT control, n=5 TRB3−/− control, n=7 WT STZ, n=12 TRB3−/− STZ. Bars represent SEM.
Absence of TRB3 Augments ER Stress in Diabetic Mice
Assessment of mRNA expression of key ER stress-associated molecules in the renal cortices revealed higher GRP78/BiP and activating transcription factor-4 (ATF4) and a trend for augmented C/EBP homologous protein (CHOP) in the diabetic TRB3−/− mice compared with their WT controls (Figure 3, G–I).
We have previously shown that ER stress inducers (thapsigargin, tunicamycin), oxidative stress, and palmitate augment TRB3 expression in podocytes.4 Supplemental Figure 1 demonstrates that palmitate, the most abundant free fatty acid in the sera of diabetic patients, potently and rapidly upregulates TRB3 and other ER stress-associated factors, including ATF4, CHOP, phosphorylation of eukaryotic translation initiation factor 2α, and splicing of X-box binding protein-1 in podocytes. Next, we isolated glomeruli and again observed a significant upregulation of TRB3 mRNA expression in the glomeruli derived from WT diabetic mice compared with their controls (Figure 4A). Figure 4, B–D, demonstrates a significant increase in GRP78/BiP, CHOP, and Lcn2/Ngal in the glomeruli of the diabetic TRB3−/− mice compared with the WT diabetic mice.
Figure 4.

Absence of TRB3 augments ER stress in the glomeruli of diabetic mice. Real-time PCR on glomerular preparations comparing diabetic with nondiabetic controls revealed expression of (A) TRB3, (B) GRP78/BiP, (C) Lcn2/Ngal, and (D) CHOP mRNA. *P<0.05 versus diabetic WT mice; n=4 WT control, n=3 TRB3−/− control, n=5 WT STZ, n=3 TRB3−/− STZ. Bars represent SEM.
Podocytes and tubular cells secrete Lcn2/Ngal.11 To localize the site of Lcn2/Ngal production, we performed in situ RNA hybridization and observed glomerular expression of Lcn2/Ngal in the diabetic WT and TRB3−/− mice (Supplemental Figure 2).
Phosphorylation of AKT at Ser473 Is Enhanced in Renal Cortices of Diabetic TRB3−/− mice
AKT is activated and phosphorylated at Ser473 and Thr308, and studies have shown that in certain cell types TRB3 inhibits the phosphorylation of AKT.3,12 Therefore, we evaluated phosphorylation of AKT in the renal cortices of the experimental mice. Indeed, we observed higher phosphorylation of AKT at Ser473 in the TRB3−/− diabetic mice compared with the WT diabetic mice (Figure 5, A and B). In contrast, phosphorylation of AKT at Thr308 was significantly reduced in the diabetic TRB3−/− mice compared with the diabetic WT mice (Figure 5, A and C).
Figure 5.

Knockout of TRB3 increases phosphorylation of AKT at Ser473 and reduces the phosphorylation at Thr308 in renal cortices of diabetic mice. (A) Western blotting of pAKTSer473, pAKTThr308, and total AKT in the experimental mice. Densitometry of (B) pAKTSer473/AKT and (C) pAKTThr308/AKT. *P<0.05 versus WT control mice, **P<0.05 versus TRB3−/− control mice; n=5 WT control, n=5 TRB3−/− control, n=8 WT STZ, n=8 TRB3−/− STZ. Bars represent SEM.
TRB3 May Inhibit Phosphorylation of AKT at Ser473 by Binding to Rictor and mTORC2
To investigate how TRB3 inhibits the phosphorylation of AKT, we transfected transformed podocytes with pcDNA3-HA (hemagglutinin)-TRB3 (HA-tagged TRB3 expression vector) and consistently observed a reduction in phosphorylation of pAKTSer473 (Figure 6, A and B). Studies have suggested that TRB3 inhibits AKT by binding and preventing its phosphorylation,3 and recent work has suggested that phosphorylation of AKT at Ser473 is mediated by the Rictor-mTORC2 complex.13 Therefore, we transfected both podocytes and murine proximal tubular cells (MCTs) with pcDNA3-HA-TRB3 or the plasmid control (pcDNA3) and immunoprecipitated the TRB3-containing complexes with anti-HA beads. Figure 6C illustrates our results in MCT cells, which presented more efficient transfection of HA-TRB3 than podocytes. Our studies demonstrate that TRB3 binds to Rictor and mTOR. In contrast to previous studies,3 we did not observe direct binding of TRB3 to AKT or the phosphorylated forms of AKT (Figure 6C).
Figure 6.

TRB3 may inhibit phosphorylation of AKTSer473 by binding to Rictor and mTORC2. (A) Fully differentiated transformed podocytes were transfected with pcDNA3-HA-TRB3 (HA-TRB3), which consistently reduced phosphorylation of AKT at Ser473 (pAKTSer473) (B) Densitometry results of transfection of HA-TRB3 or pcDNA3 in transformed podocytes (study performed three times). (C) MCT cells were transfected with HA-TRB3 or pcDNA3, and the TRB3-containing complexes were immunoprecipitated with anti-HA beads. TRB3 binds to Rictor and mTOR, but not directly to AKT or pAKT. This study is representative of studies performed three times. (D) Primary podocytes were grown from WT and TRB3−/− mice. Western blotting revealed higher phosphorylation of pAKTSer473 in primary podocytes grown from TRB3−/− mice (study is representative of Western blotting performed twice). (E) MCT cells were transfected with pcDNA3 or HA-TRB3, and Western blotting for targets of mTORC2 and AKT was assessed. Bars represent SEM.
In primary podocytes grown from TRB3−/− mice, phosphorylation of pAKTSer473 was higher and, surprisingly, phosphorylation of AKTThr308 increased modestly (Figure 6D). Mammalian TORC2 has many downstream targets, including signal transducer and activator of transcription 3 (STAT3),14 and Forkhead box protein/forkhead in rhabdomyosarcoma O1 (FOXO1).15 In tubular cells transfected with TRB3, we observed lower phosphorylation of AKTSer473, STAT3Tyr705 and FOXO1Thr24, but TRB3 did not inhibit the phosphorylation of another AKT target GSK3α/β (Figure 6E).
TRB3 Inhibits Expression of IL-6 in Tubular Cells
We have previously shown that TRB3 inhibits expression of the chemoattractant MCP-1 in transformed podocytes.4 IL-6, like MCP-1, is proinflammatory in diabetic kidney disease. Here we transfected MCTs with pcDNA3-HA-TRB3 or pcDNA3 and assessed supernatant IL-6 expression. Figure 7A demonstrates lower IL-6 secreted by TRB3-transfected cells, in both normal-glucose (5.5 mM) and high-glucose conditions (30 mM). Thus, TRB3 inhibits basal expression of IL-6, and this effect appears to be independent of high-glucose conditions. In Figure 7B, we normalized IL-6 to total supernatant protein and again observed lower IL-6 expression in the cells overexpressing TRB3. Interestingly, IL-6 mRNA expression decreased 20% in the TRB3-transfected cells, suggesting that TRB3 alters IL-6 expression at both the mRNA and protein levels (data not shown).
Figure 7.

TRB3 inhibits IL-6 expression in MCT cells. MCT cells were transfected with pcDNA3 or pcDNA3-HA-TRB3 and grown in normal-glucose (5.5 mM) and high-glucose (30.5 mM) conditions. Eighteen hours after transfection, supernatants were collected, and ELISAs detected (A) IL-6 expression pg/ml supernatant and (B) IL-6 pg/mg (protein). *P<0.05 versus normal glucose, pcDNA3; **P<0.05 versus pcDNA3 high glucose. This is representative of studies performed at least three times. Bars represent SEM.
Discussion
We provide the first direct evidence that TRB3 reduces albuminuria and inflammatory gene expression in diabetic kidney disease. Moreover, absence of TRB3 increases ER stress in the diabetic kidney. In accordance with TRB3 blocking the phosphorylation of the signal transducer AKT at Ser473 residues, we show enhanced renal cortical phosphorylation of AKT at Ser473 residues in diabetic TRB3 knockout mice. We also demonstrate that TRB3 binds to Rictor and mTOR and reduces the phosphorylation of other downstream targets of mTORC2, including STAT3 and FOXO-1.
AKT is a serine-threonine kinase that regulates multiple cellular processes, including glucose uptake, proliferation, differentiation, inflammation, and survival. Early studies suggested that full activation of AKT requires phosphorylation of the activation loop (Thr308 residues) by 3-phosphoinositide–dependent protein kinase-1 and phosphorylation at the hydrophobic motif (HM; Ser473) by mTORC2.13,16 Phosphorylation of AKT at Thr450 residues may also affect kinase activity and stability.17,18 However, recent studies suggest that downstream substrates may require different levels of AKT activity and that dual phosphorylation of AKT at the activation and HM may not be required for all of AKT actions.19,20 Indeed, differential phosphorylation of AKT on the HM and activation loop may confer substrate specificity and facilitate splitting of the upstream insulin signal into discrete outputs.21–23
In cortices of the diabetic kidney, angiotensin II and reactive oxygen species induce the phosphorylation of AKT.24–29 Most studies used nonspecific phospho-AKT antibodies; thus, differential phosphorylation of AKT at the HM and activation loops has not been rigorously evaluated in the kidney. Our study indicates that TRB3 differentially affects the phosphorylation of AKT. TRB3 consistently reduces the phosphorylation of AKTSer473. In contrast, in vitro TRB3 has a variable effect on the phosphorylation of AKT at Thr308 residues (Figure 6, C–E), but we and others have not observed higher phosphorylation of AKTThr308 associated with TRB3 expression.30 Thus, the lower phosphorylation of AKTThr308 in the renal cortices of the diabetic TRB3 knockout mice is likely an indirect effect of TRB3 knockout. These findings are reminiscent of studies by Kiss-Toth and colleagues, demonstrating that TRB3 can function as both an activator or inhibitor of mitogen-activated protein kinase activity; they hypothesize that this depends on the ratio of TRB3 to its substrates.5 Additionally, TRB3 inhibits p70 S6 kinase activity.31 We observed higher phosphorylation of p70 S6 kinase in the renal cortices of the diabetic TRB3−/− mice (not shown), which can reduce insulin-related substrate-1 activity and subsequent activation of 3-phosphoinositide–dependent protein kinase-1, which phosphorylates AKTThr308.32,33 Alternatively, TRB3 could regulate Pleckstrin homology domain leucine-rich repeat protein phosphatase, which dephosphorylates AKT at its hydrophobic motif34; another possibility is that GRP78/BiP induced by ER stress also regulates phosphorylation of AKT.35 We are currently investigating these hypotheses.
We observed augmented expression of ER stress markers in the glomeruli and renal cortices of diabetic TRB3−/− knockout mice. This is not surprising because chemical chaperones that modulate ER stress attenuate diabetic nephropathy,10,36 and absence of a key endogenous chaperone can impair the protective and recovery phases of ER stress. ER stress induces inflammation,37 and the diabetic TRB3−/− mice had higher expression of IL-6, MCP-1, and Lcn2/Ngal. High levels of IL-6 have been observed in rodent models38–42 and patients with diabetic nephropathy and are likely pathogenic.43–46 Interestingly, AKT can induce IL-6 expression through NF-κB–dependent pathways.47–49 Alternatively, IL-6 can activate AKT,50,51 driving a positive-feedback loop. In podocytes and tubular cells, high-glucose conditions increase IL-6 expression, which promotes apoptosis and tubular inflammation,52,53 although we did not observe more apoptosis in the tubular or glomerular regions of the diabetic TRB3−/− mice (not shown). TRB3 reduced expression of IL-6 (Figure 7), similar to its effects on MCP-1.4 It is likely that the higher albuminuria observed in the TRB3−/− mice was related to augmented expression of inflammatory cytokines and chemokines.
Lcn2/Ngal is upregulated by ER stress and is considered an ER stress marker.54,55 In situ RNA hybridization studies demonstrate glomerular expression of Lcn2/Ngal in the diabetic WT and TRB3−/− mice (Supplemental Figure 2). Although contribution from both tubular and glomerular compartments is likely, our studies suggest that glomerular production of Lcn2/Ngal is probably higher than tubular expression. ER stress was augmented in the glomeruli and tubules, suggesting that they are both sensitive to ER stress. The relevance of podocyte production of Lcn2/Ngal remains unknown, and it will be interesting to determine whether it independently regulates ER stress.
We used the STZ model of diabetes because we first demonstrated upregulation of TRB3 in the kidneys of these mice.4 It is the most commonly used model of type 1 diabetes and has been most studied in relation to the association between ER stress and diabetic kidney disease. Albuminuria is an independent risk factor for all-cause and cardiovascular mortality and a significant risk factor for ESRD, AKI, and progressive kidney disease.56–58 Although the changes are subtle, we argue that they are clinically relevant.
AKT transduces insulin receptor signals; thus, blockade of AKT by TRB3 could cause insulin resistance.59 However, in our study of type 1 diabetes, fasting glucose in the diabetic and nondiabetic WT and TRB3−/− mice were similar, in keeping with a previously published study in another strain of TRB3−/− mice.60 Recently, two groups have shown that partial TRB3 gene silencing (by RNAi technology) alleviates diabetic cardiomyopathy and atherosclerosis. In both studies the efficiency of TRB3 knockout in the heart and aorta was about 70%, and serum glucose levels were dramatically improved. Thus, improvements in diabetic cardiomyopathy and atherosclerosis in type 2 diabetes could have been related to improved metabolic control.61,62
mTOR is a conserved serine/threonine kinase modulated by growth factors and cellular energy status. It forms two distinct molecular complexes known as mTOR complex 1 (mTORC1) and mTORC2. mTORC1 regulates growth, autophagy, survival, and metabolism, whereas the role of mTORC2 in cellular biology is incompletely understood. Sarbassov elegantly demonstrated that the Rictor mTORC2 directly phosphorylates AKT/PKB at Ser473 residues.13 Before the discovery that mTORC2 phosphorylates AKT at Ser473, studies suggested that TRB3 directly binds to AKT.3 Because we observed differential effects of TRB3 on phosphorylation of AKT at Ser473 and Thr308, we hypothesized that TRB3 binds to mTORC2. Indeed, as shown in Figure 6 we report for the first time that TRB3 binds to both Rictor and mTOR, and not directly to AKT.
Activation of mTORC1 in podocytes promotes the development of diabetic nephropathy.63,64 However, the effect of mTORC2 activation in diabetic kidney disease remains unknown. In addition to regulating the phosphorylation of AKT at Ser473, mTORC2 also phosphorylates STAT3,14 FOXO-1,15 serum/glucocorticoid-regulated kinase-165,66 and protein kinase C and regulates actin cytoskeletal dynamics, metabolism, motility, survival; our studies suggest that it also affects kidney inflammation.67,68 Exogenously expressed TRB3 reduces the phosphorylation of STAT3 and FOXO-1 but did not have an effect on other AKT targets, including GSK3α/β (Figure 6E). Lower phosphorylation of STAT3 in STZ-induced diabetes reduces albuminuria and inflammation,39 and FOXO-1 increases expression of catalase, thereby reducing proinflammatory H2O2.69 In stress-associated conditions, podocyte-specific knockout of Rictor (a component of mTORC2) causes proteinuria,64 and in podocytes curcumin inhibits the phosphorylation of Akt at Ser473 and reduces albumin permeability.70 These studies all support the importance of tight regulation of mTORC2 and AKT phosphorylation. Similar to our study, recent work suggests that inhibition of mTORC2 function inhibits inflammation in rodent inflammatory models71 and reduces IL-6 expression in stem cells.72 Thus, TRB3 may provide a novel means of blocking aberrant mTORC2 function in chronic inflammatory and neoplastic diseases (Figure 8).
Figure 8.

Proposed mechanisms of protective role of TRB3 in diabetic kidney disease. The diabetic milieu activates the ER stress pathway, including protein kinase RNA–like ER kinase (PERK). PERK phosphorylates eukaryotic translation initiation factor-α to inhibit protein translation with selective translation of ATF4, which induces expression of CHOP and drives TRB3 expression. We demonstrate that in kidney cells TRB3 binds to Rictor and mTORC2 to inhibit phosphorylation of pAKTSer473, which can reduce inflammatory gene expression, including IL-6, MCP-1, and Lcn2/Ngal. ROS, reactive oxygen species.
In conclusion, our study demonstrates that TRB3 reduces albuminuria in the diabetic kidney, and this may be related to its ability to block mTORC2 function, modulate ER stress and reduce inflammatory mediators of renal disease. As our understanding of mTORC2 function grows, augmentation of TRB3 expression may be a novel therapeutic target in diabetic kidney disease.
Concise Methods
We obtained TRB3−/− mice from Dr. Shizuo Akira’s laboratory at Osaka University in Japan. The targeting vector was prepared by replacing exons 2 and 3 of TRB3 with a neomycin-resistance gene cassette (Figure 9).73 The TRB3−/− mice were obtained by homologous recombination in micro-injected mice, and heterozygous F1 progeny were backcrossed more than eight times onto a C57BL/6 background. Mice were housed and handled in accordance with Department of Veterans Affairs and National Institutes of Health guidelines under Institutional Animal Care and Use Committee–approved protocols. Type 1 diabetes was induced in 10-week-old TRB3−/− and littermate WT male mice by low-dose STZ injection (50 mg/kg×5 days; n=9 WT, n=17 TRB3−/−), and controls were injected with sodium citrate buffer (n=5 WT controls, n=6 TRB3−/−). Blood glucose levels were assessed at baseline and every 4 weeks, and mice were euthanized 12 weeks after induction of type 1 diabetes. Twenty-four-hour urine collections were performed every 4 weeks in metabolic cages after appropriate acclimatization. Urinary albumin and creatinine levels were assessed by ELISA using the Albuwell M and Creatinine Companion kits (Exocell, Philadelphia, PA). Serum creatinine was assessed by HPLC.74
Figure 9.
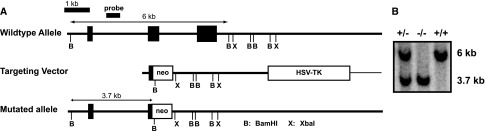
Strategy to generate TRB3 knockout mouse. (A) Representation of targeted disruption of the mouse TRB3 gene. (B) Southern blot of BamHl digested genomic DNA from TRB3+/− −/− and +/+ mice.
Histology
PBS-perfused kidneys were fixed in 10% formalin, sectioned, and stained with PAS. Twenty randomly selected glomeruli in the outer cortex of each kidney section were evaluated in a blinded manner. An image of each glomerulus was overlaid with grids. Each grid intersection was determined for the following features: PAS positive mesangial area and nuclear number. The accumulation of all the grid intersections was calculated for overall glomerular area.75 In situ RNA hybridization was performed on cryopreserved kidneys, as we have previously described using sense and antisense probes for Lcn2/Ngal.11
Cultured Cells
Conditionally immortalized podocytes were grown and differentiated as described.11,76 Primary podocytes were propagated from kidneys from 6- to 10-week-old WT and TRB3−/− mice with Dynabeads (Life Technologies, Grand Island, NY) as we have previously reported. Passage 2 cells were used, and for validation they were grown on cover slips and stained with antisynaptopodin antibody (clone G1D4; Fitzgerald Industries, Acton, MA).11 MCT cells were a kind gift from Eric G. Neilson (Northwestern University, Feinberg School of Medicine, Chicago, IL).77 Glomerular preparations were also performed using Dynabeads, and RNA was immediately isolated with RNeasy columns (Qiagen, Valencia, CA).
Plasmids and Transient Transfections
Fully differentiated podocytes were harvested from 10-cm plates with Accutase (Innovative Cell Technologies, San Diego, CA), replated, and transfected in 12-well plates with Opti-MEM (Life Technologies) and Lipofectamine 2000 (Life Technologies) using pcDNA3-HA-TRB3 and its control pcDNA3.12 MCT cells were transfected as we have previously described.78
Real-Time PCR
RNA, cDNA, and real-time PCR studies were performed with TaqMan Gene Expression Assays (TRB3: Mm00454879_m1, mIL-6 Mm00446190_m1, MCP-1 Mm00441242, FN1 Mm01256744_m1, TGFβ Mm01178820_m1, BiP Mm00517690_g1; Life Technologies) and CHOP, Lcn2/Ngal and rpl19 SYBR green primers were used.79 ATF4 primers were F-5′ATGGCGTATTAGAGGCAGCAGTGC, B-5′CGAAGGTATCTTTGTCCGT-TACAGC.
Western Blotting
Cellular lysates were prepared with cell lysis buffer (Cell Signaling Technology, Beverly, MA) with protease inhibitors.12 Samples were run on NuPAGE Bis-Tris gels (Life Technologies) and transferred onto nitrocellulose membranes. The following antibodies were used: TRB3 (Calbiochem, La Jolla, CA), AKTSer473 (4060), AKTThr308 (2965), AKT (4691), Rictor (2114), mTOR (2983), pSTAT3 (9145), STAT3 (9132), pFOXO1 (9464) FOXO1 (2880), pGSK3α/β(9331), Cell Signaling Technology (Danvers MA), actin (SC-1616) (Santa Cruz Biotechnology, Santa Cruz, CA), and HA (MMS-101) (Covance, Princeton, NJ). Detection was performed with ECL Prime Detection Reagents (GE Healthcare, Piscataway, NJ).
Immunoprecipitation
MCT cells were transfected with pcDNA3 or pcDNA3-HA-TRB3.77 The next day, cellular lysates were prepared and sonicated (3×10 seconds each), then centrifuged at 4°C at high speed for 15 minutes. One thousand micrograms of lysate was incubated overnight at 4°C with 30 μl HA-Tag (C29F4, CST) rabbit mAb (Sepharose bead conjugate), and the next day the beads were washed five times and resuspended in 2× sample buffer and boiled for 5 minutes. Input was 4.5% of lysate. Samples were run on Bis-Tris gels and high molecular weight proteins transferred for 2 hours and 15 minutes.
Statistical Analyses
Differences were analyzed using the t test, ANOVAs with post hoc Tukey tests for pair-wise comparisons, and Kruskal–Wallis tests for nonparametric studies. Analysis was accomplished with SPSS software, version 20 (IBM, Armonk, NY).
Supplementary Material
Acknowledgments
MCT cells were a kind gift from Eric G. Neilson. We also thank Timo Rieg and Nazila Sabri for excellent technical advice and assistance. We would also like to thank Marc Montminy, Salk Institute, and Alexandra Newton, University of California San Diego, for their helpful discussions.
These studies were performed with the support of the Department of Veterans Affairs Career Development Transition Award and Merit Award and the University of California San Diego Senate and National Institutes of Health (NIH)/National Institute of Diabetes and Digestive and Kidney Diseases DRC Pilot and Feasibility Grant P30 DK063491, awarded to R.C. V.V. was supported by NIH grant RO1-DK56248 and the O’Brien Core Center for Acute Kidney Injury Research NIH grant P30-DK079337.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2013070811/-/DCSupplemental.
References
- 1.Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, Saran R, Wang AY, Yang CW: Chronic kidney disease: global dimension and perspectives. Lancet 382: 260–272, 2013 [DOI] [PubMed] [Google Scholar]
- 2.Cunard R, Sharma K: The endoplasmic reticulum stress response and diabetic kidney disease. Am J Physiol Renal Physiol 300: F1054–F1061, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Du K, Herzig S, Kulkarni RN, Montminy M: TRB3: A tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 300: 1574–1577, 2003 [DOI] [PubMed] [Google Scholar]
- 4.Morse E, Schroth J, You YH, Pizzo DP, Okada S, Ramachandrarao S, Vallon V, Sharma K, Cunard R: TRB3 is stimulated in diabetic kidneys, regulated by the ER stress marker CHOP, and is a suppressor of podocyte MCP-1. Am J Physiol Renal Physiol 299: F965–F972, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiss-Toth E, Bagstaff SM, Sung HY, Jozsa V, Dempsey C, Caunt JC, Oxley KM, Wyllie DH, Polgar T, Harte M, O’neill LA, Qwarnstrom EE, Dower SK: Human tribbles, a protein family controlling mitogen-activated protein kinase cascades. J Biol Chem 279: 42703–42708, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Wu M, Xu LG, Zhai Z, Shu HB: SINK is a p65-interacting negative regulator of NF-kappaB-dependent transcription. J Biol Chem 278: 27072–27079, 2003 [DOI] [PubMed] [Google Scholar]
- 7.Kuo CH, Morohoshi K, Aye CC, Garoon RB, Collins A, Ono SJ: The role of TRB3 in mast cells sensitized with monomeric IgE. Exp Mol Pathol 93: 408–415, 2012 [DOI] [PubMed] [Google Scholar]
- 8.Prudente S, Hribal ML, Flex E, Turchi F, Morini E, De Cosmo S, Bacci S, Tassi V, Cardellini M, Lauro R, Sesti G, Dallapiccola B, Trischitta V: The functional Q84R polymorphism of mammalian Tribbles homolog TRB3 is associated with insulin resistance and related cardiovascular risk in Caucasians from Italy. Diabetes 54: 2807–2811, 2005 [DOI] [PubMed] [Google Scholar]
- 9.Saltiel AR: Putting the brakes on insulin signaling. N Engl J Med 349: 2560–2562, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Luo ZF, Feng B, Mu J, Qi W, Zeng W, Guo YH, Pang Q, Ye ZL, Liu L, Yuan FH: Effects of 4-phenylbutyric acid on the process and development of diabetic nephropathy induced in rats by streptozotocin: Regulation of endoplasmic reticulum stress-oxidative activation. Toxicol Appl Pharmacol 246: 49–57, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Lee SJ, Borsting E, Declèves AE, Singh P, Cunard R: Podocytes express IL-6 and lipocalin 2/ neutrophil gelatinase-associated lipocalin in lipopolysaccharide-induced acute glomerular injury. Nephron, Exp Nephrol 121: e86–e96, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morse E, Selim E, Cunard R: PPARalpha ligands cause lymphocyte depletion and cell cycle block and this is associated with augmented TRB3 and reduced Cyclin B1 expression. Mol Immunol 46: 3454–3461, 2009 [DOI] [PubMed] [Google Scholar]
- 13.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM: Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307: 1098–1101, 2005 [DOI] [PubMed] [Google Scholar]
- 14.Hong SM, Park CW, Cha HJ, Kwon JH, Yun YS, Lee NG, Kim DG, Nam HG, Choi KY: Rapamycin inhibits both motility through down-regulation of p-STAT3 (S727) by disrupting the mTORC2 assembly and peritoneal dissemination in sarcomatoid cholangiocarcinoma. Clin Exp Metastasis 30: 177–187, 2013 [DOI] [PubMed] [Google Scholar]
- 15.Yuan M, Pino E, Wu L, Kacergis M, Soukas AA: Identification of Akt-independent regulation of hepatic lipogenesis by mammalian target of rapamycin (mTOR) complex 2. J Biol Chem 287: 29579–29588, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA: Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15: 6541–6551, 1996 [PMC free article] [PubMed] [Google Scholar]
- 17.Oh WJ, Wu CC, Kim SJ, Facchinetti V, Julien LA, Finlan M, Roux PP, Su B, Jacinto E: mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J 29: 3939–3951, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, Lowry C, Newton AC, Mao Y, Miao RQ, Sessa WC, Qin J, Zhang P, Su B, Jacinto E: The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J 27: 1932–1943, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Logie L, Ruiz-Alcaraz AJ, Keane M, Woods YL, Bain J, Marquez R, Alessi DR, Sutherland C: Characterization of a protein kinase B inhibitor in vitro and in insulin-treated liver cells. Diabetes 56: 2218–2227, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Moore SF, Hunter RW, Hers I: mTORC2 protein complex-mediated Akt (Protein Kinase B) Serine 473 Phosphorylation is not required for Akt1 activity in human platelets. J Biol Chem 286: 24553–24560, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B: SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127: 125–137, 2006 [DOI] [PubMed] [Google Scholar]
- 22.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM: Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 11: 859–871, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Tan SX, Ng Y, Meoli CC, Kumar A, Khoo PS, Fazakerley DJ, Junutula JR, Vali S, James DE, Stöckli J: Amplification and demultiplexing in insulin-regulated Akt protein kinase pathway in adipocytes. J Biol Chem 287: 6128–6138, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feliers D, Duraisamy S, Faulkner JL, Duch J, Lee AV, Abboud HE, Choudhury GG, Kasinath BS: Activation of renal signaling pathways in db/db mice with type 2 diabetes. Kidney Int 60: 495–504, 2001 [DOI] [PubMed] [Google Scholar]
- 25.Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, Abboud HE: Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J Biol Chem 280: 39616–39626, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Lloberas N, Cruzado JM, Franquesa M, Herrero-Fresneda I, Torras J, Alperovich G, Rama I, Vidal A, Grinyó JM: Mammalian target of rapamycin pathway blockade slows progression of diabetic kidney disease in rats. J Am Soc Nephrol 17: 1395–1404, 2006 [DOI] [PubMed] [Google Scholar]
- 27.Gorin Y, Kim NH, Feliers D, Bhandari B, Choudhury GG, Abboud HE: Angiotensin II activates Akt/protein kinase B by an arachidonic acid/redox-dependent pathway and independent of phosphoinositide 3-kinase. FASEB J 15: 1909–1920, 2001 [DOI] [PubMed] [Google Scholar]
- 28.Rane MJ, Song Y, Jin S, Barati MT, Wu R, Kausar H, Tan Y, Wang Y, Zhou G, Klein JB, Li X, Cai L: Interplay between Akt and p38 MAPK pathways in the regulation of renal tubular cell apoptosis associated with diabetic nephropathy. Am J Physiol Renal Physiol 298: F49–F61, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagai K, Matsubara T, Mima A, Sumi E, Kanamori H, Iehara N, Fukatsu A, Yanagita M, Nakano T, Ishimoto Y, Kita T, Doi T, Arai H: Gas6 induces Akt/mTOR-mediated mesangial hypertrophy in diabetic nephropathy. Kidney Int 68: 552–561, 2005 [DOI] [PubMed] [Google Scholar]
- 30.Salazar M, Lorente M, García-Taboada E, Hernández-Tiedra S, Davila D, Francis SE, Guzmán M, Kiss-Toth E, Velasco G: The pseudokinase tribbles homologue-3 plays a crucial role in cannabinoid anticancer action. Biochim Biophys Acta 1831: 1573–1578, 2013 [DOI] [PubMed] [Google Scholar]
- 31.Matsushima R, Harada N, Webster NJ, Tsutsumi YM, Nakaya Y: Effect of TRB3 on insulin and nutrient-stimulated hepatic p70 S6 kinase activity. J Biol Chem 281: 29719–29729, 2006 [DOI] [PubMed] [Google Scholar]
- 32.Haruta T, Uno T, Kawahara J, Takano A, Egawa K, Sharma PM, Olefsky JM, Kobayashi M: A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol Endocrinol 14: 783–794, 2000 [DOI] [PubMed] [Google Scholar]
- 33.Liu J, Zhang W, Chuang GC, Hill HS, Tian L, Fu Y, Moellering DR, Garvey WT: Role of TRIB3 in regulation of insulin sensitivity and nutrient metabolism during short-term fasting and nutrient excess. Am J Physiol Endocrinol Metab 303: E908–E916, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warfel NA, Newton AC: Pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP): a new player in cell signaling. J Biol Chem 287: 3610–3616, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yung HW, Charnock-Jones DS, Burton GJ: Regulation of AKT phosphorylation at Ser473 and Thr308 by endoplasmic reticulum stress modulates substrate specificity in a severity dependent manner. PLoS ONE 6: e17894, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cunard R, Sharma K: The endoplasmic reticulum stress response and diabetic kidney disease. Am J Physiol Renal Physiol 300: F1054–F1061, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang K, Kaufman RJ: From endoplasmic-reticulum stress to the inflammatory response. Nature 454: 455–462, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Navarro JF, Milena FJ, Mora C, León C, García J: Renal pro-inflammatory cytokine gene expression in diabetic nephropathy: Effect of angiotensin-converting enzyme inhibition and pentoxifylline administration. Am J Nephrol 26: 562–570, 2006 [DOI] [PubMed] [Google Scholar]
- 39.Lu TC, Wang ZH, Feng X, Chuang PY, Fang W, Shen Y, Levy DE, Xiong H, Chen N, He JC: Knockdown of Stat3 activity in vivo prevents diabetic glomerulopathy. Kidney Int 76: 63–71, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu Q, Prabhu A, Xu S, Manigrasso MB, Maric C: Dose-dependent effects of dihydrotestosterone in the streptozotocin-induced diabetic rat kidney. Am J Physiol Renal Physiol 297: F307–F315, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Min D, Lyons JG, Bonner J, Twigg SM, Yue DK, McLennan SV: Mesangial cell-derived factors alter monocyte activation and function through inflammatory pathways: Possible pathogenic role in diabetic nephropathy. Am J Physiol Renal Physiol 297: F1229–F1237, 2009 [DOI] [PubMed] [Google Scholar]
- 42.Sanchez-Nino MD, Bozic M, Cordoba-Lanus E, Valcheva P, Gracia O, Ibarz M, Fernandez E, Navarro-Gonzalez JF, Ortiz A, Valdivielso JM: Beyond proteinuria: VDR activation reduces renal inflammation in experimental diabetic nephropathy. Am J Physiol Renal Physiol 302: F647–F657, 2012 [DOI] [PubMed] [Google Scholar]
- 43.Sekizuka K, Tomino Y, Sei C, Kurusu A, Tashiro K, Yamaguchi Y, Kodera S, Hishiki T, Shirato I, Koide H: Detection of serum IL-6 in patients with diabetic nephropathy. Nephron 68: 284–285, 1994 [DOI] [PubMed] [Google Scholar]
- 44.Suzuki D, Miyazaki M, Naka R, Koji T, Yagame M, Jinde K, Endoh M, Nomoto Y, Sakai H: In situ hybridization of interleukin 6 in diabetic nephropathy. Diabetes 44: 1233–1238, 1995 [DOI] [PubMed] [Google Scholar]
- 45.Shikano M, Sobajima H, Yoshikawa H, Toba T, Kushimoto H, Katsumata H, Tomita M, Kawashima S: Usefulness of a highly sensitive urinary and serum IL-6 assay in patients with diabetic nephropathy. Nephron 85: 81–85, 2000 [DOI] [PubMed] [Google Scholar]
- 46.Saraheimo M, Teppo AM, Forsblom C, Fagerudd J, Groop PH: Diabetic nephropathy is associated with low-grade inflammation in Type 1 diabetic patients. Diabetologia 46: 1402–1407, 2003 [DOI] [PubMed] [Google Scholar]
- 47.Hwang SY, Kim JY, Kim KW, Park MK, Moon Y, Kim WU, Kim HY: IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-kappaB- and PI3-kinase/Akt-dependent pathways. Arthritis Res Ther 6: R120–R128, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tang CH, Lu DY, Yang RS, Tsai HY, Kao MC, Fu WM, Chen YF: Leptin-induced IL-6 production is mediated by leptin receptor, insulin receptor substrate-1, phosphatidylinositol 3-kinase, Akt, NF-kappaB, and p300 pathway in microglia. J Immunol 179: 1292–1302, 2007 [DOI] [PubMed] [Google Scholar]
- 49.Cheng A, Dong Y, Zhu F, Liu Y, Hou FF, Nie J: AGE-LDL activates Toll like receptor 4 pathway and promotes inflammatory cytokines production in renal tubular epithelial cells. Int J Biol Sci 9: 94–107, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen RH, Chang MC, Su YH, Tsai YT, Kuo ML: Interleukin-6 inhibits transforming growth factor-beta-induced apoptosis through the phosphatidylinositol 3-kinase/Akt and signal transducers and activators of transcription 3 pathways. J Biol Chem 274: 23013–23019, 1999 [DOI] [PubMed] [Google Scholar]
- 51.Hodge DR, Hurt EM, Farrar WL: The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer 41: 2502–2512, 2005 [DOI] [PubMed] [Google Scholar]
- 52.Surnamekim GI, Surnamemin GS, Surnamepark GH: Sequential signaling cascade of IL-6 and PGC-1alpha is involved in high glucose-induced podocyte loss and growth arrest. Biochem Biophys Res Commun 435: 702–707, 2013 [DOI] [PubMed] [Google Scholar]
- 53.Tang SC, Chan LY, Leung JC, Cheng AS, Chan KW, Lan HY, Lai KN: Bradykinin and high glucose promote renal tubular inflammation. Nephrol Dial Transplant 25: 698–710, 2010 [DOI] [PubMed] [Google Scholar]
- 54.Mahadevan NR, Rodvold J, Almanza G, Pérez AF, Wheeler MC, Zanetti M: ER stress drives Lipocalin 2 upregulation in prostate cancer cells in an NF-κB-dependent manner. BMC Cancer 11: 229, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hsin IL, Hsiao YC, Wu MF, Jan MS, Tang SC, Lin YW, Hsu CP, Ko JL: Lipocalin 2, a new GADD153 target gene, as an apoptosis inducer of endoplasmic reticulum stress in lung cancer cells. Toxicol Appl Pharmacol 263: 330–337, 2012 [DOI] [PubMed] [Google Scholar]
- 56.Matsushita K, van der Velde M, Astor BC, Woodward M, Levey AS, de Jong PE, Coresh J, Gansevoort RT, Chronic Kidney Disease Prognosis Consortium : Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: A collaborative meta-analysis. Lancet 375: 2073–2081, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van der Velde M, Matsushita K, Coresh J, Astor BC, Woodward M, Levey A, de Jong P, Gansevoort RT, van der Velde M, Matsushita K, Coresh J, Astor BC, Woodward M, Levey AS, de Jong PE, Gansevoort RT, Levey A, El-Nahas M, Eckardt KU, Kasiske BL, Ninomiya T, Chalmers J, Macmahon S, Tonelli M, Hemmelgarn B, Sacks F, Curhan G, Collins AJ, Li S, Chen SC, Hawaii Cohort KP, Lee BJ, Ishani A, Neaton J, Svendsen K, Mann JF, Yusuf S, Teo KK, Gao P, Nelson RG, Knowler WC, Bilo HJ, Joosten H, Kleefstra N, Groenier KH, Auguste P, Veldhuis K, Wang Y, Camarata L, Thomas B, Manley T, Chronic Kidney Disease Prognosis Consortium : Lower estimated glomerular filtration rate and higher albuminuria are associated with all-cause and cardiovascular mortality. A collaborative meta-analysis of high-risk population cohorts. Kidney Int 79: 1341–1352, 2011 [DOI] [PubMed] [Google Scholar]
- 58.Gansevoort RT, Matsushita K, van der Velde M, Astor BC, Woodward M, Levey AS, de Jong PE, Coresh J, Chronic Kidney Disease Prognosis Consortium : Lower estimated GFR and higher albuminuria are associated with adverse kidney outcomes. A collaborative meta-analysis of general and high-risk population cohorts. Kidney Int 80: 93–104, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koo SH, Satoh H, Herzig S, Lee CH, Hedrick S, Kulkarni R, Evans RM, Olefsky J, Montminy M: PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat Med 10: 530–534, 2004 [DOI] [PubMed] [Google Scholar]
- 60.Okamoto H, Latres E, Liu R, Thabet K, Murphy A, Valenzeula D, Yancopoulos GD, Stitt TN, Glass DJ, Sleeman MW: Genetic deletion of Trb3, the mammalian Drosophila tribbles homolog, displays normal hepatic insulin signaling and glucose homeostasis. Diabetes 56: 1350–1356, 2007 [DOI] [PubMed] [Google Scholar]
- 61.Wang ZH, Shang YY, Zhang S, Zhong M, Wang XP, Deng JT, Pan J, Zhang Y, Zhang W: Silence of TRIB3 suppresses atherosclerosis and stabilizes plaques in diabetic ApoE-/-/LDL receptor-/- mice. Diabetes 61: 463–473, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ti Y, Xie GL, Wang ZH, Bi XL, Ding WY, Wang J, Jiang GH, Bu PL, Zhang Y, Zhong M, Zhang W: TRB3 gene silencing alleviates diabetic cardiomyopathy in a type 2 diabetic rat model. Diabetes 60: 2963–2974, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Inoki K, Mori H, Wang J, Suzuki T, Hong S, Yoshida S, Blattner SM, Ikenoue T, Rüegg MA, Hall MN, Kwiatkowski DJ, Rastaldi MP, Huber TB, Kretzler M, Holzman LB, Wiggins RC, Guan KL: mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest 121: 2181–2196, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gödel M, Hartleben B, Herbach N, Liu S, Zschiedrich S, Lu S, Debreczeni-Mór A, Lindenmeyer MT, Rastaldi MP, Hartleben G, Wiech T, Fornoni A, Nelson RG, Kretzler M, Wanke R, Pavenstädt H, Kerjaschki D, Cohen CD, Hall MN, Rüegg MA, Inoki K, Walz G, Huber TB: Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest 121: 2197–2209, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stevens VA, Saad S, Chen XM, Pollock CA: The interdependence of EGF-R and SGK-1 in fibronectin expression in primary kidney cortical fibroblast cells. Int J Biochem Cell Biol 39: 1047–1054, 2007 [DOI] [PubMed] [Google Scholar]
- 66.Lu M, Wang J, Jones KT, Ives HE, Feldman ME, Yao LJ, Shokat KM, Ashrafi K, Pearce D: mTOR complex-2 activates ENaC by phosphorylating SGK1. J Am Soc Nephrol 21: 811–818, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cybulski N, Hall MN: TOR complex 2: A signaling pathway of its own. Trends Biochem Sci 34: 620–627, 2009 [DOI] [PubMed] [Google Scholar]
- 68.Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, Hall MN: Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 6: 1122–1128, 2004 [DOI] [PubMed] [Google Scholar]
- 69.Venkatesan B, Mahimainathan L, Das F, Ghosh-Choudhury N, Ghosh Choudhury G: Downregulation of catalase by reactive oxygen species via PI 3 kinase/Akt signaling in mesangial cells. J Cell Physiol 211: 457–467, 2007 [DOI] [PubMed] [Google Scholar]
- 70.Vassiliadis J, Bracken C, Matthews D, O’Brien S, Schiavi S, Wawersik S: Calcium mediates glomerular filtration through calcineurin and mTORC2/Akt signaling. J Am Soc Nephrol 22: 1453–1461, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mascarenhas D, Routt S, Singh BK: Mammalian target of rapamycin complex 2 regulates inflammatory response to stress. Inflamm Res 61: 1395–1404, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fan W, Cheng K, Qin X, Narsinh KH, Wang S, Hu S, Wang Y, Chen Y, Wu JC, Xiong L, Cao F: mTORC1 and mTORC2 play different roles in the functional survival of transplanted adipose-derived stromal cells in hind limb ischemic mice via regulating inflammation in vivo. Stem Cells 31: 203–214, 2013 [DOI] [PubMed] [Google Scholar]
- 73.Satoh T, Kidoya H, Naito H, Yamamoto M, Takemura N, Nakagawa K, Yoshioka Y, Morii E, Takakura N, Takeuchi O, Akira S: Critical role of Trib1 in differentiation of tissue-resident M2-like macrophages. Nature 495: 524–528, 2013 [DOI] [PubMed] [Google Scholar]
- 74.Dunn SR, Qi Z, Bottinger EP, Breyer MD, Sharma K: Utility of endogenous creatinine clearance as a measure of renal function in mice. Kidney Int 65: 1959–1967, 2004 [DOI] [PubMed] [Google Scholar]
- 75.Declèves AE, Mathew AV, Cunard R, Sharma K: AMPK mediates the initiation of kidney disease induced by a high-fat diet. J Am Soc Nephrol 22: 1846–1855, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shankland SJ, Pippin JW, Reiser J, Mundel P: Podocytes in culture: past, present, and future. Kidney Int 72: 26–36, 2007 [DOI] [PubMed] [Google Scholar]
- 77.Haverty TP, Kelly CJ, Hines WH, Amenta PS, Watanabe M, Harper RA, Kefalides NA, Neilson EG: Characterization of a renal tubular epithelial cell line which secretes the autologous target antigen of autoimmune experimental interstitial nephritis. J Cell Biol 107: 1359–1368, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Borsting E, Cheng VP, Glass CK, Vallon V, Cunard R: Peroxisome proliferator-activated receptor-γ agonists repress epithelial sodium channel expression in the kidney. Am J Physiol Renal Physiol 302: F540–F551, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Archer DC, Frkanec JT, Cromwell J, Clopton P, Cunard R: WY14,643, a PPARalpha ligand, attenuates expression of anti-glomerular basement membrane disease. Clin Exp Immunol 150: 386–396, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.