

Abstract
We report a novel approach to a new class of bioengineered, monodispersed, self-assembling vault nanoparticles consisting of a protein shell exterior with a lipophilic core interior designed for drug and probe delivery. Recombinant vaults were engineered to contain a small amphipathic α-helix derived from the nonstructural protein 5A of hepatitis C virus, thereby creating within the vault lumen a lipophilic microenvironment into which lipophilic compounds could be reversibly encapsulated. Multiple types of electron microscopy showed that attachment of this peptide resulted in larger than expected additional mass internalized within the vault lumen attributable to incorporation of host lipid membrane constituents spanning the vault waist (>35 nm). These bioengineered lipophilic vaults reversibly associate with a sample set of therapeutic compounds, including all-trans retinoic acid, amphotericin B, and bryostatin 1, incorporating hundreds to thousands of drug molecules per vault nanoparticle. Bryostatin 1 is of particular therapeutic interest because of its ability to potently induce expression of latent HIV, thus representing a preclinical lead in efforts to eradicate HIV/AIDS. Vaults loaded with bryostatin 1 released free drug, resulting in activation of HIV from provirus latency in vitro and induction of CD69 biomarker expression following intravenous injection into mice. The ability to preferentially and reversibly encapsulate lipophilic compounds into these novel bioengineered vault nanoparticles greatly advances their potential use as drug delivery systems.
Keywords: vaults, nanoparticles, drug delivery systems, bryostatin 1, HIV latency
Chemotherapy relies heavily on small therapeutic compounds, many of which are difficult to formulate and administer or suffer from biodistribution problems. Three main strategies have evolved to address these issues: direct drug modification to create a new drug with altered physicochemical properties, drug solubilization with excipients, and/or drug encapsulation within delivery systems.1,2 Of special importance is the ability of drug delivery systems to effectively deliver therapeutic agents to targeted tissues, thereby reducing the required dosage and preventing the toxic effects of excipients and drug interactions with off-target tissues. Nanoparticle (NP) delivery systems focus predominantly on dendrimers, polymers, viruses, and liposomal formulations.3−6 However, these technologies often rely upon materials that can vary greatly in size, composition, ease of preparation, dispersion, and immunogenicity. To address these issues, we have been studying the feasibility of bioengineering vaults as a next-generation drug delivery system given their collectively unique advantages of being naturally occurring, monodispersed, biocompatible, and nonimmunogenic.7
Vaults, the largest known ribonucleoprotein complex in eukaryotic organisms, derive their name from their multiple-arched morphology reminiscent of vaulted ceilings in gothic cathedrals.8−10 Native vaults appear as hollow, thin-walled sub-100 nm ellipsoidal structures with well-defined monodispersity, uniform NP size of 67 nm × 40 nm × 40 nm, and mass of ∼13 MDa.11,12 Native vaults consist of multiple copies of three different proteins: the major vault protein (MVP, 100 kDa), vault poly(ADP-ribose) polymerase (VPARP, 193 kDa), and telomerase-associated protein 1 (TEP1, 290 kDa) along with several copies of one or more small untranslated vault-associated RNA (vRNAs).8,9 MVP constitutes ∼75% of the native vault particle mass, and exogenous expression of MVP alone in insect cells is both necessary and sufficient for the formation of hollow vault-like particles that are morphologically identical to native vaults, but lack VPARP, TEP1, and vRNA.13 Native and recombinant vaults consist of 78 copies of MVP (39 per half vault), which begin by protruding internally into the vault lumen at the NP waistline and then proceed to run lengthwise toward either vault cap, where they become externally solvent exposed.11,14,15 Novel recombinant MVP-only vaults have been created that not only retain native vault architecture but also incorporate a variety of functionalities internally and/or externally.7 Additionally, the use of an MVP interaction domain (INT, 17 kDa) derived from the C-terminus of VPARP has allowed for novel vault formation. This INT domain associates noncovalently with an internally facing MVP binding site and can be used to internally package other proteins of interest into the vault lumen via recombinant covalent attachment to INT.16,17
Initial bioengineering of the vault for sequestering small lipophilic compounds made use of a smaller NP known as a nanodisk (ND).18−21 NDs (10–20 nm discoidal lipoprotein complexes) were incorporated into the vault lumen, where they encapsulated all-trans retinoic acid (ATRA) for treatment against hepatocellular carcinoma cells.22 However, this approach is hindered by lipopolysaccharide-associated cytotoxicity and cumbersome synthesis.23 As such, we set out to refine and simplify our approach for vault-mediated drug delivery. Our search for a suitable candidate yielded an interesting peptide derived from hepatitis C virus (HCV). The nonstructural protein 5A (NS5A) is a 56 kDa viral phosphoprotein involved in HCV replication.24−27 NS5A associates with the cytoplasmic leaflet of host-cell endoplasmic reticulum membranes along with other HCV proteins involved in viral replication. NS5A interaction with host membranes has been mapped to the first 31 amino acids of the protein.28,29 NMR analysis of this region characterized it as an amphipathic α-helix (residues 5–25) of <4 nm in length that functions as a monotopic (in-plane) membrane anchor domain via lipophilic interactions between strictly conserved α-helix tryptophan residues and the acyl chains of the neighboring host phospholipids (Figure 1A). Furthermore, the polar face of this amphipathic α-helix also contains strictly conserved charged residues arranged in an asymmetrical distribution believed to have a putative functional role via interactions with other components of the viral replication complex.30,31 Interestingly, these properties also allow for their self-association in water when membrane surfaces are absent. As such, NS5A1-31 was selected for recombinant attachment to the N-terminus of MVP with the assessment that the peptide would self-associate to form an internalized protein-based lipophilic ring stabilized by complementation of ionic and van der Waals forces of adjacent copies of the amphipathic α-helix within the vault lumen (Figure 1B). Herein we detail the covalent attachment of the NS5A1-31 amphipathic α-helix to MVP, which results in the formation of a new class of nanoparticles consisting of a self-assembling protein shell with an internalized lipophilic core that is capable of reversibly sequestering small lipophilic compounds of interest for vault-mediated drug delivery (Figure 1C).
Figure 1.

(A) NMR-resolved three-dimensional structure of the hepatitis C NS5A1-31 amphipathic α-helix domain highlighting its asymmetric charge distribution along the polar face (PDB 1R7E). Hydrophobic residues are in black, while polar residues are in yellow with acidic (Glu/Asp) and basic (Arg/Lys) functional groups in red and blue, respectively. The cysteine residue SH is in green. (B) Simplified cartoon cutaway schematic of an AH vault depicting the attachment of NS5A1-31 amphipathic α-helix to MVP with subsequent vault self-assembly and self-association of adjacent NS5A1-31 amphipathic α-helices to form an internalized lipophilic ring for sequestering small therapeutic compounds. (C) Three sample therapeutic compounds that become reversibly associated within AH vaults.
Results and Discussion
Amphipathic α-Helix Vault as a Self-Assembling Protein Shell–Lipophilic Core Complex
Development of the vault as a potentially viable drug delivery system is motivated by the particles’ inherent advantages: small size, self-assembly, uniform composition, monodispersity, nonimmunogenicity, and relative ease of bioengineering. However, while encapsulation of recombinant proteins within vaults has allowed for exploitation of novel functionalities, the ability to successfully incorporate and retain small molecules of therapeutic value has been relatively unexplored. Covalent attachment of a small amphipathic α-helix from the hepatitis C viral protein NS5A was proposed to yield vaults containing a localized ring of lipophilic density lining the internal circumference of the NP as adjacent monomeric copies of NS5A1-31 self-interact due to their intrinsic properties.
Covalent attachment of NS5A1-31 to the N-terminus of MVP did not inhibit MVP expression nor interfere with vault self-assembly. During purification, the majority of these amphipathic α-helix-containing (AH) vaults were collected within the 40–45% layer of a discontinuous sucrose gradient as well-defined, intact monodispersed NPs, consistent with previous vault studies. When examined by uranyl acetate negatively stained transmission electron microscopy (TEM), AH vaults are morphologically distinct from both native and recombinant MVP-only vaults due to the presence of a significantly large region within the vault lumen that uncharacteristically lacks normal uranyl acetate staining (Figure 2A). While the absence of dark, uranyl acetate stained areas under TEM is reflective of structural features of the vault (i.e., the protein shell itself), the high intensity of this nonstaining region within most of the AH vaults is inconsistent with the amount of additional mass conferred onto the NP via covalent attachment of the 31 amino acid peptide NS5A1-31 to the N-terminus of MVP despite all 78 copies of the peptide being localized internally into the vault lumen at the NP waist.15 As such, we suspected that this additional internalized mass within AH vaults might represent additional cellular components that coassociated within the AH vault lumen via interactions with the NS5A1-31 amphipathic α-helices. As the intensity of this non-uranyl acetate staining region within AH vaults appeared to vary from vault to vault, we further hypothesized that it is dynamic in nature and could therefore be altered. When purified AH vaults were treated with 5% Tween 20 detergent followed by repurification, this nonstaining region of additional mass within AH vaults experienced a significant loss in intensity, suggesting that a sizable portion of the novel mass sequestered within the AH vault lumen is detergent soluble and therefore not part of the AH vault itself (Figure 2B).
Figure 2.

(A) Negative stain TEM of AH vaults showing significant internalized mass within a majority of the NPs (white arrowheads). (B) Negative stain TEM of AH vaults following treatment with 5% Tween 20 indicating a loss in the internalized mass prominence and frequency, suggesting a dynamic, detergent-soluble nature. (C) CryoET tilt image showing the internalized additional mass associated with AH vaults can traverse the entire vault lumen (complete, white arrowheads; incomplete, white arrows).
Since TEM imaging of vaults does not faithfully represent their true 3D structure, we turned to cryo-electron tomography (cryoET) to further characterize this additional mass within AH vaults in their frozen hydrated state in three dimensions. CryoET tilt cross-section slices revealed that a majority of AH vaults contained internalized mass that completely traverses the vault lumen (white arrowheads), a significant distance of >35 nm with an apparent thickness of ∼3–5 nm (Figure 2C). Additionally, a minority of AH vaults showed incomplete luminal coverage (white arrows). CryoET tomography tilt slices and segmentation of the 3D reconstructions of various well-defined AH vaults further confirmed that this novel additional mass sequestered within the lumen retains a dynamic morphology (Figure 3A–E). Additionally, AH vaults lacking such internalized mass (before or after detergent treatment) remain morphologically identical to a wide variety of recombinant vaults containing short peptide sequences of various length/composition/functionality at the N-terminus of the MVP, implying that the unique identity of NS5A1-31 itself must be responsible for mediating the encapsulation of this unknown additional internalized mass within these AH vaults (Figure 3F–H).
Figure 3.

CryoET tilt slices of three AH vaults with varying levels of internalized density: (A) z-plane cross section and (B) xy-plane cross section. CryoET segmented 3D reconstructions of the three different AH vaults: (C) exterior view, (D) xy-plane cross section, and (E) z-plane cross section. (F) CryoET tilt-slice cross section along the z-plane of a single AH vault identified as lacking internalized mass. (G) CryoET segmented 3D reconstruction of the same empty AH vault with z-plane cross sectioning. (H) Prior cryoET 3D reconstructions sectioned along the z-plane of MVP-only vaults (left) and His-T7-tagged MVP vaults (right). His-T7 is a 31 amino acid unstructured peptide that does not span the entire vault lumen and is shown here to illustrate the contrasting ability of the NS5A1-31 peptide (adapted from Mikyas etal.).15
The ability of this novel internalized mass to span the entire vault waist must be therefore attributable to the presence of additional cellular components, which we hypothesized to be host membrane lipid constituents (phospholipids, glycolipids, sterols, fatty acids, etc.) given NS5A1-31’s natural in vivo function as a monotopic lipid membrane anchor. The lack of contaminating proteins, DNA, or RNA within purified AH vaults, as well as dynamic morphological nature, also helps support the interpretation that this internally associated mass is constituted by various lipophilic material interacting with the 78 copies of NS5A1-31 that line the inner circumference of the vault waistline. To test this hypothesis, AH vaults were subsequently incubated with the lipophilic dye 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate (DiD), a long-chain dialkylcarbocyanine analogue that increases fluorescent intensity in the presence of a lipophilic environment.32 DiD showed a distinct qualitative increase in fluorescence upon co-incubation with AH vaults compared to free dye or dye incubated with control vaults, thereby corroborating an improved lipophilic nature associated with AH vaults. Such an internalized lipophilic core of 3–5 nm in thickness and >35 nm in diameter would represent ∼4800 nm3 of lipophilic volume within the NP available for absorption of lipophilic compounds of interest.
Drug-Binding Properties of AH Vaults
To test the ability of purified AH vaults to bind therapeutic compounds, we examined their ability to selectively retain either doxorubicin (DOX), amphotericin B (AMB), or ATRA, three small therapeutic molecules of varying lipophilicity and aqueous solubilities. The AH vaults and drugs were co-incubated, and the resulting complexes separated over an overnight discontinuous sucrose gradient. While AH vaults sediment into the denser layers of the gradient due to their large molecular mass, non-vault-associated materials remain in the lighter layers of the gradient and are subsequently lost during the final ultracentrifugation at 100000g as the NPs are pelleted. In the case of DOX, a fluorescent cytotoxic anthracycline antibiotic, neither control nor AH vaults showed any detectable retention of compound in the collected vault fraction, which is in general agreement with DOX’s high water solubility of >50 mg/mL despite a lipophilicity partition constant (LogP) of 1.27.33
Conversely, AMB, an antifungal amphipathic polyene antibiotic with poor water solubility at physiological pH of less than 0.75 mg/mL despite a LogP of 0.8, was selectively retained by AH vaults during separation with 45 molecules of AMB per single AH vault, while control vaults showed no detectable AMB association (Figure 4A).34 Similarly, ATRA, a highly toxic vitamin A-derived retinoid that regulates gene transcription with near absolute water insolubility and correlated LogP of 6.30, coassociated with AH vaults at 182 molecules of the drug per single AH vault (Figure 4B).22 However, ATRA appears to have some nonspecific association with the NP itself, as seen by its low level of association with the control vaults. The higher loading efficacy of ATRA (∼75%) over AMB (∼50%) might be reflective of the innate physicochemical properties between these compounds and consistent with the greater lipophilicity of ATRA. Since the potential volume of such a lipophilic microenvironment internalized within AH vaults could be as large as ∼4800 nm3, it is plausible for thousands to tens of thousands of hydrophobic molecules to be encapsulated within a single AH vault. As such, our initial experimental values represent only a small fraction of their theoretical maximum capacity.
Figure 4.

Spectrophotometric analysis showing the adsorption spectra for AMB (A) and ATRA (B) after incubation with either control (solid line) or AH vaults (dotted line) with subsequent repurification of vaults via ultracentrifugation over a discontinuous sucrose gradient. Spectra represent the 40–45% layer where vaults sediment.
To better address maximal loading of AH vaults, we conducted smaller scale drug-loading experiments with higher titrated ratios of drug to vault. Instead of the previous 1 μg to 100 μg ratio used above, we incubated 100 μg of control or AH vaults with 10 or 50 μg of AMB (Table 1). Following incubation, vaults and their associated drug cargo were recovered from excess, unbound material by passage through a microscale gel filtration spin column. Control vaults showed low levels of drug retention of 264 and 431 molecules of AMB per single control vault for the two respective loading conditions. In contrast, AH vaults showed considerably higher AMB retention following post column separation with 1213 and 2071 molecules of AMB per single AH vault for the two conditions. It seems likely that the AMB seen in the recovered control vault samples represent a weak and/or transient nonspecific drug–vault interaction that occurs at these higher AMB to vault ratios. Meanwhile, the 4–5-fold increase in AMB associated with the AH vaults is apparently due to sequestration of the drug in their large lipophilic microenvironment. The calculated values from the 10 and 50 μg AMB:100 μg AH vault samples do not violate the starting ratio of 815:1. Rather the observed 1213 and 2071 AMB per single AH vault stems from inefficient vault recovery over the gel filtration spin column, which leads to an apparent overcalcultion of the loading efficiency.
Table 1. AH Vault Drug-Loading Capacity Using Amphotericin B vs Control Vaults.
| AMB:vault starting ratio (M) | AMB:vault ending ratio (M) | AMB:vault ending ratio (post 7 days) (M) | % change | |
|---|---|---|---|---|
| control vault | 10 μg:100 μg (815:1) | 32.6 ng:1 μg (264:1) | 26.8 ng:1 μg (217:1) | 18% ↓ |
| 50 μg:100 μg (4075:1) | 53.0 ng:1 μg (431:1) | 28.0 ng:1 μg (228:1) | 47% ↓ | |
| AH vault | 10 μg:100 μg (815:1) | 149.2 ng:1 μg (1213:1) | 133.6 ng:1 μg (1085:1) | 11% ↓ |
| 50 μg:100 μg (4075:1) | 254.7 ng:1 μg (2071:1) | 240.8 ng:1 μg (1958:1) | 6% ↓ |
Following a 1-week storage at 4 °C, the drug-bound vaults were re-examined. Control vault samples experienced a mild to severe drop in AMB content (18% loss for the 10 μg sample and 47% for the 50 μg sample, respectively). Conversely, AH vaults experienced only a minor drop in associated drug with 90% or greater of the original AMB being retained (11% loss for the 10 μg sample and 6% for the 50 μg sample, respectively). This loss of AMB in the control vault samples may be indicative of poor protection from the aqueous environment, thereby allowing faster molecular decomposition in line with AMB’s general stability. Meanwhile, the negligible loss of AMB in the AH vault samples likely results from the drug molecules being sequestered within the NP’s lipophilic core, which provides greater overall stability and protection. The ability of AH vaults to encapsulate >2000 drug molecules while potentially offering a more hospitable microenvironment is an important milestone in their development as a novel excipient reservoir for solubilizing lipophilic therapeutic compounds for drug delivery.
AH Vaults for Treating HIV Latency Using Bryostatin 1
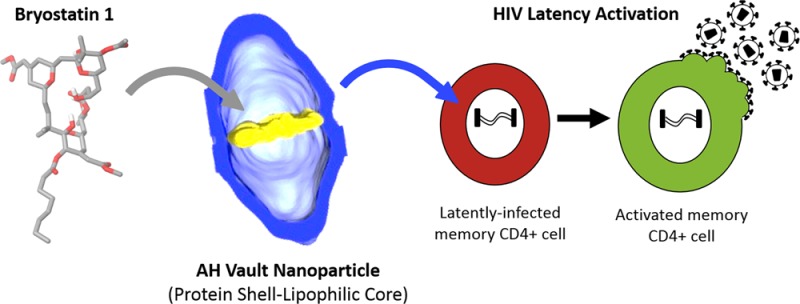
As an initial clinical application for the AH vaults, we focused on the delivery of bryostatin 1, a promising therapeutic lead for the treatment of Alzheimer’s disease, cancer, and HIV/AIDS eradication. Bryostatins are a family of macrolide lactone compounds with potent neurobiochemical, anticancer, and immuno-modulating properties both in vitro and in vivo, which operate putatively through modulation of protein kinase C (PKC).35,36 While practical access to the bryostatins from natural sources (e.g., Bugula neritina), engineered biosynthesis, and total synthesis are still works in progress, more accessible, designed, and highly potent analogues are now becoming available.37 However, little attention has been directed at the formulation and delivery of these agents, issues that are of huge significance to preclinical studies on both the natural and designed analogues.
Our specific interest in bryostatin and their analogues (bryologues) is driven in part from their potential use in the eradication of HIV/AIDS. In the span of a generation, AIDS/HIV has undergone a remarkable transformation from a lethal infection to a largely manageable chronic disease. However, over 33 million people remain infected with the virus. Highly active antiretroviral therapy (HAART) currently reduces viral activity to undetectable levels, thereby slowing disease progression. However, HAART does not address the issue of provirus latency within cellular reservoirs, which can resupply active virus. Latently infected CD4+ T cells can persist for decades in HAART-treated individuals and therefore represent a key barrier to disease eradication.38 One potential method for eliminating the latent reservoir of HIV is to induce the dormant provirus to express viral proteins, which would make the host cell susceptible to immune effector mechanisms, viral cytopathic effects, and additional therapies directed toward viral proteins such as anti-HIV envelope immunotoxins.39 Delivery of latency activators using nanoparticles could afford a variety of benefits, including the combinatorial and targeted introduction of these agents specifically to the cell type of choice, leading to less toxicity associated with activation of nontarget cell types. To explore this possibility, we previously introduced bryostatin 1 into lipid nanoparticles and demonstrated that these particles activate latent HIV and can also be targeted toward CD4+ T cells using monoclonal antibodies.40 Here we have incorporated bryostatin 1 into AH vault particles and evaluated their properties in vitro and in vivo.
Bryostatin 1 (LogP of 4.25–5.40, predicted) incorporation into AH vaults was assessed by high-performance liquid chromatography (HPLC) coupled with multiple reaction monitoring (MRM) tandem mass spectrometry (MS/MS) in lieu of spectrophotometric analysis.41 MRM-LC-MS/MS allowed for sensitive detection (>0.009 ng/μL) of the sodiated bryostatin 1 ion at m/z 927.4, consistent with previous reports.42 AH vaults were co-incubated for 30 min at 4 °C with bryostatin 1 and subsequently collected from solution using ultracentrifugation at 100000g. Aliquots of the starting material, spin supernatant, and the resuspended vaults were analyzed by MRM-LC-MS/MS, and the bryostatin 1 concentration was measured using a standard curve generated using known concentrations of bryostatin 1. Approximately 10.6 ± 1.4 ng of bryostatin 1 was measured in the incubated, precentrifuged sample and is in accordance with the expected value of 10 ng/μL (1 μg of bryostatin 1 per 100 μg of AH vault in 100 μL of PBS–). Meanwhile, the 13.4 ± 2.3 ng of bryostatin 1 measured in the resuspended vault pellet is within experimental error and indicates that approximately 100% of the starting bryostatin 1 was retained through association with the AH vaults with ∼83 molecules of the drug per single AH vault. However, because of the low availability and high cost associated with bryostatin 1, only a few initial studies were conducted using control vaults.
Compared to empty AH vaults, AH vaults containing bryostatin 1 showed clear dose-dependent activation of latent HIV provirus in the J-Lat 10.6 cell line, a well-characterized model for the main T-lymphocyte cell reservoir, with activity starting at 1 ng/μL of AH vaults (Figure 5A). Alternatively, stimulation of T cells with PKC-activating compounds, such as bryostatin 1, induces cell surface expression of CD69, which occurs at similar concentrations to those required to activate HIV from latency.43,44 As such, CD69 expression can be used as a biomarker for evaluating whether bryostatin 1 delivered via association with AH vaults remains bioactive in the desired T cell type. When tested for activity in this way, bryostatin 1-loaded AH vaults activated CD69 expression in primary human peripheral blood mononuclear cells (PBMCs) obtained from four different donors in a dose-dependent manner with stimulation occurring at concentrations as low as 0.1 ng/μL AH vaults as analyzed by flow cytometry (Figure 5B). To evaluate whether these drug-loaded vaults are also bioactive in vivo, they were injected into C57/bl6 mice at 1 μg of bryostatin 1 per 100 μg of AH vaults per mouse. At 24 h postinjection, over 90% of CD4+ T cells present within harvested splenocytes had been induced to express CD69, demonstrating that the bryostatin 1-loaded AH vaults can successfully deliver compounds in vivo (Figure 5C). Notably, these untargeted vaults also induced >70% of non-CD4+ T cells (primarily CD8+ T cells) and ∼40% of non-T cells to express CD69. This activation of a broad spectrum of cell types illustrates the potential benefits of targeting the vaults more selectively to the cell type of interest (in this case, to CD4+ T cells). Methods for targeting vaults are available and are currently being explored.45 Further improvements to the HIV provirus latency activating AH vaults could also be achieved by using more potent analogues of bryostatin or prostratin.36,43,46 Second-generation AH vaults could therefore include both targeting moieties and enhanced latency-activating compounds.
Figure 5.

(A) In vitro latent HIV activation using bryostatin 1-loaded AH vaults on J-Lat 10.6 cells for 48 h. In these assays 50 nM bryostatin 1 compound (without vaults) was used as a positive control and induced GFP expression in 30.6% (±0.6%) of cells. Error bars indicate ±1 SD (N = 3). NS not significant, **p < 0.0001 empty vault vs vault + bryostatin 1 (2-sided t test). (B) In vitro stimulation of CD69 (HIV provirus latency activation biomarker) activation using bryostatin 1-loaded AH vaults on primary human PBMCs for 24 h. Positive control stimulations with 50 nM bryostatin 1 compound (without vaults) induced CD69 expression in 69.4% (±31.3%) of cells. Error bars indicate ±1 SD (N = 4 different cell donors). NS not significant, *p < 0.01 empty vault vs vault + bryostatin 1 (2-sided t test). (C) In vivo CD69 stimulation from harvested C57/bl6 mouse splenocytes at 24 h postinjection of either media control, bryostatin 1, bryostatin 1-loaded AH vaults, or empty AH vaults. Error bars indicate ±1 SD (3–5 mice per group).
Conclusions
These experiments demonstrate the novel ability of AH vaults to encapsulate various therapeutic compounds, up to >2000 molecules per single AH vault, through reversible association with a highly lipophilic interior that results from the covalent attachment of an amphipathic α-helical peptide to the MVP (summarized in Table 2). Furthermore, these drug-loaded AH vaults can deliver their therapeutic cargo, such as bryostatin 1, in vitro using both cultured and primary cell lines, as well as in vivo. The formation of these self-assembling protein shell–lipophilic core NPs benefits not only from the key properties of vaults such as monodispersity, cell specific targeting, and biocompatibility, but also from being a vault-drug formulation based approach that can completely internalize their cargo, thereby offering an additional layer of protection. As such, these novel AH vaults represent an interesting “proof of concept” approach to functionalizing the vault for potential use in drug delivery.
Table 2. Summarization of Bioengineered AH Vaults’ Ability to Selectively Associate with a Small Sample Set of Therapeutic Compounds with Different Physicochemical Properties via Noncovalent, Reversible Association within a Novel Internalized Lipophilic Core Microenvironment Generated by the Covalent Attachment of a Unique Amphipathic α-Helix Peptide (NS5A1-31) to the MVP.
| therapeutic compound | solubilityH2O (mg/mL) | LogP (octanol/water) | loading condition | drug:vault starting ratio (M) | drug:vault ending ratio (M) | detection method |
|---|---|---|---|---|---|---|
| doxorubicin (580 g/mol) | 50 | 1.27 (PubChem) | 0.5 h at 4 °C, 20 h centrifugation | 10 μg:1000 μg (129:1) | none detected | UV/vis spectra, DOXε480nn = 11 500 M–1 cm–1 |
| amphotericin B (924 g/mol) | 0.75 | 0.8 (DrugBank) | 0.5 h at 4 °C, 20 h centrifugation | 10 μg:1000 μg (80:1) | 5.6 ng:l μg (45:1) | UV/vis spectra, AMBε406 nm = 150 000 M–1 cm–1 |
| 0.5 h at 4 °C, 5 min spin column | 10 μg:100 μg (815:1) | 149.2 ng:l μg (1213:1) | UV/vis spectra, AMBε406 nm = 150 000 M–1 cm–1 | |||
| 0.5 h at 4 °C, 5 min spin column | 50 μg:100 μg (4075:1) | 254.7 ng:l μg (2071:1) | UV/vis spectra, AMBε406 nm = 150 000 M–1 cm–1 | |||
| all-trans retinoic acid (300 g/mol) | insoluble | 6.30 (DrugBank) | 0.5 h at 4 °C, 20 h centrifugation | 10 μg:1000 μg (250:1) | 7.3 ng:l μg (182:1) | UV/vis spectra, ATRAε350 nm = 44,300 M–1 cm–1 |
| bryostatin 1 (905 g/mol) | insoluble | 4.25–5.40 (predicted, ChEMBL and ChemSpyder) | 0.5 h at 4 °C, 1 h centrifugation | 1 μg:100 μg (83:1) | 13.4 ng:l μg (83:1) | mass spec (MRM-LC-MS/MS) |
Materials and Methods
AH Vault Cloning, Expression, and Purification
NS5A1-31 was PCR amplified from a construct containing HCV NS5A1-31 fused to GFP generously provided by Darius Moradpour M.D. at the Centre Hospitalier Universitaire Vaudois, University of Lausanne, Switzerland. The following primers were used to generate a ∼100 bp PCR fragment with flanking Nco I restriction sites (underlined). Forward: 5′GAATTCACCATGGCCGGTTCCTGGC3′, Reverse: 5′CCTTGCTCACCCATGGTTGGCATGAG3′. The resulting PCR fragment contained the amino acid residue sequence MAGSWLRDIWDWICEVLSDFKTWLKAKLMPTM, which incorporates a start Met, as well as two codon sequence changes (Ser1Ala and Gln31Thr, underlined) expected to behave as silent mutations with no anticipated consequence on the structural folding of the amphipathic helix domain contained from residues 5–25. Residue Leu31 is replaced with the start Met of MVP (underlined, bold). Subsequent Nco I digestion was used to insert NS5A1-31 immediately upstream, in-frame to rat MVP cDNA previously cloned into pFastBac-1 vector using standard molecular biology techniques. This construct was subsequently used to establish a baculovirus used in Sf9 (Spodoptera frugiperda) cell expression, as previously detailed elsewhere.13 Purification of AH vaults was conducted using standard vault purification protocol with isolated vaults collected within the 40% or 45% layer of a semidiscontinuous sucrose gradient and stored in 1× PBS– at 4 °C. Roughly 10–20% of total AH vaults assembled into larger structures called vaultimers, located primarily in the 50% sucrose gradient fraction, which have been previously observed.13 Detergent treatment of AH vaults was carried out by incubating purified vaults in 1× PBS– + 5% Tween 20 for 1 h at 4 °C with rotation and repurified according to standard methods.
AH Vault Transmission Electron Microscopy, Cryo-electron Tomography, and Segmentation
Negative staining and TEM were conducted using a JEM 1200EX microscope (JEOL) equipped with a Bioscan 600 W digital camera (Gatan Inc.). Cryogenically frozen samples were prepared on Quantifoil grids by plunge freezing. An FEI Tecnai F20 transmission cryo-electron microscope was used to acquire tomography tilt series from −70° to +70° with the batch tomography program (FEI). The tilt series were recorded on a 16 megapixel TVIPS CCD camera with a total electron dosage of about 100 electrons on each imaged sample area and a magnification that gives a pixel size of 0.45 nm per pixel at the specimen level. The tomography tilt series were processed with a suite of programs to generate a segmented 3D reconstruction as follows. First, the series were precisely aligned using the eTomo program from the Imod package. Rough alignment was performed by cross-correlation of images in the tilt series, followed by fine alignment using 10 nm gold beads as fiducial markers. A boundary model was created to adjust the tilt axis before a final, precisely aligned tilt stack was produced for each series. The aligned tilt series was then used to make a 3D reconstruction using GPU-based SIRT (simultaneous iterative reconstruction technique) reconstruction implemented in the Inspect3D program from FEI. The 3D reconstructions were saved as a stack of x–y sections stacked along the z direction. This 3D tomographic reconstruction was further processed by median filtering and pixel binning to enhance structural contrast. Individual vault particles were then segmented from the 3D density maps and displayed by volume renderings of the vault particles with packaged material with Amira (Visage Inc.). For densities corresponding to the vault particle, the known 39-fold symmetry was imposed to improve the signal-to-noise ratio for volume rendering.
AH Vault Drug Packaging and Spectrophotometric Analysis
AH vault lipophilicity was initially assayed using 5 μL of a 10 μg/μL 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate (DiD(644 ex/665 em), Invitrogen Molecular Probes), dimethyl sulfoxide (DMSO) stock added to 1000 μg of prepurified AH vault, control vault, bovine serum albumin (Sigma), or alone in 1× PBS– buffer for 30 min at 4 °C with rotation and protection from light. Samples were overlaid onto 1 mL of 1× PBS– buffer in TLA100.1 rotor tubes (Beckman Coulter) and ultracentrifuged at 100000g for 1 h at 4 °C. Pellets were resuspended in 100 μL of 1× PBS– buffer, and fluorescence was measured at 665 nm.
Retention of either doxorubucin (Sigma), amphotericin B (Sigma), or all-trans retinoic acid (Sigma) was conducted as follows: 1000 μg of prepurified AH or CP vaults was incubated with 10 μg of each respective compound from appropriate stocks into 1 mL of 1× PBS– buffer for 30 min at 4 °C with rotation and protection from light. Samples were overlaid onto a semidiscontinuous sucrose gradient and centrifuged at 25 000 rpm (77000g) using a Beckman SW41 Ti swinging bucket rotor for 16 h at 4 °C on a Beckman-Coulter ultracentrifuge. Gradient fractions were collected and ultracentrifuged for 2 h at 40 000 rpm (100000g) to pellet vaults. Collected pellets were resuspended in 1× PBS– buffer and assayed for purity by either SDS-PAGE with Coomassie blue staining or by Western blotting and quantitated by bicinchoninic acid (BCA). Collected fraction samples were standardized to [vault] = 1 μg/μL and further diluted 1:10 for UV/vis absorbance spectroscopy in a 100 μL microquartz cuvette from 300 to 450 or 300 to 600 nm with the long wavelength of vault-only control samples adjusted to baseline. Compound concentration was calculated using known molar extinction coeffecients (DOX ε480 nm = 11 500 M–1 cm–1(water), AMBε406 nm = 150 000 M–1 cm–1(methanol), ATRAε350 nm = 44 300 M–1 cm–1(ethanol)).
Alternatively, small-scale drug loading in control or AH vaults was conducted in a semianalogous manner. Either 10 or 50 μg of AMB was added to 100 μg of prepurified control or AH vaults in PBS– buffer to a total volume of 100 μL for 30 min at 4 °C with rotation and protection from light. Samples were then applied directly to Micro Bio-Spin P6 gel columns (Bio-Rad) that were pre-equilibrated with four washes of 400 μL of 1× PBS– buffer and centrifuged for 4 min at 1000g at room temperature to collect vaults and associated AMB within the filtrate. Control AMB in 1× PBS– buffer alone was completely (100%) removed from the filtrate following passage over the gel filtration column. Collected filtrates were analyzed for vault concentration using BCA, while AMB concentration was assayed using the previously mentioned known molar extinction coefficient using a Nanodrop 1000 (Thermo Scientific). Samples were stored at 4 °C and reassayed after 1 week.
AH Vault Packaging of Bryostatin 1
A 1 μg amount of bryostatin-1 (Tocris Bioscience) in ethanol was incubated with 100 μg of prepurified AH vaults (1 wt %/wt) in 100 μL of 1× PBS– for 30 min at 4 °C. A small aliquot of this starting material was saved for analysis, and the remaining suspension treated with ultracentrifugation at 53 000 rpm (100000g) for 1 h at 4 °C in TLA100.1 rotor tubes on a Beckman Coulter tabletop ultracentrifuge. Supernatant, containing any unbound bryostatin 1, was carefully aspirated and saved for analysis. The AH vault pellet was resuspended in 50 μL of 1× PBS– and incubated overnight at 4 °C with gentle rotation. Aliquots of the starting material (bryostatin 1 and prepurified AH vaults prior to centrifugation), centrifugation supernatant, and resuspended centrifugation pellet were prepared for HPLC-MRM-MS/MS analysis.
AH Vault Bound Bryostatin 1 Quantitation by Liquid Chromatography–Mass Spectrometry
Bryostatin 1 AH vault starting material, supernatant, and resuspended pellet were analyzed to determine binding of bryostatin 1 to AH vaults. A 1 μL sample of each of the three samples was diluted 1:200 in pure methanol and transferred to an autosampler vial containing a 0.2 mL glass insert. Injections of 25 μL were made onto an Agilent 1200 HPLC system using an Agilent Zorbax 300SB-C3 (3 μm, 2.1 cm × 150 mm) column. Samples were eluted using a linear gradient of 100% solvent A (98% water, 2% acetonitrile, 0.1% formic acid) to 100% solvent B (98% acetonitrile, 0.1% formic acid) over 10 min with a 4 min isocratic hold at 100% solvent B at a flow rate of 250 μL/min. Then 25 μL injections of 0.1% formic acid were inserted throughout the run to assess and address bryostatin 1 carryover in sequential analyses. The sodiated bryostatin 1 [M + Na]+ ion was monitored on an AB Sciex 4000 QTRAP triple quadrupole mass spectrometer outfitted with a Turbo Spray ESI source. The instrument was operated with 5500 V applied to the ESI source and a capillary temperature of 250 °C. Bryostatin 1 was analyzed in MRM mode, measuring three separate precursor–product ion transitions: m/z 927.4 → 867.4, 927.4 → 593.3, and 927.4 → 549.3. The average peak area of all three bryostatin 1 transitions was determined for each analysis using Analyst 1.4.2 software for transition peak integration. Each sample was run in triplicate except for the supernatant, which was analyzed only once. A stock solution of bryostatin 1 in pure methanol was prepared at a concentration of 905 ng/μL. This stock solution was then serially diluted in methanol to construct a standard curve at the following concentrations: 0.009, 0.045, 0.090, 0.225, and 0.450 ng/μL. Standard bryostatin 1 samples were analyzed using the same instrument and method parameters listed above. All measurements were performed in triplicate.
AH Vault Packaged Bryostatin 1 in Vitro HIV Latency Stimulation and Analysis
J-Lat clone 10.6 cells were obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. These cells were cultured in RPMI medium 1640 (Invitrogen) containing 10% fetal bovine serum (Omega Scientific) and 100 units/mL of penicillin and 100 μg/mL of streptomycin (Invitrogen). During stimulation cells were seeded at a concentration of 50 000 cells/well in a V-bottomed 96-well plate containing the relevant concentration of vaults in 100 μL of RPMI media. Cells were incubated for 48 h and then harvested by washing with fresh media and resuspending pellets in 2% paraformaldehyde. The percentage of GFP+ cells was quantified by flow cytometry using a LSRFortessa machine (BD Biosciences).
AH Vault Packaged Bryostatin 1 Human PBMC in Vitro CD69 Stimulation and Analysis
Peripheral blood mononuclear cells isolated using Ficoll-Paque Plus separation (GE Healthcare) from HIV sero-negative donors were obtained from the UCLA Virology Core. Cells were placed in V-bottomed 96-well plates at a density of 100 000 cells per well in a 200 μL volume of RF10 media containing the indicated vaults. Media only and stimulation with 50 nM bryostatin 1 compound alone were used as controls. Cells were incubated for 48 h before harvesting for analysis using flow cytometry.
AH Vault Packaged Bryostatin 1 in Vivo Animal CD69 Stimulation and Analysis
Animal experiments were approved by the UCLA animal research committee and conformed to all local and national guidelines and regulations. C57/bl6 mice were obtained from the UCLA Department of Radiation Oncology. A 100 μg amount of AH vaults at 10 ng of bryostatin 1 per 1 μg of vault was resuspended in 150 μL of RPMI media and then administered intravenously via retro-orbital injection. At 24 h postinjection mice were sacrificed and the spleens isolated. Spleens were disaggregated by forcing tissue through a steel mesh and then filtered through a 40 μm filter to produce a single-cell suspension. These cells were then stained for flow cytometry to analyze surface expression of relevant markers.
Flow Cytometry Analysis of Cell Surface Markers
Cells were stained by first suspending them in 50 μL of a 1:1 mix of 1× PBS– and human AB serum (Sigma). For human PBMC analysis the cells were stained with anti-human CD69-phycoerythrin (Beckman Coulter). For mouse splenocyte analysis the cells were stained with a combination of anti-mouse CD3-fluorescein isothiocyanate (eBioscience), CD69-phycoerythrin (eBioscience), and CD4-peridinin chlorophyll protein (eBioscience). Following antibody addition, the mixture was incubated at 4 °C for 15 min. The cells were then washed, resuspended in 1× PBS– containing 2% paraformaldehyde, and stored at 4 °C. Fixed cells were analyzed using an LSRFortessa (BD Biosciences). The resultant list mode files were processed using FlowJo software (version 7.6).
Acknowledgments
The authors gratefully acknowledge support from the National Institutes of Health (AI70010 to J.Z., U19AI096113, Project 3.4 to J.Z., GM071940 and AI094386 to Z.H.Z., S10RR024605 and R01GM103479 to J.A.L., and R01 CA031841 and R01 CA031845 to P.A.W.); the UCLA CFAR (AI28697) for core support and a UCLA CFAR Seed Grant to L.H.R.; and a Grand Challenges Explorations Grant (OPP1032668 to J.Z.) from the Bill and Melinda Gates Foundation.
The authors declare the following competing financial interest(s): V.A.K. and L.H.R. have financial interest in a startup company, Vault Nano Inc., that has licensed intellectual property used in this study from the Regents of the University of California.
Author Contributions
△ D. C. Buehler and M. D. Marsden contributed equally to this work.
Funding Statement
National Institutes of Health, United States
References
- Fauman E. B.; Rai B. K.; Huang E. S. Structure-Based Druggability Assessment-Identifying Suitable Targets for Small Molecule Therapeutics. Curr. Opin. Chem. Biol. 2011, 15, 463–468. [DOI] [PubMed] [Google Scholar]
- Uskokovic V. Entering the Era of Nanoscience: Time To Be So Small. J. Biomed. Nanotechnol. 2013, 9, 1441–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y.; Nolte R. J.; Cornelissen J. J. Virus-Based Nanocarriers for Drug Delivery. Adv. Drug Delivery Rev. 2012, 64, 811–825. [DOI] [PubMed] [Google Scholar]
- Allen T. M.; Cullis P. R. Drug Delivery Systems: Entering the Mainstream. Science 2004, 303, 1818–1822. [DOI] [PubMed] [Google Scholar]
- Felnerova D.; Viret J. F.; Gluck R.; Moser C. Liposomes and Virosomes as Delivery Systems for Antigens, Nucleic Acids and Drugs. Curr. Opin. Biotechnol. 2004, 15, 518–529. [DOI] [PubMed] [Google Scholar]
- Duncan R. The Dawning Era of Polymer Therapeutics. Nat. Rev. Drug Discovery 2003, 2, 347–360. [DOI] [PubMed] [Google Scholar]
- Rome L. H.; Kickhoefer V. A. Development of the Vault Particle as a Platform Technology. ACS Nano 2013, 7, 889–902. [DOI] [PubMed] [Google Scholar]
- van Zon A.; Mossink M. H.; Scheper R. J.; Sonneveld P.; Wiemer E. A. The Vault Complex. Cell. Mol. Life Sci. 2003, 60, 1828–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suprenant K. A. Vault Ribonucleoprotein Particles: Sarcophagi, Gondolas, or Safety Deposit Boxes?. Biochemistry 2002, 41, 14447–14454. [DOI] [PubMed] [Google Scholar]
- Kedersha N. L.; Rome L. H. Isolation and Characterization of a Novel Ribonucleoprotein Particle: Large Structures Contain a Single Species of Small RNA. J. Cell Biol. 1986, 103, 699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H.; Kato K.; Yamashita E.; Sumizawa T.; Zhou Y.; Yao M.; Iwasaki K.; Yoshimura M.; Tsukihara T. The Structure of Rat Liver Vault at 3.5 Angstrom Resolution. Science 2009, 323, 384–388. [DOI] [PubMed] [Google Scholar]
- Kedersha N. L.; Heuser J. E.; Chugani D. C.; Rome L. H. Vaults. III. Vault Ribonucleoprotein Particles Open into Flower-Like Structures with Octagonal Symmetry. J. Cell Biol. 1991, 112, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen A. G.; Raval-Fernandes S.; Huynh T.; Torres M.; Kickhoefer V. A.; Rome L. H. Assembly of Vault-Like Particles in Insect Cells Expressing Only the Major Vault Protein. J. Biol. Chem. 2001, 276, 23217–23220. [DOI] [PubMed] [Google Scholar]
- Kong L. B.; Siva A. C.; Rome L. H.; Stewart P. L. Structure of the Vault, a Ubiquitous Cellular Component. Structure 1999, 7, 371–379. [DOI] [PubMed] [Google Scholar]
- Mikyas Y.; Makabi M.; Raval-Fernandes S.; Harrington L.; Kickhoefer V. A.; Rome L. H.; Stewart P. L. Cryoelectron Microscopy Imaging of Recombinant and Tissue Derived Vaults: Localization of the MVP N-Termini and VPARP. J. Mol. Biol. 2004, 344, 91–105. [DOI] [PubMed] [Google Scholar]
- Kickhoefer V. A.; Garcia Y.; Mikyas Y.; Johansson E.; Zhou J. C.; Raval-Fernandes S.; Minoofar P.; Zink J. I.; Dunn B.; Stewart P. L.; etal. H. Engineering of Vault Nanocapsules with Enzymatic and Fluorescent Properties. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 4348–4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poderycki M. J.; Kickhoefer V. A.; Kaddis C. S.; Raval-Fernandes S.; Johansson E.; Zink J. I.; Loo J. A.; Rome L. H. The Vault Exterior Shell Is a Dynamic Structure that Allows Incorporation of Vault-Associated Proteins into Its Interior. Biochemistry 2006, 45, 12184–12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayburt T. H.; Sligar S. G. Membrane Protein Assembly into Nanodiscs. FEBS Lett. 2010, 584, 1721–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civjan N. R.; Bayburt T. H.; Schuler M. A.; Sligar S. G. Direct Solubilization of Heterologously Expressed Membrane Proteins by Incorporation into Nanoscale Lipid Blayers. BioTechniques 2003, 35, 556–560562–563. [DOI] [PubMed] [Google Scholar]
- Ryan R. O. Nanodisks: Hydrophobic Drug Delivery Vehicles. Expert Opin. Drug Delivery 2008, 5, 343–351. [DOI] [PubMed] [Google Scholar]
- Tufteland M.; Ren G.; Ryan R. O. Nanodisks Derived from Amphotericin B Lipid Complex. J. Pharm. Sci. 2008, 97, 4425–4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buehler D. C.; Toso D. B.; Kickhoefer V. A.; Zhou Z. H.; Rome L. H. Vaults Engineered for Hydrophobic Drug Delivery. Small 2011, 7, 1432–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erridge C.; Bennett-Guerrero E.; Poxton I. R. Structure and Function of Lipopolysaccharides. Microbes Infect. 2002, 4, 837–851. [DOI] [PubMed] [Google Scholar]
- Pawlotsky J. M.; Germanidis G. The Non-Structural 5A Protein of Hepatitis C Virus. J. Viral Hepat. 1999, 6, 343–356. [DOI] [PubMed] [Google Scholar]
- Macdonald A.; Harris M. Hepatitis C Virus NS5A: Tales of a Promiscuous Protein. J. Gen. Virol. 2004, 85, 2485–2502. [DOI] [PubMed] [Google Scholar]
- McLauchlan J. Hepatitis C virus: Viral Proteins on the Move. Biochem. Soc. Trans. 2009, 37, 986–990. [DOI] [PubMed] [Google Scholar]
- Khabar K. S.; Polyak S. J. Hepatitis C Virus-Host Interactions: The NS5A Protein and the Interferon/Chemokine Systems. J. Interferon Cytokine Res. 2002, 22, 1005–1012. [DOI] [PubMed] [Google Scholar]
- Penin F.; Brass V.; Appel N.; Ramboarina S.; Montserret R.; Ficheux D.; Blum H. E.; Bartenschlager R.; Moradpour D. Structure and Function of the Membrane Anchor Domain of Hepatitis C Virus Nonstructural Protein 5A. J. Biol. Chem. 2004, 279, 40835–40843. [DOI] [PubMed] [Google Scholar]
- Moradpour D.; Brass V.; Penin F. Function Follows Form: The Structure of the N-Terminal Domain of HCV NS5A. Hepatology 2005, 42, 732–735. [DOI] [PubMed] [Google Scholar]
- Brass V.; Bieck E.; Montserret R.; Wolk B.; Hellings J. A.; Blum H. E.; Penin F.; Moradpour D. An Amino-Terminal Amphipathic Alpha-Helix Mediates Membrane Association of the Hepatitis C Virus Nonstructural Protein 5A. J. Biol. Chem. 2002, 277, 8130–8139. [DOI] [PubMed] [Google Scholar]
- Tellinghuisen T. L.; Marcotrigiano J.; Rice C. M. Structure of the Zinc-Binding Domain of an Essential Component of the Hepatitis C Virus Replicase. Nature 2005, 435, 374–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H.; Larson R. G. Preparation of Fluorescent Particles with Long Excitation and Emission Wavelengths Dispersible in Organic Solvents. Langmuir 2004, 20, 7436–7443. [DOI] [PubMed] [Google Scholar]
- Zeman S. M.; Phillips D. R.; Crothers D. M. Characterization of Covalent Adriamycin-DNA Adducts. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 11561–11565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill R. J. Amphotericin B Formulations: A Comparative Review of Efficacy and Toxicity. Drugs 2013, 73, 919–934. [DOI] [PubMed] [Google Scholar]
- Mehla R.; Bivalkar-Mehla S.; Zhang R.; Handy I.; Albrecht H.; Giri S.; Nagarkatti P.; Nagarkatti M.; Chauhan A. Bryostatin Modulates Latent HIV-1 Infection via PKC and AMPK Signaling but Inhibits Acute Infection in a Receptor Independent Manner. PLoS One 2010, 5, e11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeChristopher B. A.; Loy B. A.; Marsden M. D.; Schrier A. J.; Zack J. A.; Wender P. A. Designed, Synthetically Accessible Bryostatin Analogues Potently Induce Activation of Latent HIV Reservoirs in Vitro. Nat. Chem. 2012, 4, 705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender P. A.; Baryza J. L.; Brenner S. E.; DeChristopher B. A.; Loy B. A.; Schrier A. J.; Verma V. A. Design, Synthesis, and Evaluation of Potent Bryostatin Analogs that Modulate PKC Translocation Selectivity. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 6721–6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden M. D.; Zack J. A. Establishment and Maintenance of HIV Latency: Model Systems and Opportunities for Intervention. Future Virol. 2010, 5, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden M. D.; Zack J. A. HIV/AIDS Eradication. Bioorg. Med. Chem. Lett. 2013, 23, 4003–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovochich M.; Marsden M. D.; Zack J. A. Activation of Latent HIV Using Drug-Loaded Nanoparticles. PLoS One 2011, 6, e18270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bignami G. S.; Wagner F.; Grothaus P. G.; Rustagi P.; Davis D. E.; Kraft A. S. Biological Activity of 26-Succinylbryostatin 1. Biochim. Biophys. Acta 1996, 1312, 197–206. [DOI] [PubMed] [Google Scholar]
- Nelson T. J.; Sen A.; Alkon D. L.; Sun M. K. Adduct Formation in Liquid Chromatography-Triple Quadrupole Mass Spectrometric Measurement of Bryostatin 1. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2013, 944C, 55–62. [DOI] [PubMed] [Google Scholar]
- Beans E. J.; Fournogerakis D.; Gauntlett C.; Heumann L. V.; Kramer R.; Marsden M. D.; Murray D.; Chun T. W.; Zack J. A.; Wender P. A. Highly Potent, Synthetically Accessible Prostratin Analogs Induce Latent HIV Expression in Vitro and ex Vivo. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 11698–11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear H. D.; McFadden A. W.; Kostuchenko P. J.; Lipshy K. A.; Hamad G. G.; Turner A. J.; Roberts J. D.; Carr M.; Carr S.; Grant S. Bryostatin 1 Activates Splenic Lymphocytes and Induces Sustained Depletion of Splenocyte Protein Kinase C Activity in Vivo after a Single Intravenous Administration. Anticancer Drugs 1996, 7, 299–306. [DOI] [PubMed] [Google Scholar]
- Kickhoefer V. A.; Han M.; Raval-Fernandes S.; Poderycki M. J.; Moniz R. J.; Vaccari D.; Silvestry M.; Stewart P. L.; Kelly K. A.; Rome L. H. Targeting Vault Nanoparticles to Specific Cell Surface Receptors. ACS Nano 2009, 3, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender P. A.; Kee J. M.; Warrington J. M. Practical Synthesis of Prostratin, DPP, and Their Analogs, Adjuvant Leads against Latent HIV. Science 2008, 320, 649–652. [DOI] [PMC free article] [PubMed] [Google Scholar]