

Abstract

DNA polymerases slide on DNA during replication, and the interface must be mobile for various conformational changes. The role of lubricant interfacial water is not understood. In this report, we systematically characterize the water dynamics at the interface and in the active site of a tight binding polymerase (pol β) in its binary complex and ternary state using tryptophan as a local optical probe. Using femtosecond spectroscopy, we observed that upon DNA recognition the surface hydration water is significantly confined and becomes bound water at the interface, but the dynamics are still ultrafast and occur on the picosecond time scale. These interfacial water molecules are not trapped but are mobile in the heterogeneous binding nanospace. Combining our findings with our previous observation of ultrafast water motions at the interface of a loose binding polymerase (Dpo4), we conclude that the binding interface is dynamic and the water molecules in various binding clefts, channels, and caves are mobile and even fluid with different levels of mobility for loose or tight binding polymerases. Such a dynamic interface should be general to all DNA polymerase complexes to ensure the biological function of DNA synthesis.
Polymerases synthesize DNA one base at a time through a series of dynamic motions, including global and local conformational changes and chemical reactions.1−5 For processive DNA replication, the polymerases must slide on DNA6−8 and the water molecules at binding interfaces should play an active role.9−17 From extensive analyses of many protein–DNA complexes,9−17 the interfacial water at a large binding area seems to behave as a molecule lubricant to increase structural flexibility and adaptability and to lubricate protein sliding for fast translocation. Such interfacial dynamics are difficult to study experimentally.18 We recently reported our direct probing of solvent accessibility and mobility at the binding interface of a loose binding polymerase (Dpo4)–DNA complex and observed ultrafast interfacial water dynamics, concluding a fluid binding interface and mobile active site and implying a lower fidelity of the polymerase.19 However, for a tight polymerase–DNA binding complex,20−22 it is unknown how flexible confined water molecules are at the interface and what their dynamic role is in DNA replication.
Here, we choose the polymerase β (pol β)–DNA complex to study the water dynamics at its tight binding interface. Pol β has 335 amino acids with a molecular mass of 39 kDa and belongs to the X-family of DNA polymerases with lyase and polymerase domains (Figure 1).22−26 The enzyme has been extensively studied using biochemical,27−29 nuclear magnetic resonance (NMR),30−32 and X-ray33−35 methods as well as molecular dynamics (MD) simulations.36−38 The N-terminal lyase domain (8 kDa) has an enzymatic activity to remove the 5′-deoxyribose phosphate intermediate during base excision repair. The polymerase domain (31 kDa) is composed of three subdomains: a double-stranded DNA (dsDNA) binding D subdomain (also called the thumb subdomain), a nucleotidyl-transfer C subdomain (palm), and a dNTP selection N subdomain (finger). Upon dsDNA recognition, the polymerase wraps around the DNA substrate and forms a “donut” shape with a compact configuration (Figure 1). With an incoming nucleotide, the ternary complex further changes its conformation from the “open” to “closed” form to sandwich the nucleotide by moving an α-helix closer to the nascent base pairs.23,24 Accompanying these major structural changes are a series of local side chain conformational changes that are also important for polymerase function.23,24,28,34 For both binary and ternary structures, the active site is still solvent accessible and water molecules were recently proposed to be involved in the local active-site reorganization and catalytic nucleotidyl-transfer reactions.39,40
Figure 1.
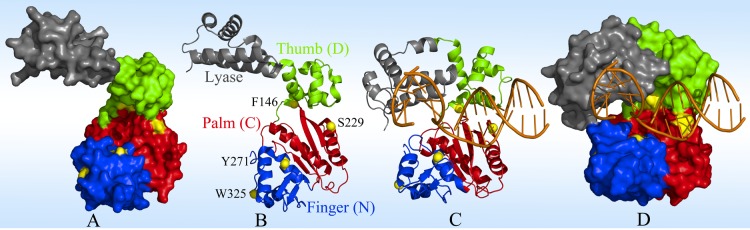
Surface and ribbon representations of pol β in apo (A and B, Protein Data Bank entry 1BPD) and binary (C and D, Protein Data Bank entry 1BPX) states. Pol β consists of a lyase domain (gray) and three polymerase subdomains, thumb (green), palm (red), and finger (blue), arranged in a right-hand configuration. The definition of the subdomains is that of Steitz et al.67 The yellow patches and spheres show the wild-type and mutant tryptophan positions. The DNA backbone and base pairs are colored orange.
In this report, we used a methodology41 similar to that in our previous study of the loose binding Dpo4–DNA complex19 to characterize the water dynamics at the binding interface for the binary and ternary complexes of pol β with femtosecond resolution. Because the single intrinsic W325 is located on the finger subdomain and is far from the binding interface, it was used as a control site. Using the W325F mutant as a template, we designed two additional mutants, F146W and S229W, at the binding sites on the thumb and palm subdomains, respectively, and one mutant, Y271W, near the active site on the finger subdomain. We determined the time scales of water motion around the protein surface, at the DNA binding interface of the complex, and in the active site with and without binding to the incoming nucleotide. These results were directly compared with those from the loose binding Dpo4–DNA complex,19 allowing us to elucidate the molecular mechanism of the interfacial water dynamics in different polymerase–DNA binding complexes.
Materials and Methods
Sample Preparation
Rat pol β, encoded in plasmid PET17b, was overexpressed in Escherichia coli BL21(DE3), and the protein purification followed the procedures reported previously.29 The protein sample was finally stored in a buffer containing 100 mM Tris-HCl (pH 8.0), 150 mM KCl, 2 mM DTT, 3% glycerol, and 1.5 mM MgCl2. All three double mutants (F146W, S299W, and Y271W) were designed and created using site-directed mutagenesis. They were overexpressed and purified via a similar procedure and finally showed structures similar to that of the wild type based on their circular dichroism (CD) spectra. We also examined their enzymatic activities42 and found that all three mutants were enzymatically active. The oligomers of the DNA substrate were purchased from Integrated DNA Technologies (Coralville, IA) and annealed in our lab to form two-nucleotide-gapped dsDNA. The sequences for the oligomers are as follows: 36mer (template), 5′-TGGATTGAGGTGACTATGGTTGGACGGCTGCGAGGC-3′; 19mer (prime), 5′-GCCTCGCAGCCGTCCAACddC-3′; 15mer (downstream oligo), 5′-/5Phos/AGTCACCTCAATCCA-3′. The mixing concentrations of the enzyme, DNA substrate, and dATP were 400 μM, 450 μM, and 1 mM, respectively. On the basis of the known dissociation constants of binary and ternary complexes,43 there should be more than 98% of the enzyme complexes in our samples.
Femtosecond Fluorescence Spectroscopy
All the femtosecond-resolved fluorescence transients were obtained using the fluorescence upconversion method as reported previously.44 Briefly, an 800 nm pulsed laser beam was generated from a femtosecond amplifier system with a repetition rate at 1 kHz. Then it was split into two separated beams. One beam was used to generate the pump beam at 590 nm using an optical parametric amplifier (OPA). This beam was frequency doubled to produce the final 295 nm pump pulse. The other 800 nm beam was attenuated as a gating pulse. The pump–pulse energy was ∼140 nJ before the pulse was focused into the motor-controlled rotating sample cell with a thickness of 1 mm. Using 295 nm as the pump wavelength can minimize the tyrosine absorption in pol β. The fluorescence emission from excited-state tryptophan was collected by a pair of parabolic mirrors and focused into a 0.2 mm BBO crystal to mix with the gating beam in a nonlinear configuration. The up-converted signal ranging from 223 to 253 nm was detected with a monochromator with a photomultiplier. The instrument response time, determined by measuring the Raman signal of water at 327 nm, is around 450 fs under the current nonlinear configuration. All the experiments were conducted at the magic angle by setting the pump–pulse polarization at 54.7° relative to the acceptance axis of the mixing crystal and the probe pulse polarization parallel to the axis. All fluorescence transients were taken in a time window of up to 3 ns.
Results and Discussion
Steady-State Emission, Ultrafast Fluorescence Transients, and Local Polarity
Figure 2A shows the steady-state emission spectra of three mutants and the wild type (WT) in the apo state. The emission peaks range from 336 to 346 nm, indicating that S229W and W325 (WT) are solvent-exposed and F146W and Y271W are partially buried. When DNA binds, the emission spectra for all three mutants show noticeable changes. The emission peak shifts from 336 to 338.7 nm for F146W, from 342.7 to 339.9 nm for S229W, and from 337.6 to 340 nm for Y271W. With further binding of an incoming nucleotide, F146W and S229W show negligible changes because both mutants are away from the active site. For Y271W at the active site, the emission peak shifts back to 338 nm. For the WT, because the control site W325 is away from the binding interface, the fluorescence emission remains the same in all three states with a peak at 346 nm. Figure 2B shows a comparison of the emission spectra of Y271W at the active site with the control site W325 before and after binding to DNA. The changes in all these emission spectra in the three mutants reflect the local environmental variations upon binding of DNA and incorporation of the incoming nucleotide at the active site. We also noticed that besides the spectral shifts the emission profiles were altered. All the emission spectra of the mutants follow a smooth log-normal shape in the apo state but show certain structures in the complex states (Figure 2B), possibly because of a more rigid local environment after binding DNA. Compared with the Dpo4 polymerase that shows nearly no changes in the emission spectra at the binding interface in the complexed states,19 the pol β complex shows spectral changes and suggests tighter recognition. Nevertheless, the emission peaks of all mutants are still located at >338 nm, indicating a polar binding interface and a significant number of water molecules around the probing sites.41,45
Figure 2.
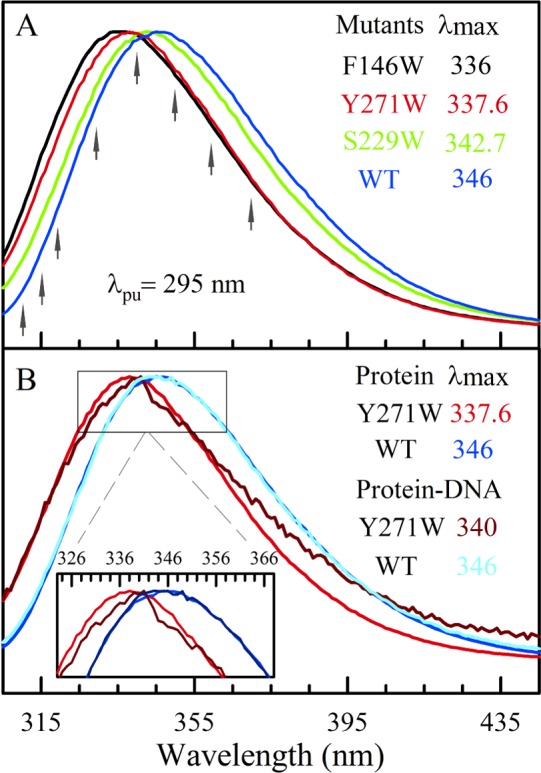
(A) Normalized steady-state fluorescence spectra of three double mutants (F146W, Y271W, and S229W using W325F as the double-mutant template) and WT (W325) in the apo state. The black arrows indicate the gating wavelengths for the fluorescence transients. (B) Normalized steady-state fluorescence spectra of Y271W and WT in the apo and binary states. The inset is a close-up of the emission peaks. WT, far from the binding interface, is used as a control in the experiment.
Figure 3 shows a series of fluorescence transients gated from the blue to red sides of the emission spectra for the Y271W mutant in the apo and binary states. The signals of dsDNA alone in buffer solution, resulting from ultrafast deactivation of the excited state via conical intersections,46,47 are also shown in Figure 3. For the sake of clarity, the contribution of DNA in the transients of the binary Y271W mutant has been carefully subtracted. In the complex states, the DNA signals can be obtained from the difference between the signals with and without DNA for the control site of WT. Alternatively, we can directly measure the fluorescence signals of the W325F mutant–DNA complex because the mutant has no Trp residue. Also, we did not observe the difference in the signals between dsDNA alone in buffer solution and in the complex (Figure S1 of the Supporting Information), indicating that the hydration and photophysics of DNA were minimally perturbed in the complex states even with the 90° kink of the DNA structure upon recognition. The fluorescence transients of Y271W in the ternary state, WT in the apo state, and the two other mutants of F146W and S229W in the apo and binary states are given in Supporting Information (Figures S2–S5).
Figure 3.

Normalized fluorescence transients of Y271W in the apo state (left), gated from the blue to red sides of the emission spectra. Transients of Y271W in the binary state after the DNA signals had been removed (middle). Transients of DNA in buffer (right), showing the dominant ultrafast decays in <1 ps. The symbols are the experimental data, and the solid lines show the best exponential fits.
All fluorescence transients show typical solvation dynamics with the decays at the blue side and rises at the red end as we reported for surface hydration dynamics for many proteins.48−53 In the apo state, the solvation process directly reflects the relaxation of water molecules around the Trp probe within a distance of ∼10 Å.54,55 For example, the transients of the apo Y271W mutant show three decay components (400–600 fs, 4.1–7.5 ps, and 40–59 ps) and one rise component (3.7 ps) at the blue and red sides, respectively, similar to the pattern observed in apo Dpo4 polymerase. The subpicosecond component represents the bulk-type water motion at the outer hydration layers, while the component in a few picoseconds reflects the relaxation of the local water networks in the inner hydration layers near the protein surface.48−53 The component in tens of picoseconds is from the water network rearrangements coupled with protein fluctuations.49,51−53 Although certain MD simulations showed that the long component in tens of picoseconds was mainly from local protein (or DNA) motions,56−58 our extensive studies both in experiments and in MD simulations showed that the long component is from water–protein coupled motions, mainly driven by local water network relaxation.41,49,51−55,59,60 Significantly, when DNA binds, the subpicosecond decay components in the three mutants disappear and the other two decays on the picosecond time scale slow down (6.2–8.4 and 62–90 ps in the Y271W binary state). With the incoming nucleotide, the dynamics of Y271W further slow down (6.5–11.5 and 65–105 ps). This observation is striking. In Dpo4 polymerase, the ultrafast components are still present, though they are strongly suppressed upon DNA recognition.19 In contrast, the absence of ultrafast water motions in pol β indicates that the outer-layer mobile water molecules have been either squeezed out or converted to “bound” water like inner-layer water networks upon binding DNA. The slowdown of the picosecond components reflects more rigid water networks at the binding interface and in the active site.
Hydration Dynamics, Interfacial Water Flexibility, and Mobile Water Networks
Using the methodology we developed,41,44,48,61 we can derive the solvation energies and corresponding time scales. Figure 4 shows the constructed correlation functions with the local apo structures within 12 Å of the Trp probes, and Figure 5 gives the resulting solvation energies, time scales, and solvation speeds (also see Table S1 of the Supporting Information). Overall, the patterns of hydration correlation dynamics are completely consistent with our numerous previous observations.48−53 Among all the mutants and WT, the F146W mutant in the apo state shows the slowest hydration dynamics with only two decay components at 6.3 and 103 ps. The F146W mutation, located on an α-helix in the thumb subdomain, has a local concave structure and densely charged surrounding (Figure 4A) and is partially buried in the enzyme with an emission peak at 336 nm. Thus, such a partially buried Trp probe can sense the inner-layer hydration dynamics and cannot detect the outer-layer mobile water motions.53 The observed slow hydration dynamics is consistent with the local structural and chemical properties, as extensively reported for apomyoglobin.52,53 All other enzymes in the apo state show three decay components with one ultrafast dynamics in hundreds of femtoseconds, consistent with the observation in Dpo419 and their emission peaks at >336 nm with certain exposure to solvent. The two dynamics on the picosecond time scales are faster than that of F146W, consistent with their local structural and chemical properties (Figure 4B–D).52,53
Figure 4.

Solvation (hydration) correlation functions for apo (black), binary (red), and ternary (blue) states with the insets shown on the short time scale. The two binding-site mutants F146W (A) and S229W (B) show the slower dynamics from the apo to binary states, with similar dynamics from the binary to ternary states. The dynamics of the active-site mutant Y271W (C) gradually slow down in the three states. The WT (D) exhibits the same dynamics as a control. The circles are the values derived from the experiment data, and the solid lines show the best exponential fits. The corresponding local structures within 12 Å of tryptophan probes (shown as the yellow spheres and patches) are also shown in ribbon and surface representations.
Figure 5.

Detailed analyses of the solvation dynamics of the three mutants and WT in the apo (gray), binary (green), and ternary (red) states. Panels A–C show the relaxation energies, relaxation times, and solvation speeds, respectively. When the DNA and nucleotide bind, the solvation energies are redistributed according to the local water network change (see context). F146W and S229W, located at the DNA binding sites, show significant slowdown in both relaxation times and solvation speeds from the apo to binary states. The relaxation of the active-site mutant Y271W gradually slows down from the apo to complex states. Notice that the ultrafast components completely disappear in the complex states of S229W and Y271W because of the confinement of water at the interface. As a control, the WT keeps similar solvation dynamics in all states.
The observed dynamics upon binding of DNA are striking. First, the ultrafast components in mutants S229W and Y271W disappear (Figure 4B,C), and we did not detect bulk-type mobile water molecules, indicating that such water molecules either are squeezed out of the interface or become more rigid, sandwiched by the enzyme and DNA surfaces. Second, the two dynamics in picoseconds significantly slow down, especially the long component by a factor of 2, indicating more rigid water networks at the enzyme–DNA interface. With the incoming nucleotide (dATP), we did not observe further changes for the two binding sites of F146W and S229W (Figure 4A,B), but for Y271W at the active center, the dynamics further slow down (Figure 4C), clearly indicating local environmental changes upon transitions from the apo to the binary and to the ternary complexes. These dramatic changes in the binding sites and at the active site were not clearly observed in Dpo4 and reflect the nature of the binding interface in the pol β–DNA complex. It is equally important that even though the binding site is clearly tighter than that of the Dpo4 complex, the observed water dynamics are still on the picosecond time scale, not nanoseconds, which would be expected for water trapped inside proteins,62−64 reflecting the fact that the interfacial water molecules are still mobile and the sandwiched water networks at the interface are flexible. Finally, for the control site of the WT, the dynamics in three states are the same (Figure 4D), showing no interactions with DNA or dATP.
We further analyzed these correlation dynamics and ascertained the hydration dynamics with more details (Figure 5 and Table S1 of the Supporting Information). The simplest system is the WT with similar dynamics in the three states and W325, mostly exposed to solvent with an emission peak at 346 nm, probes local surface hydration water motions with the largest solvation energy of ∼1550 cm–1, consisting of three components, E1 (41%), E2 (31%), and E3 (28%), with the three corresponding relaxation times being τ1 (600 fs), τ2 (4.3 ps), and τ3 (∼70 ps), respectively. Such patterns were observed for the other mutants and many other systems reported previously,48−53,59,60,65,66 representing the ultrafast surface hydration water motions and coupled water–protein fluctuations. The key studies here are to observe the changes in solvation energy and relaxation time in the binding complex. For F146W, we observed that E2 decreases (43 cm–1) and E3 increases (144 cm–1) (Figure 5A), indicating that the inner-layer water molecules become more rigid after DNA binding as reflected by the increase in the relaxation times from 6.3 to 7.4 ps and from 103 to 192 ps (Figure 5B) and corresponding slower solvation speeds (Figure 5C). For S299W, after DNA recognition, the bulk-type mobile water motion vanishes but the energies of the two components in picoseconds increase significantly (∼200 cm–1) (Figure 5A), indicating that more mobile out-layer hydration water molecules become confined at the interface, leading to greater solvation energy release, which is also reflected by the slowdown of the relaxation times from 5.0 to 8.5 ps and from 54 to 111 ps (Figure 5B) and of the corresponding solvation speeds (Figure 5C). For the active-site Y271W mutant, changes similar to those of S229W were observed upon binding to DNA. Interestingly, with the incoming nucleotide in the ternary complex, the second component, E2, decreases by 73 cm–1 after the increase from the apo to binary states by 111 cm–1 and E3 increases again by 36 cm–1 with an addition of 61 cm–1 in the binary state. The relaxations keep slowing down [5.1, 7.7, and 8.7 ps and 64, 111, and 113 ps (Figure 5B) for the apo, binary, and ternary states, respectively], and so do the corresponding solvation speeds (Figure 5C).
These observations for Y271W in the three states reflect the active-site environment changes upon binding of DNA and recognition of dATP. From the binary to ternary structures, the finger domain undergoes a series of rearrangements.23,24,33 The most critical conformational change is the α-helix N switching from the “open” to the “closed” configuration.23,24 Meanwhile, the local residues of the finger domain also readjust their positions ready for catalysis.24 One of the obvious adjustments is that Y271 moves closer to DNA to form a hydrogen bond with the primer terminal base instead of hydrogen bonding with the template base facing the incoming nucleotide.24 Apparently, the local environment of Y271 is different in all three states. Mutation of tyrosine to tryptophan minimally perturbed the function of pol β as examined by its CD spectrum and enzymatic activity. Thus, Y271W directly probed the local solvation (hydration) dynamics and the changes upon binding DNA and recognition of the incoming nucleotide at the active site.
On the basis of the fluorescence transients, the detailed analyses of the solvation correlation functions, i.e., the relaxation energies and times, and the solvation speeds, we revealed a unique picture of the hydration dynamics at the pol β–DNA binding interface and in the active site. On one side, the interfacial water is clearly not of the bulk water type. The relaxation of interfacial water has ∼2-fold slower dynamics than the surface hydration water, certainly confined by the relatively tighter binding of DNA. On the other side, unlike any trapped water molecules inside proteins observed by X-ray structures33−35 or NMR studies,30−32 these interfacial water molecules still relax on the picosecond time scale, maintaining a certain level of flexibility that is critical to the enzyme–DNA interactions and pol β function.
Heterogeneous Interface Caves, Confined Water Clusters, and Dynamic Water Interface
X-ray structures show how pol β interacts with DNA and the incoming nucleotide.23,34 As shown in panels C and D of Figure 1, the thumb, palm, and finger subdomains hold DNA mainly by interacting with the DNA backbone. The lyase domain not only binds downstream of the DNA duplex but also protects the active site in the finger subdomain. These structures are static and reveal only the binding architecture. The interacting surfaces of the polymerase and DNA are of a heterogeneous roughness, and at the interface, various shallow clefts and deep caves are formed. Water molecules mediate these interactions and also often behave as “fillers” for maintaining packing densities. Thus, water molecules fill these interfacial holes, and the interface is heterogeneously distributed with various confined water clusters. These large water clusters are dynamic and mobile, resulting in the difficulty in observing them in the X-ray structures. MD simulations may provide insights into water motions at the interface.
Figure 6 shows the results from our 4 ns MD simulations for the pol β–DNA binary complex with a fixed DNA configuration. All MD simulation methods were the same as before, but in the binding complex, the DNA was fixed to maintain the reasonable recognition.54,55 The simple MD simulations were used to estimate the number of water molecules at the interface and around the probe. With the addition of distances to pol β and DNA from these water molecules of <10 Å, we found that on the average, ∼270 water molecules are located at the interface of the complex (Figure 6A). Apparently, the interface of the complex is full of water molecules to “lubricate” the interactions between pol β and DNA. Panels B–E of Figure 6 show the number of water molecules within 10 Å of the Trp probes and their distributions with distances (Tables S2 and S3 of the Supporting Information), along with the snapshots of the local structures at 4 ns. In Figure 6E, among 79 water molecules within 10 Å in the WT, only 10 water molecules are in direct contact with Trp325 at 3.5 Å in the first layer but 33 water molecules are directly bound to the enzyme; 56 water molecules were 5 Å from the enzyme surface forming the first two hydration layers, while only 23 water molecules in the outer hydration layers were close to bulk water. From the apo to binary states for the three mutants, we observed that for F146W the total number of water molecules within 10 Å is 60 and shows nearly no change, consistent with the buried and concave location of Trp and the probing of only inner-layer hydration water. For S229W, the number of waters significantly changes from 132 to 54, a dramatic decrease with more than half of the water molecules squeezed out during DNA binding, consistent with recognition in the minor groove. For Y271W, the number of waters changes from 81 to 61, consistent with a relatively open active site. Strikingly, after DNA recognition, nearly all interfacial water molecules are within 5 Å of the enzyme or DNA and become the inner-layer hydration water, and most are in the first hydration layer within 3.5 Å. Clearly, there is no ultrafast mobile water, and all water molecules are bound to the enzyme or DNA, consistent with our observation of the two relaxation components for typical bound water. Careful examination of interfacial structures shows that these water molecules are located in clefts and caves (Figure 6B–D) that are connected to outside enzyme or DNA surface, and thus, these interfacial water clusters are not trapped in nanochannels, are mobile, and even can exchange with bulk water on a relatively long time scale.
Figure 6.
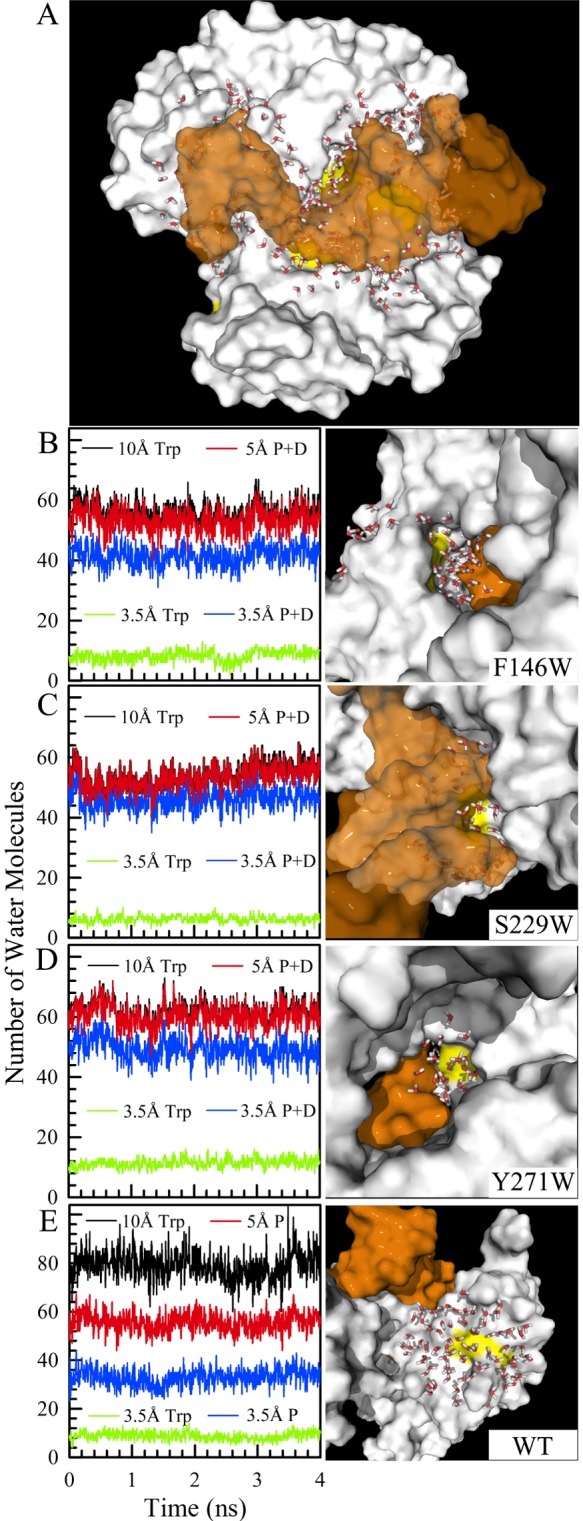
(A) Snapshot of a 4 ns MD simulation with 257 water molecules at the interface of the complex of pol β (white) and dsDNA (orange). The yellow patches indicate the tryptophan probes. (B–E) Distributions of water at different distances from the probe Trp and among these water molecules the distributions to the surfaces of polymerase and DNA. The local structures are also shown at the right. The WT represents the typical protein surface hydration from the inner to outside layers of water and to bulk solution.
The MD results show that the protein–DNA interface and active site of the pol β–DNA complex are not dry and are solvent accessible. For a tighter binding complex of pol β with DNA, a significant amount of water molecules remain at the complex interface. These interfacial molecules are not completely trapped and are still mobile in a dynamic equilibrium with bulk water. Significantly, for the pol β–DNA complex, the interfacial water molecules all become bound water with pol β or DNA to form one or two layers of hydration. Such a few layers of water are probably critical to polymerase–DNA interactions by maintaining a certain level of flexibility for the binding interface, including helping pol β quickly sample different dNTPs at the active site. By comparing these findings with the observations for the loose binding Dpo4–DNA complex (Figure 7), we observed dramatically different changes in the water–protein coupling dynamics (τ3) and related solvation speeds (S3) for the two polymerases. For Dpo4, we only observed a 15–20% increase in the long relaxation times but also an increase in the solvation speeds, consistent with the loose binding interface and more interfacial water molecules. For pol β, the long relaxation times significantly increase by a factor of 1.7–2, and all the corresponding solvation speeds decrease by 30%, consistent with the tight recognition and mostly bound water molecules at the interface. Both interfaces for the two different systems, loose or tight, are full of mobile water molecules and are dynamic in nature. Such a dynamic interface can be general for all polymerase–DNA complexes for their DNA replication or repair function.
Figure 7.

Comparison of the key third components of Dpo4 and pol β. τ3 (top) and the corresponding S3 (bottom) are directly related to the flexibility of the binding interface and the mobility of the interfacial water molecules. For pol β, the relaxation significantly slows down, but for Dpo4, it shows slight changes, reflecting the different binding properties of the two polymerases. However, all water at the interface for the two polymerases is still mobile.
Conclusions
We report here our systematic characterization of the water dynamics at several specific sites of polymerase β in its apo form, the binary complex with DNA, and the ternary state with DNA and an incoming nucleotide. Using tryptophan as an optical probe placed at strategic locations through site-directed mutagenesis, we studied two binding sites, one (F146W) near the DNA backbone in the thumb D subdomain and the other (S229W) at the DNA minor groove in the palm C subdomain, and the active site (Y271W) in the finger N subdomain. We used the wild type (W325) as a control site far from the binding interface also in the finger N subdomain. Using femtosecond fluorescence spectroscopy, we determined all hydration (solvation) dynamics in the three states of pol β. All the dynamics in the apo form completely reflect hydration water motions around a protein surface, i.e., the water relaxations from inner (a few picoseconds) and outer (subpicoseconds) layers and the coupled inner water–protein fluctuations (tens of picoseconds). Significantly, in the binding complexes, the dynamics significantly slow down by a factor of nearly 2 but still occur in picoseconds, revealing a mobile interface even for the tight binding complex. These dynamic behaviors completely reflect bound hydration water molecules interacting with the enzyme or DNA at the interface.
Combining these observed dynamics, our MD simulations, X-ray structures, and our previous studies of the loose binding Dpo4 polymerase, we can paint a general picture of polymerase–DNA binding interactions and dynamics. The heterogeneous recognition surfaces form a binding interface with various clefts, channels, and caves of different sizes and shapes. These empty nanospaces are full of water molecules with various interactions with the binding partners to mediate recognition or simply as fillers to maintain local densities. These confined water clusters are not static but are dynamic in nature, fluctuating with bulk water and lubricating polymerase–DNA interactions, leading to a dynamic interface with a different level of mobility for a loose or tight binding interface. Such a dynamic property of the interface should be general to all polymerase–DNA binding complexes to ensure the biological function of DNA synthesis.
Acknowledgments
We thank Drs. Michelle Roettger and Chih-Wei Chang for help with the experiments. This work was supported in part by an allocation of computing time by the Ohio Supercomputer Center.
Supporting Information Available
A description of data analyses of fluorescence transients in the complex states, the fluorescence transients of DNA and the three mutants in the three states, and the related fitting results. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
¶ Y.Y. and Y.Q. contributed equally to this work.
The work is supported in part by the National Institutes of Health (Grant GM095997), the National Science Foundation (Grant CHE0748358), the Camille Dreyfus Teacher-Scholar, and a Guggenheim Fellowship (to D.Z.) and by funding from Academia and MOST of Taiwan (to M.-D. T.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Beard W. A.; Wilson S. H. (2014) Structure and mechanism of DNA polymerase β. Biochemistry 53, 2768–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai M. D. (2014) How DNA polymerases catalyze DNA replication, repair, and mutation. Biochemistry 53, 2749–2451. [DOI] [PubMed] [Google Scholar]
- Florian J.; Goodman M. F.; Warshel A. (2003) Computer simulation of the chemical catalysis of DNA polymerases: Discriminating between alternative nucleotide insertion mechanisms for T7 DNA polymerase. J. Am. Chem. Soc. 125, 8163–8177. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan R.; Schlick T. (2004) Orchestration of cooperative events in DNA synthesis and repair mechanism unraveled by transition path sampling of DNA polymerase β’s closing. Proc. Natl. Acad. Sci. U.S.A. 101, 5970–5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoso Y.; Joyce C. M.; Potapova O.; Le Reste L.; Hohlbein J.; Torella J. P.; Grindley N. D. F.; Kapanidis A. N. (2010) Conformational transitions in DNA polymerase I revealed by single-molecule FRET. Proc. Natl. Acad. Sci. U.S.A. 107, 715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelch B. A.; Makino D. L.; O’Donnell M.; Kuriyan J. (2011) How a DNA polymerase clamp loader opens a sliding clamp. Science 334, 1675–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blainey P. C.; Luo G. B.; Kou S. C.; Mangel W. F.; Verdine G. L.; Bagchi B.; Xie X. S. (2009) Nonspecifically bound proteins spin while diffusing along DNA. Nat. Struct. Mol. Biol. 16, 1224–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X. P.; Onrust R.; Odonnell M.; Kuriyan J. (1992) Three-dimensional structure of the β subunit of E. coli DNA polymerase III holoenzyme: A sliding DNA clamp. Cell 69, 425–437. [DOI] [PubMed] [Google Scholar]
- Reddy C. K.; Das A.; Jayaram B. (2001) Do water molecules mediate protein-DNA recognition?. J. Mol. Biol. 314, 619–632. [DOI] [PubMed] [Google Scholar]
- Jayaram B.; Jain T. (2004) The role of water in protein-DNA recognition. Annu. Rev. Biophys. Biomol. Struct. 33, 343–361. [DOI] [PubMed] [Google Scholar]
- Levy Y.; Onuchic J. N. (2006) Water mediation in protein folding and molecular recognition. Annu. Rev. Biophys. Biomol. Struct. 35, 389–415. [DOI] [PubMed] [Google Scholar]
- Janin J. (1999) Wet and dry interfaces: The role of solvent in protein-protein and protein-DNA recognition. Struct. Folding Des. 7, 277–279. [DOI] [PubMed] [Google Scholar]
- Davey C. A.; Sargent D. F.; Luger K.; Maeder A. W.; Richmond T. J. (2002) Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J. Mol. Biol. 319, 1097–1113. [DOI] [PubMed] [Google Scholar]
- Kosztin D.; Bishop T. C.; Schulten K. (1997) Binding of the estrogen receptor to DNA. The role of waters. Biophys. J. 73, 557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billeter M.; Guntert P.; Luginbuhl P.; Wuthrich K. (1996) Hydration and DNA recognition by homeodomains. Cell 85, 1057–1065. [DOI] [PubMed] [Google Scholar]
- Sunnerhagen M.; Denisov V. P.; Venu K.; Bonvin A. M. J. J.; Carey J.; Halle B.; Otting G. (1998) Water molecules in DNA recognition I: Hydration lifetimes of trp operator DNA in solution measured by NMR spectroscopy. J. Mol. Biol. 282, 847–858. [DOI] [PubMed] [Google Scholar]
- Schwabe J. W. R. (1997) The role of water in protein DNA interactions. Curr. Opin. Struct. Biol. 7, 126–134. [DOI] [PubMed] [Google Scholar]
- Zhong D.; Pal S. K.; Zewail A. H. (2001) Femtosecond studies of protein-DNA binding and dynamics: Histone I. ChemPhysChem 2, 219–227. [DOI] [PubMed] [Google Scholar]
- Qin Y.; Yang Y.; Zhang L.; Fowler J. D.; Qiu W.; Wang L.; Suo Z.; Zhong D. (2013) Direct probing of solvent accessibility and mobility at the binding interface of polymerase (Dpo4)-DNA complex. J. Phys. Chem. A 117, 13926–13934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin M. C.; Wang J. M.; Steitz T. A. (2001) Structure of the replicating complex of a pol α family DNA polymerase. Cell 105, 657–667. [DOI] [PubMed] [Google Scholar]
- Kelman Z.; Odonnell M. (1995) DNA polymerase III holoenzyme: Structure and function of a chromosomal replicating machine. Annu. Rev. Biochem. 64, 171–200. [DOI] [PubMed] [Google Scholar]
- Yamtich J.; Sweasy J. B. (2010) DNA polymerase family X: Function, structure, and cellular roles. Biochim. Biophys. Acta 1804, 1136–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya M. R.; Prasad R.; Wilson S. H.; Kraut J.; Pelletier H. (1997) Crystal structures of human DNA polymerase β complexed with gapped and nicked DNA: Evidence for an induced fit mechanism. Biochemistry 36, 11205–11215. [DOI] [PubMed] [Google Scholar]
- Beard W. A.; Wilson S. H. (2006) Structure and mechanism of DNA polymerase β. Chem. Rev. 106, 361–382. [DOI] [PubMed] [Google Scholar]
- Pelletier H.; Sawaya M. R.; Kumar A.; Wilson S. H.; Kraut J. (1994) Structures of ternary complexes of rat DNA polymerase β, a DNA template-primer, and ddCTP. Science 264, 1891–1903. [PubMed] [Google Scholar]
- Pelletier H.; Sawaya M. R.; Wolfle W.; Wilson S. H.; Kraut J. (1996) Crystal structures of human DNA polymerase β complexed with DNA: Implications for catalytic mechanism, processivity, and fidelity. Biochemistry 35, 12742–12761. [DOI] [PubMed] [Google Scholar]
- Abbotts J.; Sengupta D. N.; Zmudzka B.; Widen S. G.; Notario V.; Wilson S. H. (1988) Expression of human DNA polymerase β in Escherichia coli and characterization of the recombinant enzyme. Biochemistry 27, 901–909. [DOI] [PubMed] [Google Scholar]
- Zhong X.; Patel S. S.; Werneburg B. G.; Tsai M. D. (1997) DNA polymerase β: Multiple conformational changes in the mechanism of catalysis. Biochemistry 36, 11891–11900. [DOI] [PubMed] [Google Scholar]
- Werneburg B. G.; Ahn J.; Zhong X.; Hondal R. J.; Kraynov V. S.; Tsai M. D. (1996) DNA polymerase β: Pre-steady-state kinetic analysis and roles of arginine-283 in catalysis and fidelity. Biochemistry 35, 7041–7050. [DOI] [PubMed] [Google Scholar]
- Kraynov V. S.; Werneburg B. G.; Zhong X.; Lee H.; Ahn J. W.; Tsai M. D. (1997) DNA polymerase β: Analysis of the contributions of tyrosine-271 and asparagine-279 to substrate specificity and fidelity of DNA replication by pre-steady-state kinetics. Biochem. J. 323, 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejewski M. W.; Liu D.; Prasad R.; Wilson S. H.; Mullen G. P. (2000) Backbone dynamics and refined solution structure of the N-terminal domain of DNA polymerase β. Correlation with DNA binding and dRP lyase activity. J. Mol. Biol. 296, 229–253. [DOI] [PubMed] [Google Scholar]
- Berlow R. B.; Swain M.; Dalal S.; Sweasy J. B.; Loria J. P. (2012) Substrate-dependent millisecond domain motions in DNA polymerase β. J. Mol. Biol. 419, 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenthal B. D.; Beard W. A.; Shock D. D.; Wilson S. H. (2013) Observing a DNA polymerase choose right from wrong. Cell 154, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya M. R.; Pelletier H.; Kumar A.; Wilson S. H.; Kraut J. (1994) Crystal structure of rat DNA polymerase β: Evidence for a common polymerase mechanism. Science 264, 1930–1935. [DOI] [PubMed] [Google Scholar]
- Freudenthal B. D.; Beard W. A.; Wilson S. H. (2012) Structures of dNTP intermediate states during DNA polymerase active site assembly. Structure 20, 1829–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.; Beard W. A.; Wilson S. H.; Broyde S.; Schlick T. (2004) Highly organized but pliant active site of DNA polymerase β: Compensatory mechanisms in mutant enzymes revealed by dynamics simulations and energy analyses. Biophys. J. 86, 3392–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.; Arora K.; Beard W. A.; Wilson S. H.; Schlick T. (2004) Critical role of magnesium ions in DNA polymerase β’s closing and active site assembly. J. Am. Chem. Soc. 126, 8441–8453. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan R.; Arora K.; Wang Y.; Beard W. A.; Wilson S. H.; Schlick T. (2006) Regulation of DNA repair fidelity by molecular checkpoints: “Gates” in DNA polymerase β’s substrate selection. Biochemistry 45, 15142–15156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Yu X.; Hu P.; Broyde S.; Zhang Y. (2007) A water-mediated and substrate-assisted catalytic mechanism for Sulfolobus solfataricus DNA polymerase IV. J. Am. Chem. Soc. 129, 4731–4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Schlick T. (2008) Quantum mechanics/molecular mechanics investigation of the chemical reaction in Dpo4 reveals water-dependent pathways and requirements for active site reorganization. J. Am. Chem. Soc. 130, 13240–13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong D. (2009) Hydration dynamics and coupled water-protein fluctuations probed by intrinsic tryptophan. Adv. Chem. Phys. 143, 83–149. [Google Scholar]
- Yang Y. (2009) Ultrafast hydration dynamics on protein surface and at protein-DNA interface. Ph.D. Thesis, The Ohio State University, Columbus, OH. [Google Scholar]
- Ahn J. W.; Kraynov V. S.; Zhong X.; Werneburg B. G.; Tsai M. D. (1998) DNA polymerase β: Effects of gapped DNA substrates on dNTP specificity, fidelity, processivity and conformational changes. Biochem. J. 331, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Kao Y.; Qiu W.; Wang L.; Zhong D. (2006) Femtosecond studies of tryptophan fluorescence dynamics in proteins: Local solvation and electronic quenching. J. Phys. Chem. B 110, 18097–18103. [DOI] [PubMed] [Google Scholar]
- Lakowicz J. R. (2007) Principles of Fluorescence Spectroscopy, Springer, Berlin. [Google Scholar]
- Perun S.; Sobolewski A. L.; Domcke W. (2005) Ab initio studies on the radiationless decay mechanisms of the lowest excited singlet states of 9H-adenine. J. Am. Chem. Soc. 127, 6257–6265. [DOI] [PubMed] [Google Scholar]
- Middleton C. T.; de La Harpe K.; Su C.; Law Y. K.; Crespo-Hernandez C. E.; Kohler B. (2009) DNA excited-state dynamics: From single bases to the double helix. Annu. Rev. Phys. Chem. 60, 217–239. [DOI] [PubMed] [Google Scholar]
- Qiu W.; Zhang L.; Kao Y. T.; Lu W.; Li T.; Kim J.; Sollenberger G. M.; Wang L.; Zhong D. (2005) Ultrafast hydration dynamics in melittin folding and aggregation: Helix formation and tetramer self-assembly. J. Phys. Chem. B 109, 16901–16910. [DOI] [PubMed] [Google Scholar]
- Qiu W.; Kao Y. T.; Zhang L.; Yang Y.; Wang L.; Stites W. E.; Zhong D.; Zewail A. H. (2006) Protein surface hydration mapped by site-specific mutations. Proc. Natl. Acad. Sci. U.S.A. 103, 13979–13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W.; Zhang L.; Okobiah O.; Yang Y.; Wang L.; Zhong D.; Zewail A. H. (2006) Ultrafast solvation dynamics of human serum albumin: Correlations with conformational transitions and site-selected recognition. J. Phys. Chem. B 110, 10540–10549. [DOI] [PubMed] [Google Scholar]
- Qin Y.; Chang C. W.; Wang L.; Zhong D. (2012) Validation of response function construction and probing heterogeneous protein hydration by intrinsic tryptophan. J. Phys. Chem. B 116, 13320–13330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Wang L.; Kao Y. T.; Qiu W.; Yang Y.; Okobiah O.; Zhong D. (2007) Mapping hydration dynamics around a protein surface. Proc. Natl. Acad. Sci. U.S.A. 104, 18461–18466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Yang Y.; Kao Y. T.; Wang L.; Zhong D. (2009) Protein hydration dynamics and molecular mechanism of coupled water-protein fluctuations. J. Am. Chem. Soc. 131, 10677–10691. [DOI] [PubMed] [Google Scholar]
- Li T.; Hassanali A. A.; Kao Y. T.; Zhong D.; Singer S. J. (2007) Hydration dynamics and time scales of coupled water-protein fluctuations. J. Am. Chem. Soc. 129, 3376–3382. [DOI] [PubMed] [Google Scholar]
- Li T.; Hassanali A. A.; Singer S. J. (2008) Origin of slow relaxation following photoexcitation of W7 in myoglobin and the dynamics of its hydration layer. J. Phys. Chem. B 112, 16121–16134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson L.; Halle B. (2005) Molecular origin of time-dependent fluorescence shifts in proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 13867–13872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halle B.; Nilsson L. (2009) Does the dynamic Stokes shift report on slow protwin hydration dynamics?. J. Phys. Chem. B 113, 8210–8213. [DOI] [PubMed] [Google Scholar]
- Furse K. E.; Corcelli S. A. (2008) The dynamics of water at DNA interfaces: Computational studies of Hoechst 33258 bound to DNA. J. Am. Chem. Soc. 130, 13103–13109. [DOI] [PubMed] [Google Scholar]
- Qiu W.; Wang L.; Lu W.; Boechler A.; Sanders D. A. R.; Zhong D. (2007) Dissection of complex protein dynamics in human thioredoxin. Proc. Natl. Acad. Sci. U.S.A. 104, 5366–5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong D.; Pal S. K.; Zewail A. H. (2011) Biological water: A critique. Chem. Phys. Lett. 503, 1–11. [Google Scholar]
- Yang J.; Zhang L.; Wang L.; Zhong D. (2012) Femtosecond conical intersection dynamics of tryptophan in proteins and validation of slowdown of hydration layer dynamics. J. Am. Chem. Soc. 134, 16460–16463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesenska A.; Sykora J.; Olzynska A.; Brezovsky J.; Zdrahal Z.; Damborsky J.; Hof M. (2009) Nanosecond time-dependent stokes shift at the tunnel mouth of haloalkane dehalogenases. J. Am. Chem. Soc. 131, 494–501. [DOI] [PubMed] [Google Scholar]
- Nucci N. V.; Pometun M. S.; Wand A. J. (2011) Site-resolved measurement of water-protein interactions by solution NMR. Nat. Struct. Mol. Biol. 18, 245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otting G.; Liepinsh E.; Wuthrich K. (1991) Protein hydration in aqueous solution. Science 254, 974–980. [DOI] [PubMed] [Google Scholar]
- Peon J.; Pal S. K.; Zewail A. H. (2002) Hydration at the surface of the protein monellin: Dynamics with femtosecond resolution. Proc. Natl. Acad. Sci. U.S.A. 99, 10964–10969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S. K.; Peon J.; Zewail A. H. (2002) Ultrafast surface hydration dynamics and expression of protein functionality: α-Chymotrypsin. Proc. Natl. Acad. Sci. U.S.A. 99, 15297–15302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz T. A.; Smerdon S. J.; Jager J.; Joyce C. M. (1994) A unified polymerase mechanism for nonhomologous DNA and RNA polymerases. Science 266, 2022–2025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.