

Abstract
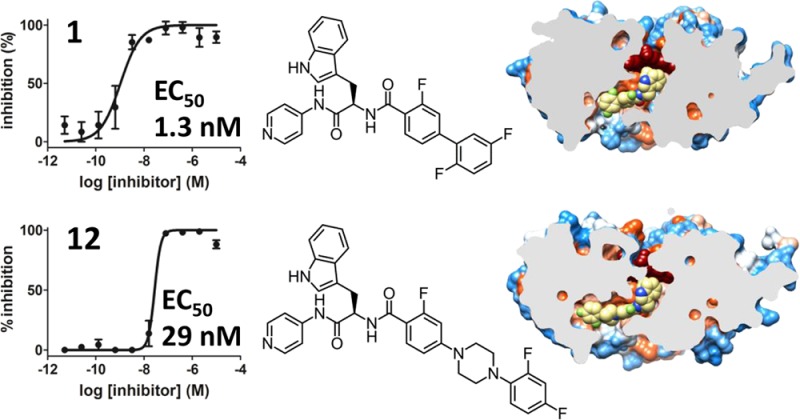
CYP51 is a P450 enzyme involved in the biosynthesis of the sterol components of eukaryotic cell membranes. CYP51 inhibitors have been developed to treat infections caused by fungi, and more recently the protozoan parasite Trypanosoma cruzi, the causative agent of Chagas disease. To specifically optimize drug candidates for T. cruzi CYP51 (TcCYP51), we explored the structure–activity relationship (SAR) of a N-indolyl-oxopyridinyl-4-aminopropanyl-based scaffold originally identified in a target-based screen. This scaffold evolved via medicinal chemistry to yield orally bioavailable leads with potent anti-T. cruzi activity in vivo. Using an animal model of infection with a transgenic T. cruzi Y luc strain expressing firefly luciferase, we prioritized the biaryl and N-arylpiperazine analogues by oral bioavailability and potency. The drug–target complexes for both scaffold variants were characterized by X-ray structure analysis. Optimization of both binding mode and pharmacokinetic properties of these compounds led to potent inhibitors against experimental T. cruzi infection.
Introduction
Chagas disease is caused by infection of mammalian host cells by the parasitic protozoon Trypanosoma cruzi, usually transmitted by blood-sucking bugs of the subfamily Triatominae.1T. cruzi colonizes the heart, gastrointestinal tract, and nervous system, causing progressive inflammation which can lead to chronic human cardiopathy and/or gastrointestinal dysfunction.2 According to the World Health Organization,3T. cruzi infects around 7–8 million people worldwide, mostly in Latin America. More recently Chagas disease has expanded beyond tropical and subtropical zones to become an emerging economic and health burden in the United States, Spain, and the UK.4 Antiparasitic treatment is recommended for all acute and chronic patients, including those with the indeterminate chronic form of Chagas disease.5,6 At the same time, chemotherapeutic options for Chagas disease are limited to benznidazole and nifurtimox, which are used during the acute phase but may cause severe gastrointestinal, dermatological, and neurological side effects.7 The efficacy of these drugs in the chronic stage is debated, and neither drug is approved by the FDA for use in the United States. A promising target for treatment of T. cruzi infection in humans and animals, identified in drug discovery efforts in academic and product development partnerships, is sterol 14-demethylase, CYP51.8
CYP51 is a clinically validated therapeutic target of antifungal azole drugs of the “conazole” pedigree.9 Their efficacy against a broad spectrum of fungal pathogens have defined azole compounds as an indispensable tool for controlling fungal diseases in humans10,11 and crop plants.12 The replicative intracellular amastigote form of T. cruzi is particularly sensitive to CYP51 inhibitors13 due to its obligate reliance on de novo synthesis of sterols as membrane building blocks. Posaconazole (Noxafil, Merck), the most recent member of the family to be marketed,14,15 demonstrates excellent anti-T. cruzi potency, high percentages of parasitological cure in animal models,13,16−18 and has a precedent of successfully curing a patient with chronic Chagas disease and systemic lupus erythematosus.19,20 Posaconazole’s efficacy against model T. cruzi infection is attributed to its favorable DMPK properties, including oral availability, long terminal half-life, and large volume of distribution.21,22 Nevertheless, the majority of azole inhibitors in clinical use or development are not powerful enough to eradicate T. cruzi from human patients or experimental animals.23−25
The similarity between sterols and their biosynthetic pathways in fungi and protozoa has led to several clinical trials of azole antifungal agents for treating Chagas disease patients. Posaconazole and ravuconazole (Eisai Co, Tokyo, Japan),26 both acquired from industry antifungal programs, have been advanced into clinical trials.20 The results thus far have been less than optimal, with only temporary efficacy over a longer time frame, indicating the need for further trials at different doses or in combination with benznidazole.27,28 Neither drug has yet been proven superior to benznidazole in treating T. cruzi infection in patients, indicating that the quest for a cure for Chagas disease must continue. It is notable that neither posaconazole nor ravuconazole were initially developed to target T. cruzi CYP51. Two next-generation promising CYP51 inhibitors, tipifarnib29−36 and fenarimol,37−39 a cancer drug candidate and a nontoxic herbicide, respectively, have been optimized to specifically target T. cruzi. Also, recently entering the anti-Chagas pipeline are azole derivatives from a collection of the Novartis Research Institute (Vienna, Austria).40
Our collaboration has focused on de novo structure-aided hit-to-lead optimization of a pyridine-based molecular scaffold discovered by a Mycobacterium tuberculosis CYP51-based screen.41−43 Structure-aided chemical tailoring, guided by a T. cruzi whole cell-based assay, has identified the favorable R-configuration of the carbon chiral center and enabled exploration of structural features contributing to enhanced microsomal stability and diminished inhibitory potential toward a panel of mammalian drug-metabolizing CYP enzymes.44,45 As a result of these earlier medicinal chemistry efforts, the EC50 of the new scaffold improved up to 4 orders of magnitude compared to that of the parental compound, LP10.45,46
We have now used a mouse model of acute T. cruzi infection to evaluate the potency and oral bioavailability of optimized CYP51 inhibitors. We are focusing on two biaryl and N-arylpiperazine scaffold variants, so named based on the structure of the longest substituent at the chiral carbon center. Using X-ray crystallography, we have characterized the binding modes of three analogues, at least one from each scaffold group, to resolutions of 2.04, 2.43, and 2.80 Å. These structures can now guide site-directed medicinal chemistry interventions aimed at further lead improvements. The structural differences between scaffolds affect the binding mode and pharmacokinetic behavior of these compounds. Using a four-day dosing regimen, we have prioritized inhibitors by efficacy in the animal model and correlated them with efficacy in the cell-based assay used to rank compounds for animal studies. Small differences in chemical structure dramatically affect the compounds’ binding modes, oral availability, terminal half-lives, and efficacy against T. cruzi. Five CYP51 inhibitors presented in this work suppressed >97% of parasite load in mice when administered orally at 50 mg/kg, bid, for four consecutive days.
Results
Chemistry
Syntheses of compounds 1–12 which were prepared for this study are summarized in Scheme 1. Palladium-mediated coupling reactions of 4-bromo-2-fluorobenzoic acid and methyl 4-bromo-2-fluorobenzoate (25) with various aryl boronic acids and N-aryl piperazines served as key steps of the syntheses of intermediates 13–19 and 26a–29a, respectively.44,45 The coupling reactions of 4-bromo-2-fluorobenzoic acid with aryl boronic acids, performed under microwave irradiation (100 °C for 1 h, or 120 °C for 2 h for the sterically hindered products such as 13 and 16), provided intermediates 13–19 in ca. 90% yields. Intermediates 26a, 27a, 28a, and 29a were prepared by the coupling of N-arylpiperazine derivatives with methyl 4-bromo-2-fluorobenzoate (25) at 60 °C in toluene, which were then hydrolyzed under basic conditions to provide carboxyl intermediates 26b, 27b, 28b, and 29b. Conversion of 19 to methyl ester 20 set the stage for replacement of the benzyl group with a 4-fluorobenzyl unit through catalytic hydrogenation (Pd/C and H2) and then O-alkylation with 4-fluorobenzyl bromide. N-Acetylation of 23(44) produced 24a, which was hydrolyzed to carboxylic acid 24b. All carboxylic acid intermediates were coupled with d-tryptophan derivative 30(45) to provide the final compounds 1–12.
Scheme 1. Syntheses of Compounds 1–12.

Reagents and conditions: (a) arylboronic acid, 5 mol % Pd2(dba)3, 10 mol % PCy3, 2M K3PO4, dioxane, 100 °C (microwave), 1 h, ca. 90%; (b) H2SO4/MeOH (1/10), 70 °C, 24 h, 91%; (c) H2 (balloon), Pd/C, MeOH–acetone, 23 °C, 24 h, 92%; (d) 4-fluorobenzyl bromide, K2CO3, acetone, 70 °C, 5 h, 95%; (e) 10% NaOH (aq), MeOH/THF (1/1), 60 °C, 3 h, ca. 95%; (f) acetic anhydride, Et3N, CH2Cl2, 0–23 °C, 1 h, 84%; (g) 10% NaOH (aq), MeOH/THF (1/1), 23 °C, 2 h, 36%; (h) 1-(aryl)piperazine, Pd(OAc)2, P(o-tolyl)3, Cs2CO3, toluene, 60 °C, 48 h, ca. 70%; (i) 13, 14, 15, 16, 17, 18, 19, 22b, 24b, 26b, 27b, 28b, or 29b (as appropriate), PyBOP, HOBt, Et3N, CH2Cl2, 23 °C, 1 h, ca. 70%.
Animal Model
To rapidly assess the effect of drugs on the parasite load in vivo, a four-day dosing mouse model utilizing transgenic T. cruzi Y luc strain expressing firefly luciferase reporter gene47 was adopted. The use of the luciferase-transfected strain enables immediate and direct reading of luminescence by imaging live animals on the detector plate. Given that the luciferase reporter carried by T. cruzi Y luc strain sustained only a limited number of passages in mammalian cell culture, we first tested the longevity of this marker in the mouse model by correlating luminescence in whole animal with parasite count in blood. The experiment was conducted in a group of five mice, each infected with 105 trypomastigotes. The impact of the transgenic T. cruzi Y luc strain was not lethal. All mice survived untreated 30 days postinfection, during which parasitemia was monitored for 21 days in live animals both by the luciferase reaction and parasite count by direct microscopic examination of fresh blood smears. The parasite count in blood dropped below the detectable level on day 21, while the luciferase signal persisted through day 21 (Figure 1A). All mice on day 21 appeared normal and agile.
Figure 1.
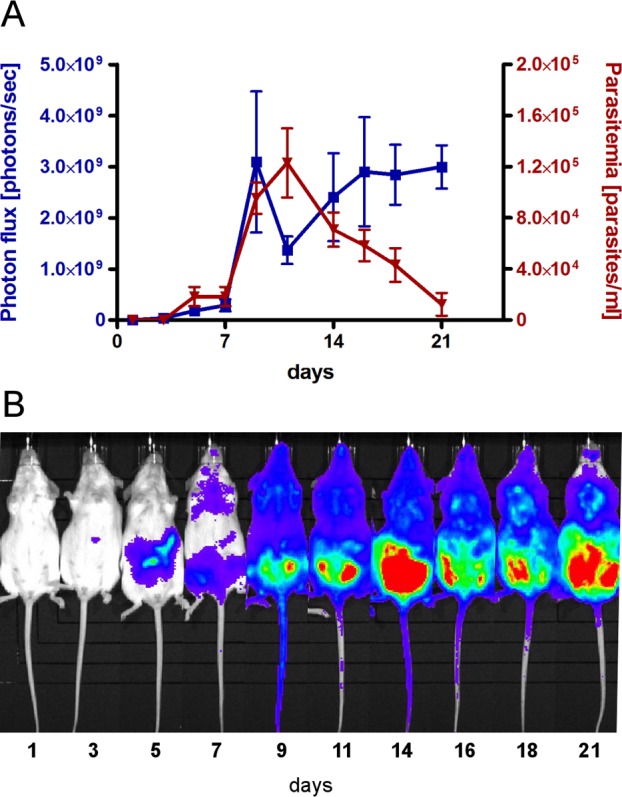
Animal model of T. cruzi Y luc infection. (A) Development of parasitemia in the untreated mice over the course of 21 days postinfection with T. cruzi Y luc parasites. Parasite count in fresh blood samples (red) was paralleled by the luminescence reading of whole amimals (blue). Each measurement is an average of five mice. (B) Evolution of the parasitemia in a single experimental animal by luminescence.
On the basis of this experiment, a four-day dosing regimen was constructed with drug administration beginning on day 3, after infection is established. If left untreated, infection quickly disseminates throughout the body and mice develop heavy parasitemia in the following 5–11 days (Figure 1B). Because trypanocidal activity of sterol biosynthesis inhibitors is time-dependent and results from complete depletion of the ergosterol pool within 72–96 h of drug exposure in vitro,13,48,49 the four-day in vivo regimen is justifiable. To assess the efficacy of test compounds, mice were infected with T. cruzi Y luc strain for three days. Starting on day 4, the infected mice were treated with test compounds either via intraperitoneal injection (ip) or oral gavage (po) for four consecutive days, twice a day (bid). At day 7 postinfection, the T. cruzi luminescent signal in the mice was read upon injection of d-luciferin.
Anti-T. cruzi Activity by ip Administration
In vivo activity of inhibitors, ranked a priori by EC50 in the cell-based assay previously described,44−46 was assessed by intraperitoneal injection in the first two independent experiments (Figure 2A,B), when the mice were treated with 40 mg/kg of test compounds, ip, bid. In the subsequent experiments, compounds were administered orally. As shown in Figure 2, the parasite load in the untreated animals significantly increased over a seven-day period. Posaconazole used as a positive control produced 94–99% inhibition of parasitemia. The parental hit, LP10, showed little efficacy under this treatment regimen, while the analogues 3, 4, and 8 suppressed parasite load by ≥97% over the four-day treatment period with a statistically significant difference (p ≤ 0.05) compared to the vehicle-treated controls. The efficacy of R-2 (52%), 1 (35%), 2 (84%), and 9 (52%) was attenuated relative to the three most effective inhibitors. The outcomes of R-2, 1, and 2 were particularly unexpected, as these compounds had superior EC50 in the T. cruzi cell-based assay (Table 1). The effect of compounds on the host cells was monitored using a high-content image-based whole cell assay used to prioritize inhibitors for testing in the animal model. No toxicity toward mouse myoblasts used to harbor parasites has been observed for any compound featured in Table 1 up to 10 μM, the highest concentration used to generate dose–response curves (Supporting Information, Figure S1)
Figure 2.

Efficacy of compounds upon ip administration. In two independent experiments, (A) and (B), compounds were administered at 40 mg/kg, ip, bid. Luminescence in mice was read upon luciferin injection on day 3 postinfection and prior to treatment (black bars) and on day 7 postinfection and after four days of treatment (gray bars). Each data point is an average of five mice; dpi-days postinfection. Posaconazole (Pos) served as a positive control. Percent inhibition for each compound is calculated relative to the untreated control on day 7 postinfection. *Values significantly different than vehicle-treated controls (p ≤ 0.05), except for LP10.
Table 1. Summary of the in Vitro and in Vivo Properties of CYP51 Inhibitorsj.
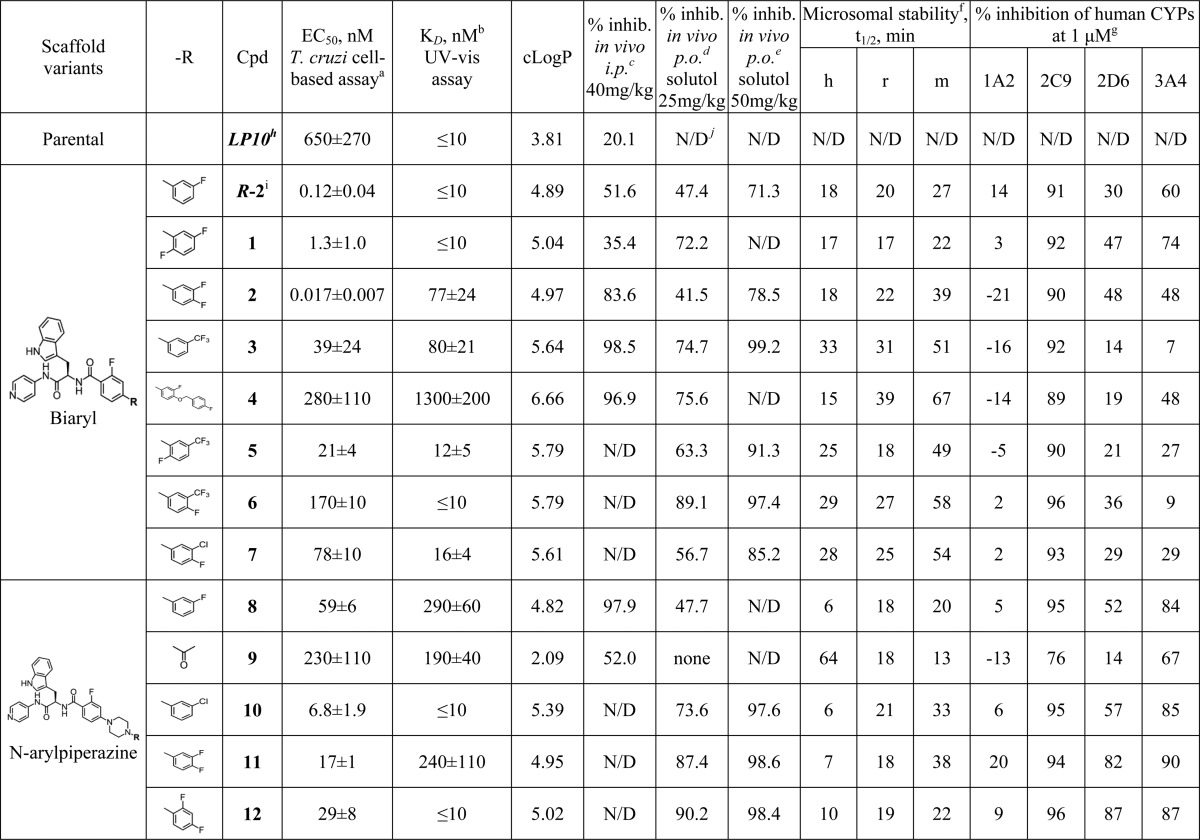
Each measurement performed in triplicate.
apparent KD values (see Supporting Information).
Each measurement is an average of five mice treated 40 mg/kg (60% DMSO), ip, bid, for 4 days.
Each measurement is an average of five mice treated 25 mg/kg (20% solutol), po, bid, for 4 days.
Each measurement is an average of five mice treated 50 mg/kg (20% solutol), po, bid, for 4 days.
Stability of compounds in human (h), rat (r), and mouse (m) liver microsomes as evaluated compared to the Sunitinib reference (see Supporting Information).
Inhibition of CYPs as evaluated in human liver microsomes using selective marker substrates for each CYP (see Supporting Information).
LP10 was previously characterized in vivo52 and close LP10 analogues were assessed for stability (4–7 min in both human and mouse liver microsomes) and cross-reactivity (21–23% inhibition of 1A2; 92% for 2C9; 62–77% for 2D6; and 75–55% inhibition of 3A4) previously.44
Compound R-2 was previously characterized structurally and in vitro.45
N/D: not determined.
The most active compounds following intraperitoneal adminstration, 3, 4, and 8, belonged to distinct scaffold variants, referred to as biaryl and N-arylpiperazine (Table 1). Both scaffolds retain the invariant N-indolyl-oxopyridinyl-4-aminopropanyl portion of the skeleton while carrying different side chain substituents at the chiral carbon center in the R-configuration: a biaryl moiety or an N-arylpiperazine moiety, in which the piperazine ring separates two aromatic rings of the biaryl structure, thus conferring flexibility to the otherwise rigid system.
Next Generation of Compounds
Six follow-up analogues, 5, 6, 7 (biaryl scaffold variant), 10, 11, and 12 (N-arylpiperazine scaffold variant), differed within each group by the halogenation pattern of the terminal phenyl ring (Table 1). Cytotoxicity of five of these new analogues was assessed against two different cell types, cardiomyocytes and hepatocytes, isolated from mouse embryos (Table 2). The biaryl compounds 5, 6, and 7 inhibited proliferation and viability of mammalian cells at concentrations below 90 μM, while N-arylpiperazine compounds 11 and 12 did not affect or only barely affected mammalian cells at 90 μM, the highest concentration applied in this experiment. Cardiomyocytes and hepatocytes were equally susceptible to compound action, with EC50 values in the same range for each of the tested compounds. The large difference in selectivity index (SI), calculated as the ratio between the EC50 values for inhibition of host cells and T. cruzi intracellular amastigotes, was due to a wider range of compound efficacy against T. cruzi. On the selectivity scale, compound 6 was ranked the lowest (<200) and compound 11 the highest (>5000). The three most selective compounds, 5, 11, and 12, belong to different scaffold variants (Table 2).
Table 2. Cytotoxicity of Compounds in Mouse Hepatocytes and Cardiomyocytes.
| hepatocytes |
cardiomyocytes |
|||
|---|---|---|---|---|
| compd | EC50 (μM) | selectivity index (SI)a | EC50 (μM) | selectivity index (SI) |
| 5 | 68.1 ± 7.2b | 3243 | 53.9 ± 8.1 | 2568 |
| 6 | 28.3 ± 4.1 | 166 | 22.2 ± 2.3 | 131 |
| 7 | 48.1 ± 12.5 | 616 | 23.3 ± 5.9 | 299 |
| 11 | >90 | >5000 | >90 | >5000 |
| 12 | >90 | >3000 | 83.2 ± 4.7 | 2869 |
Calculated as a ratio between the host cell and intracellular T. cruzi amastigote survival, both expressed as the corresponding EC50 values.
Each measurement performed in duplicate.
The anti-T. cruzi EC50 values for the new biaryl analogues, although still effective in the nanomolar range, increased compared to the immediate precursors R-2, 1, and 2 (Table 1). The N-arylpiperazine analogues retained the potency of their immediate precursors. Among the six new analogues, those belonging to the biaryl scaffold variant had a notably longer half-life in a microsome stability assay and less inhibition of human CYPs in vitro (Table 1). Inhibition of four major drug-metabolizing human CYP enzymes, 1A2, 2C9, 2D6, and 3A4, contributing to the oxidative metabolism of vast majority of drugs in clinical use,50 has been assessed. With the exception of 2C9, which was inhibited 90–96% by the majority of the tested compounds regardless of scaffold, inhibition of three other human CYPs by biaryl inhibitors dropped below 30%. At the same time, N-arylpiperazine compounds inhibited 3A4 by 85–90% and 2D6 by 14–87%. Inhibition of 1A2 dropped below 9% for both scaffold variants with an exception of 11 (20% 1A2 inhibition). For comparison, ketoconazole and posaconazole at 1 μM concentration inhibited 22/22/4/95 and −11/–3/–9/68% of human 1A2/2C9/2D6/3A4 enzymes, respectively. Thus, both ketoconazole and posaconazole hit 3A4, while other CYPs were only modestly inhibited or not inhibited at all under the experimental conditions used in these studies.
To confirm the mechanism of action, two newly synthesized compounds, 3 and 12, one for each scaffold variant, have been tested for disruption of sterol biosynthesis pathway in intracellular T. cruzi amastigotes. The assay was conducted as described elsewhere.51 Lipids extracted from intracellular amastigotes were analyzed by gas chromatography and mass spectrometry (GC-MS) and identified as described previously.52 DMSO and benznidazole were used as negative controls; posaconazole served as positive control. Cholesterol was the only peak originated from the host cells (Supporting Information, Figure S2). The major sterol observed in untreated amastigotes was episterol (peak d), followed by approximately equal amounts of fecosterol (e) and cholesta-7,24-dien-3β-ol (peak a). As a result of treatment, two 14-methylated precursors, lanosterol (f) and eburicol (h), were prominent in the GC-MS traces of the CYP51 inhibitors posaconazole and compounds 3 and 12 with concomitant decline in episterol, fecosterol, and other 14-demethylated intermediates. No changes in lipid composition have been observed in benznidazole-treated samples compared to DMSO. Benznidazole is the drug currently used to treat Chagas disease and does not target ergosterol synthesis pathway, inducing the formation of free radicals in the parasite.53 On the basis of this experiment, we conclude that in the course of hit-to-lead optimization compounds of the N-indolyl-oxopyridinyl-4-aminopropanyl-based SAR series retained the mechanism of action and specifically target T. cruzi CYP51.
Pharmacokinetic Behavior: Cyclodextrin versus Kolliphor
Most T. cruzi parasites in chronic infection reside intracellularly, largely in heart, gut, and skeletal muscles; therefore, anti-T. cruzi drugs must be lipophilic enough to penetrate cell membranes and deep tissues. On the other hand, poor water solubility leads to ineffective absorption. Aiming to establish optimal formulation for the bioavailability test, both 2-hydroxypropyl-β-cyclodextrin (HPβCD)54 and the nonionic surfactant Kolliphor55 (a mixture of polyoxyethylene esters of 12-hydroxystearic acid and free polyethylene glycol, also known as solutol) were tested as vehicles for oral administration of compounds. The plasma concentration–time curves and the tissue distribution of compounds following oral administration of a single 50 mg/kg dose in 20% Kolliphor or 20% HPβCD demonstrated remarkable differences between two vehicles (Table 3). The PK parameters extracted from the concentration–time curves showed higher oral bioavailability for the majority of compounds in Kolliphor compared to that in HPβCD. A notable exception was compound 12, which was comparable in both vehicles. When administered in Kolliphor, compound 7 demonstrated superior half-life (16.8 h), followed by 11 (4.6 h) and 12 (4.4 h). The shortest half-life was observed for 6 and 2 (<3.3 h). The highest Cmax and AUC values and the lowest clearance were achieved by 5 and 7. The lowest Cmax and highest clearance were observed for 11 and 2. The majority of tested compounds had in vivo half-lives necessitating twice a day administration.
Table 3. PK Parameters in HPβCD and Kolliphor.
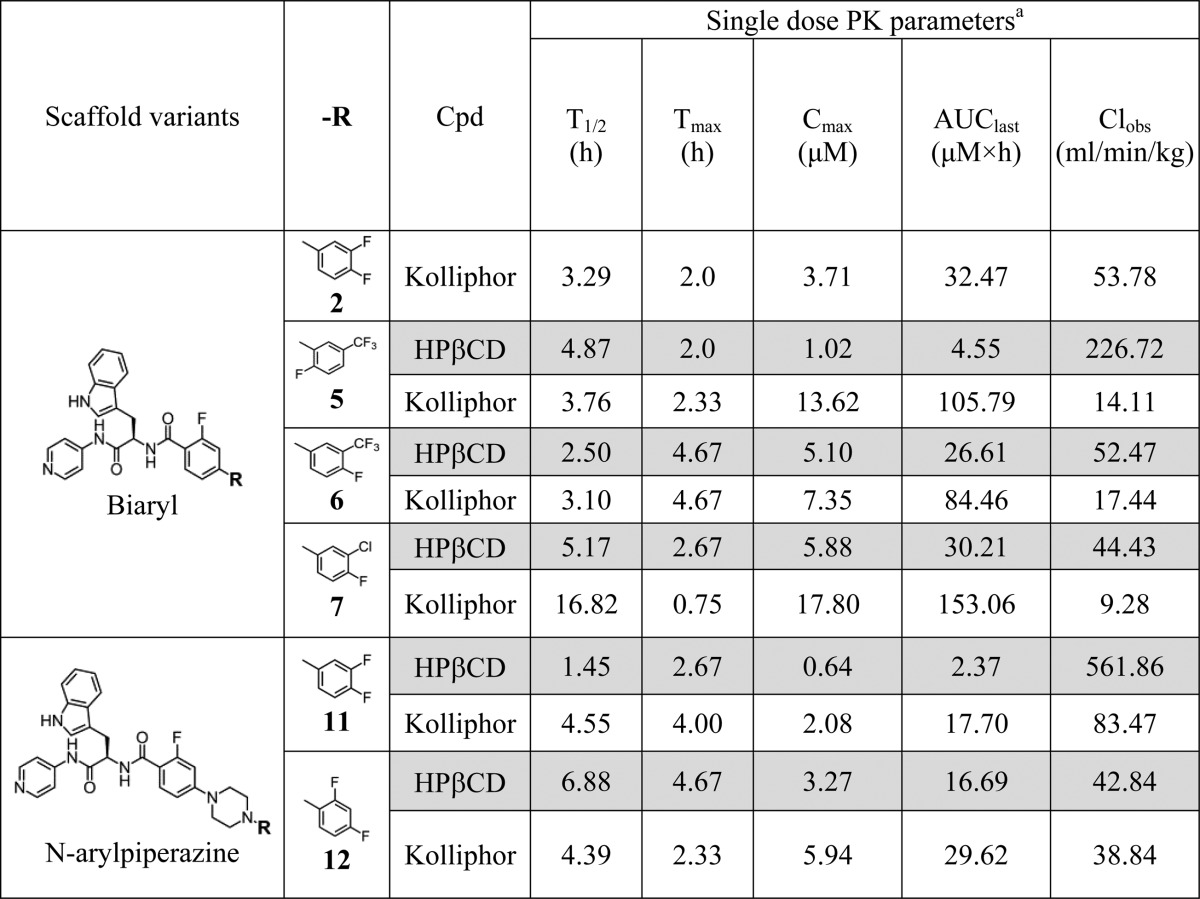
Each measurement is an average of three mice received a single 50 mg/kg oral dose of test compound as 10 mg/mL suspension in 20% HPβCD (gray fields) or 20% Kolliphor (white fields).
Tissue Tropism
Tissue tropism, an important factor defining efficacy of drugs against T. cruzi, was assessed 2 and 8 h postexposure following oral administration of compounds in Kolliphor or HPβCD at 50 mg/kg. Tabulated data are included in the Supporting Information (see Tables S1 and S2), while a graphic summary is provided in Figure 3. Tissue exposure was enhanced for a majority of the compounds when administered in Kolliphor versus HPβCD, again with the exception of 12, which was comparable in both vehicles. All compounds concentrated in intestines; intestinal concentration remained high even after 8 h exposure. Given that no nondissolved materials have been observed in the intestines, the compounds likely permeated the mucosa of the gut from the apical to the basolateral side. All compounds accumulated in liver and lung were notable in heart and fat and detectable in skeletal muscle and brain. Compound 11 crossed the blood–brain barrier and was detected in brain 2 h postexposure, although its concentration dropped to almost zero after 8 h. The concentration of 7 in all tissues remained higher after 8 h versus 2 h exposure, both in Kolliphor and HPβCD. The remaining inhibitors were detected in all the tissues after 8 h, albeit at diminished concentrations.
Figure 3.
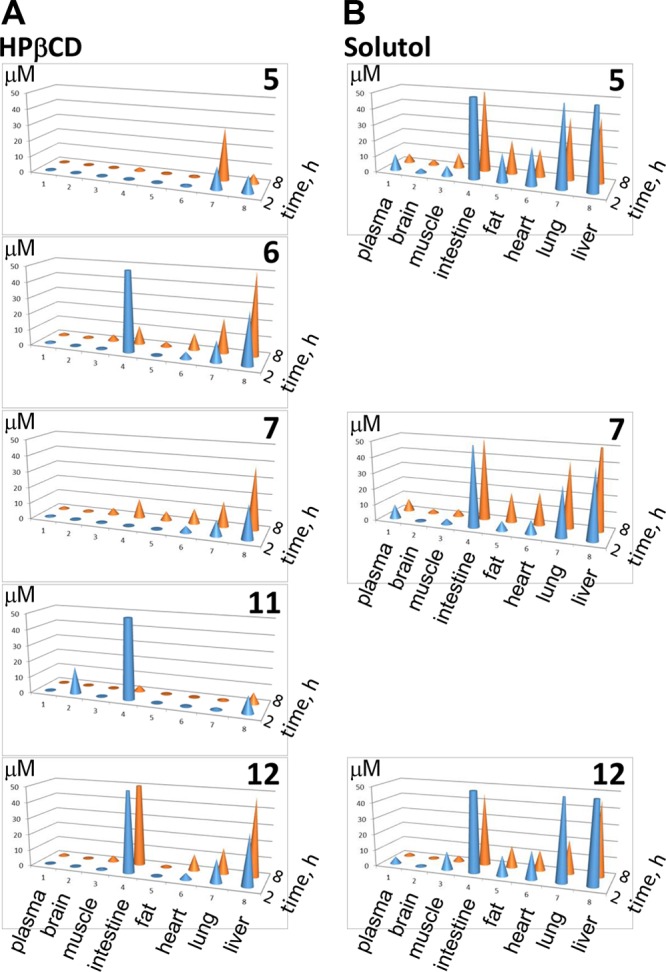
Tissue tropism of compounds. Tissue distribution of selected inhibitors administered orally as a single 50 mg/kg dose in 20% HPβCD (A) or 20% Kolliphor (B). Compound concentration detected in a tissue after 2 (blue) or 8 (orange) hours of exposure is plotted in μM. Tabulated data are presented in Supporting Information, Tables S1 and S2.
Dose Response: Cyclodextrin versus Kolliphor
To validate the choice of Kolliphor as a vehicle for the in vivo oral administration of compounds, we selected two compounds, 5 and 12, for side-by-side comparison of their efficacy using the same batch of parasites. To design each animal experiment, we have considered a number of different objectives, including time, labor, resources, and ethical aspects of animal studies, which motivated us to limit the number of experimental animals to the minimum needed to satisfy specific questions. The rationale behind the selection of this pair of inhibitors was comparable bioavailability of 12 in both vehicles and relatively low bioavailability of 5 in HPβCD compared to Kolliphor (Table 3). Thus, the enhancing effect of Kolliphor in vivo, if any, was expected to be more profound for 5. Indeed, when administered in Kolliphor, the efficacy of 5 was higher at all concentrations tested when compared to that in HPβCD, whereas the efficacy of 12 was comparable in both vehicles (Figure 4A). To establish a po administration dose, dose–response curves were obtained for four inhibitors: 5, 6, 7, and 12 (Figure 4B). The highest dose of 50 mg/kg bid was the most efficient for all the compounds, while 25 mg/kg bid dose allowed us to efficiently discriminate among and prioritize inhibitors. No signs of toxicity (weight loss, mobility issues, appearance) were observed in the four-day studies.
Figure 4.

Anti-T. cruzi efficacy of compounds in four-day mouse model of infection. (A) Dose–response in Kolliphor (KOL) versus HPβCD (CD) administration for compounds 5 (empty bars) and 12 (filled bars). Compound 5 in Kolliphor was more active than in HPβCD (p < 0.05) versus compound 12, which had comparable activity in both vehicles (P < 0.05). (B) Dose–response for Kolliphor administration of 5, 6, 7, and 12. Benznidazole (BNZ) served as a positive control.
Anti-T. cruzi Activity by po Administration
Because of its superior performance for the majority of the tested analogues, Kolliphor was chosen as a vehicle to assess bioavailability and the in vivo efficacy of compounds in four-day animal model at two different concentrations, 25 and 50 mg/kg, administered by oral gavage, bid (Table 1). All 13 compounds were tested at 25 mg/kg, and nine of them, depending on compound availability, were also tested at 50 mg/kg. Administered at 25 mg/kg, 12 showed the best efficacy (90.2%), closely followed by compounds 6 (89.1%) and 11 (87.4%), whereas analogues R-2 (47.4%), 2 (41.5%), 8 (47.7%), and particularly 9 (0%) were least potent. Compound efficacy was dose-dependent, achieving >90% inhibition for six compounds, 3 (99.2%), 5 (91.3%), 6 (97.4%), 10 (97.6%), 11 (98.6%), and 12 (98.4%), tested at 50 mg/kg, bid The efficacy of the least potent inhibitors, R-2 and 2, also increased, exceeding 70% at 50 mg/kg, bid. Compound 9 was the only one in this series lacking in vivo potency. The disconnect between the in vitro and in vivo rank order potency observed for R-2 and 2 is likely due to reduced oral bioavailability, as compound 2 has been characterized as one of the compounds, with the lowest Cmax, highest clearance and shortest half-life at 50 mg/kg (Table 3).
Collectively, the three most potent inhibitors in this series, 6, 11, and 12, belong to different scaffold variants. Compound 6 and its biaryl analogues, 3 and 5, showed >90% inhibition at 50 mg/kg, all retained 2-trifluoromethyl substituent at the terminal phenyl ring. In vivo efficacy of the biaryl analogues lacking this substituent had been notably attenuated. The two most active N-arylpiperazine derivatives 11 and 12, both retaining the 4-fluoro substituent at the terminal phenyl ring, proved critical for drug–target interactions by X-ray structure analysis.
Assessment of in Vivo Potency
The disconnect between the in vitro and in vivo rank orders of potency observed in these studies raises questions about prioritizing the CYP51 inhibitors based solely on in vitro performance without also considering PK/PD characteristics and hydrolytic stability, in conjunction with assessing susceptibility to oxidative metabolism. Inspection of the calculated log P values predicts a lowest log P of 2.09 for compound 9, which has no activity in the animal model, while log P values for compounds showing efficacy are around 5 (Table 1). A low log P value defining relative solubility of compounds in octanol versus water is generally predictive of poor membrane permeability. Another possible explanation to the lack of efficacy is that the compound 9 acetyl group may be hydrolyzed in vivo to give the piperazine derivative, which is inactive in cell-based assay.
A ratio between EC50 and the apparent KD values extrapolated from the titration curves in aqueous solution (Supporting Information, Figure S3) may provide yet another clue. The EC50 ≪ KD was a poor sign for in vivo efficacy, although this criterion is not absolute. Particularly large differences between the two parameters (about 2–3 orders of magnitude) coincided with poor in vivo efficacy of compounds R-2 and 2. EC50 ≪ KD by an order of magnitude correlated with attenuated efficacy of 4 and 8. Against this trend, compound 11 (EC50 ≪ KD by an order of magnitude) reached one of the highest efficacy values for this series in vivo. The rationale behind these observations may be in the high hydrophobicity of compounds, which interferes both with oral bioavailability and binding affinity to the target, while EC50 in cell-based assay suffers less or may even benefit to some extent from the elevated hydrophobicity.
Inhibitor Binding Modes by X-ray Crystallography
The X-ray structures determined in this work provide atomic level details of drug–target interactions both for biaryl and N-arylpiperazine derivatives. The N-arylpiperazine inhibitor, (R)-N-(3-(1H-indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4-(4-(2,4-difluorophenyl)piperazin-1-yl)-2-fluorobenzamide, compound 12, produced the highest resolution structure currently available for TcCYP51 (2.04 Å, PDB ID 4C0C). Resolution for the closely related analogue, (R)-N-(3-(1H-indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4-(4-(3,4-difluorophenyl)piperazin-1-yl)-2-fluorobenzamide, compound 11, was 2.43 Å (PDB ID 4UQH). The biaryl scaffold variant costructure with (R)-N-(3-(1H-indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2′,3,5′-trifluoro-[1,1′-biphenyl]-4-carboxamide, compound 1, (2.84 Å, PDB ID 4BMM) is a notable improvement compared to 3.1 Å resolution of the previously characterized closely related analogue R-2.45 The longest substituent at the chiral center of both scaffold variants protrudes into the β-domain (Figure 5). Being shorter than posaconazole,56 both scaffolds fit entirely within the protein interior, with the pyridinyl moiety of the inhibitor coordinated to the heme iron and the indole ring pointed at the heme macrocycle. Particularly surprising was the binding modes of 11 and 12, whose longest substituents extended along the constraint channel spanning the β-domain (Figure 5A–C) rather than hanging in the cleft between the α- and β-domain, as predicted by molecular docking45 (here and throughout, the nomenclature of the P450 structural elements is according to the generally accepted schemes57−59).
Figure 5.

Inhibitors in the active site of TcCYP51. (A,D) Slice through the binding site shows bound inhibitors (yellow spheres) and the protein surface colored by hydrophobicity, hydrophobic areas are in orange and hydrophilic areas are in blue. Heme is in dark-red spheres. (B) Piperazine group separating two phenyl rings in the 12 (yellow sticks) allows smooth bending of the long substituent along the β-sheet saddle (magenta). A fragment of the electron density map (blue mesh) contoured at 1.2 σ delineates position of 12 at 2.04 Å resolution. Protein is in ribbon, heme is in spheres. (C,F) Residues within 5 Å from the inhibitor (yellow sticks) are highlighted in blue, heme is in gray sticks. (E) Binding mode of 1 resembles that of 12 (B), with fewer contacts for the long substituent at the chiral carbon center. Electron density map at 2.84 Å is contoured at 0.8 σ. Images here and otherwise were generated using CHIMERA83 or PYMOL84
Drug–Target Interactions and SAR
On the basis of the X-ray structures now available for the 4-aminopyridine-based analogues here and elsewhere,44−46 the invariant portion of the small-molecule skeleton binds in the most conserved area of the CYP51 active site adjacent to the heme macrocycle (Figure 5; Table 4). The indole ring is bound in the energetically favorable T-shape π–π stacking mode, with the heme macrocycle tightly enclosed by the cluster of bulky electron-rich residues Y103, M106, F110, Y116, and F290, adopting a single well-defined conformation (Figure 6A). This is in contrast to the previously reported sulfonamide derivative (PDB ID 4COH), where flipping of the indole ring was observed.46 The 2-fluoro substituent of the benzamide ring, invariant in both scaffold groups, points toward a crevice formed by the α-domain residues Y103, I105, M106, and M460, residing within van der Waals distances of Y103 and I105. The 2-fluoro substituent on this ring was retained earlier in the hit-to-lead optimization for the increased half-life of compounds in the microsome extracts. No H-bonding interactions, direct or mediated by water molecules, have been established between the protein and inhibitor in any costructures reported herein. As previously observed for the sulfonamide derivative,46 a single water molecule in the active site invariantly H-bonds two amide protons of the inhibitor.
Table 4. TcCYP51 Amino Acid Point Contacts within 5 Å of the Inhibitors.
| amino acid residues | 1 (biaryl variant) | 12 (N-arylpiperazine variant) |
|---|---|---|
| invariant in CYP51 family | Y103, F110 | Y103, F110 |
| conserved across the phyla | F48, Y116, A291, T295, L356, M460 | F48, I72a, Y116, A291, T295, L356, M460 |
| substrate-specific | I105 | I105 |
| phylum-specific | M106, P210, V213, F290, | F55, I79, M106, P210, V213, F290 |
| more variable | F214, A287, M358, M360, V461 | I45, I70, V77, F214, A287, M358, M360, V461 |
Highlighted in bold are points of contact which do not overlap between two inhibitors.
Figure 6.
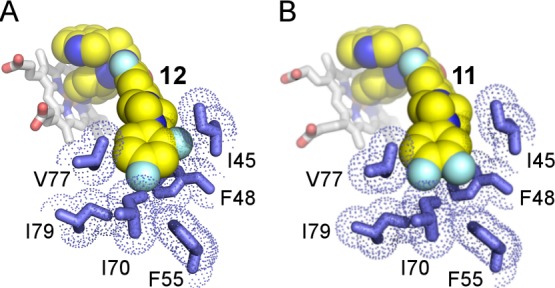
Interactions of the terminal phenyl ring in the N-arylpiperazine scaffold. Fluoro-substituted edges of the terminal phenyl ring in compounds 12 (A) and 11 (B) face residues I45, F48, F55, and I70. Inhibitors are shown in van der Waals spheres highlighted in yellow. van der Waals radii of the amino acid residues (blue sticks) are marked by blue dots. Heme is in stick mode. Heteroatoms are colored by type: oxygen in red, nitrogen in blue, fluorine in cyan, iron in ochre.
Binding of the N-Arylpiperazine Compounds 11 and 12
Despite the small differences in modification pattern of the terminal phenyl ring, both N-arylpiperazine compounds had the same binding mode. In both structures, positions of the fluorine substituents were unambiguously defined in the electron density, providing detailed information on drug–target interactions. The flexibility conferred by the piperazine ring allows 11 and 12 to adopt a smooth curvature to efficiently fill the hydrophobic channel extending into the β-domain (Figure 5A–C). The terminal phenyl ring critical for the in vivo efficacy of the N-arylpiperazine analogues reached the most remote hydrophobic cavity at the end of the channel, with the fluoro-substituted edge facing I45, F48, F55, and I70 (Figure 6). In both inhibitors, the 4-fluoro substituent points at I70 and I79 and is within 5 Å of I70, I72, V77, I79, and F55. The 2-fluoro substituent of compound 12 points at I45 and is within 5 Å of I45 and F48. Finally, the 3-fluoro substituent of compound 11 points at F55 and is within 5 Å of F48 and F55. These tight interactions of the terminal phenyl ring carrying small fluorine substituents may impose a binding challenge to N-arylpiperazine analogues featuring larger substituents, particularly at the C-4 site.
Protruding deeper into the β-domain, N-arylpiperazine moiety induces the FG-loop conformation in TcCYP51 different than that observed in the sulfonamide analogue:46 the loop shifts in an opposite direction, narrowing the gap between the α- and β-domains and virtually “squeezing” the molecules into the narrow channel. Remarkably, the twisted shape of the N-arylpiperazine moiety in the TcCYP51 active site violates a coplanar, sp2-hybridized arrangement of the nitrogen-linked aryl groups even though both nitrogen atoms of the piperazine were restrained in planar sp2 hybridization during structure refinement. As a result, the terminal phenyl ring is forced into virtually orthogonal orientation with respect to the piperazine’s nitrogen plane. Given that hybridization/conjugation at the point of ring attachment is not beneficial to the binding mode, a conformational penalty is likely imposed, which may be tolerated, at least in part, in exchange for the enthalpy gained due to the expansion of the terminal ring into the hydrophobic cavity of β-domain formed by the five hydrophobic residues: I70, I72, V77, I79, and F55.
Binding of the Biaryl Compound 1
The net contacts for 1 in TcCYP51 are less extensive than those of 11 or 12 (Figure 5D–F). Particularly notable is loss of the multiple hydrophobic interactions at the end of the constricted hydrophobic channel in the β-domain due to the smaller size of the compound 1 molecule. Attenuated interactions with the target likely account for a compromise resolution of the biaryl analogue cocrystals, as reported here and previously.44 The terminal phenyl ring of 1 is stabilized by stacking interactions with F48 and F214 and by the π–electron systems of M358 and M360. The proximity of these residues to the terminal phenyl ring of 1 may interfere with the ring modification pattern, however, spatial constraints are not nearly as tight as in N-arylpiperazine analogues, given that the ring in 1 is facing the opening to the exterior and that the FG-loop possesses a large degree of flexibility to adjust to the incoming inhibitors.46
Most of the amino acid residues concentrated around the invariant N-indolyl-oxopyridinyl-4-aminopropanyl part of the skeleton are either invariable in the CYP51 protein family (Y103 or F110) or are highly conserved across phyla (Table 4). This suggests that the N-indolyl-oxopyridinyl-4-aminopropanyl warhead loaded with different substituents could be utilized against a variety of CYP51 targets. Specificity to the target is to be defined by the structure of the variable substituent. Thus, the terminal difluorophenyl ring of N-arylpiperazine analogues binds in the remote cavity formed by the residues least conserved across the CYP51 protein family compared to the rest of the 24 exclusively hydrophobic residues constituting the binding site. This lack of conservation suggests that N-arylpiperazine inhibitors tailored to fit this cavity would be likely T. cruzi specific. Compounds constituting the biaryl scaffold may be more cross-reactive.
Discussion
The structure-aided tailoring of the N-indolyl-oxopyridinyl-4-aminopropanyl-based scaffold described here and in our previous publications42,44−46,52 is aimed at developing drug candidates to prevent or treat Chagas disease. Chagas disease is an endemic tropical infection mostly affecting people in Central and South America in areas with poor living conditions and inadequate healthcare infrastructure. However, it is now an emerging infection in the U.S., Spain, and Japan. In this work, pharmacokinetic and pharmacodynamic properties of newly developed inhibitors were monitored as part of an iterative approach toward molecular design. This involves rounds of analysis of structure–activity and structure–property relationships, compound synthesis, testing, and re-evaluation in order to obtain one or more classes of inhibitors that are orally deliverable and potent.44−46
An important observation made in this study is the disconnect between the in vitro and in vivo potency of compounds. The compounds most active in vitro, R-2 and 2, were inferior to less potent members of the scaffold group, 6, 11, and 12, in terms of in vivo performance (Table 1). A disconnect between the in vitro and in vivo compound ranking is not uncommon, being also observed for different targets due to several factors, including pharmacokinetic properties of chemicals and host metabolism.60,61 In our specific case, discrepancy in compound ranking may be also rooted in the conflict between the highly hydrophobic nature of the CYP51 binding site and the attenuated bioavailability of the hydrophobic compounds which may best satisfy the target requirement. This conflict brings into relief the shortcoming of prioritizing CYP51 inhibitors by in vitro performance alone and highlights the need for in vivo assessment in which compound potency and bioavailability are monitored at a very early stage of drug development. This is not to say that in vitro screening is not valuable. It does focus attention on a subset of compounds that are antiparasitic versus the many that are not. The shortcoming is in hit prioritization for in vivo proof of principle and is not unexpected.
To rapidly assess the effect of drugs on the parasite load in vivo, a four-day mouse model utilizing T. cruzi Y luc strain expressing firefly luciferase was adopted. To impart rigor to the test, oral bioavailability was set ab initio as a requirement. The total duration of the assay was 8 days, including establishing acute infection in mice (3 days), dosing (4 days), and evaluation of outcome (1 day). The parental compound LP10, that previously demonstrated potency in a long-term mouse model with 30 days of treatment,52 could not compete with its optimized descendants in the four-day treatment model utilized here, confirming that the bar is set high enough to ensure success of optimization steps and rapid prioritization of compounds entering longer-term dosing studies.
The transgenic T. cruzi strains that have been established in recent years accelerate drug discovery by providing several advantages over manual counting of parasites in mouse blood.62,63 However, our results also show that transgenic T. cruzi parasites expressing firefly luciferase are less virulent than the parental strain because mice survive infection with a relatively high inoculum that would be lethal with the parental strain. Similar observations have been made with transgenic T. cruzi parasites with a different background, which develop lower parasitemia when compared to wild type.64 This variability renders T. cruzi Y luc strain unsuitable either for long-term treatment schemes or evaluation of parasitological cure. Nevertheless, the rapid and sensitive detection of the luciferase-expressing parasites makes it a valuable tool for assessing compound bioavailability and efficacy in the early stages of drug discovery.
The nonionic surfactant Kolliphor, which has low reported in vivo toxicity,55 was utilized as a vehicle for oral administration to assess the solubility, efficacy, and systemic and tissue exposure of the compounds. Use of Kolliphor formulations was proven superior to HPβCD for the tested set of the compounds as far as drug solubility and exposure are concerned (Table 3). Efficacy in the four-day animal model (Figure 4A) was consistent with exposure: compound 5 formulated in Kolliphor was more active compared to the HPβCD formulation for which large difference in drug exposure was also observed between the two vehicles. Compound 12 had comparable exposure and efficacy in both formulation vehicles. No signs of toxicity or side effects have been observed in the four-day studies.
Another important observation from these studies is that small modifications in molecular structure may have dramatic effects on both in vitro and in vivo parameters of the inhibitors. Thus, modifications to the terminal phenyl ring resulted in a wide range of potencies of the biaryl scaffold (Table 1). Similarly, PK parameters of the N-arylpiperazine analogues differed significantly from one another (Table 3). Given the multiple factors affecting the efficacy of drugs, the two most efficacious and competitive compounds, 6 and 12, represented different scaffold groups. Improved microsome stability in vitro demonstrated by biaryl compound 6 contrasted with the attenuated microsome stability of N-arylpiperazine member 12. Compared to 12, compound 6 had higher Cmax and AUC, which was consistent with its low clearance. On the other hand, compound 12 had longer terminal half-life in plasma and a distinct, tight drug–target fit which distinguished it from the CYP51 inhibitors of the biaryl series. However, the near-perfect space-filling of 12 binding at the active site suggests a conformational penalty for violating the coplanar, sp2-hybridized arrangement of the nitrogen-linked aryl groups in the piperazine moiety. Finding chemical alternative(s) to the piperazine linker may alleviate the restraint and further enhance binding to TcCYP51.
The half-lives of N-indolyl-oxopyridinyl-4-aminopropanyl-based analogues tested in these rodent studies ranged from 3 to 16 h (administered in Kolliphor) (Table 3). For comparison, posaconazole and the ravuconazole prodrug E1224, used clinically in the treatment of invasive fungal infections and candidates for etiological treatment of Chagas disease, have half-lives in humans measured in days.65,66 We routinely monitored the half-lives of all the synthesized compounds in vitro using microsome extracts (Table 1). Over the course of hit-to-lead optimization, a substantial increase in the half-life has been achieved compared to the parental compound LP10,44 but room for improvement remains. In future efforts, analysis of metabolites will be undertaken to identify “weak” points that can be addressed by further medicinal chemistry optimization to alleviate metabolic liabilities that are uncovered.
The persistent cross-reactivity of compounds with CYP2C9 is noteworthy given that another human isoform, 3A4, is a major CYP off-target for the azole antifungal drugs, ketoconazole and posaconazole. The IC50 of ketoconazole and posaconazole reported against human 3A4 vary in different publications as 40 and 350 nM,35 or 8 and 120 nM.67 3A4 is highly promiscuous in substrate specificity and oxidizes over half of all administered drugs.68 Unlike 3A4, 2C9 exhibits selectivity for the oxidation of relatively small, lipophilic anionic compounds such as the nonsteroidal anti-inflammatory drugs flurbiprofen, ibuprofen, naproxen, and diclofenac as well as the hypolipidemic agent gemfibrozil. Arginine 108 is a point of specific contact stabilizing the charge of these anionic drugs.69 On the basis of the X-ray structure analysis, 2C9 has a large and hydrophobic binding site capable of simultaneously binding multiple ligands.69,70 Also, structural conformational changes upon interactions with the ligand allow 2C9 to bind molecules of various sizes and chemical structures.69,70 Given that the longest substituent at the chiral carbon center was an optimization target in the N-indolyl-oxopyridinyl-4-aminopropanyl-based SAR series, here and previously reported,44−46 persistent cross-reactivity with 2C9 may be associated with the invariable portion of the skeleton rather than variable substituent. Another pyridine-based class of CYP51 inhibitors, fenarimol analogues EPL-BS967 (also known as UDD) and EPL-BS1246 (also known as UDO),37−39 have IC50s of 8–9 μM and ≥20 μM, toward 2C9 and 3A4, respectively.67
Although both N-indolyl-oxopyridinyl-4-aminopropanyl-based and fenarimol analogues utilize a six-membered pyridine heterocycle to coordinate the heme iron (a role consistent with that of the five-membered heterocycle in azole inhibitors), the two pyridine-based scaffolds have significant differences defining their interactions with the target (Figure 7). First, the 4-pyridyl Fe-coordinating moiety of the N-indolyl-oxopyridinyl-4-aminopropanyl-based analogues contrasts to the 3-pyridyl moiety of fenarimol analogues. Second, the structure branching point (highlighted in green in Figure 7) in the fenarimol analogues is positioned three bond-lengths closer to the Fe-coordinating nitrogen atom compared to that in the N-indolyl-oxopyridinyl-4-aminopropanyl-based analogues. Both factors synergize to impose spatial constraints on the interactions of the fenarimol analogues with the heme macrocycle. Structurally, it translates to the energetically more favorable T-shape π–π stacking interactions between the indole ring and heme macrocycle in the N-indolyl-oxopyridinyl-4-aminopropanyl-based inhibitors (Figure 7A), as opposed to spatial hindrance caused by proximity of virtually coplanar aromatic systems of heme and fenarimol analogues (Figure 7B). This steric hindrance rather than the pyridine group electronic effects likely explains a weakened Fe–N coordination bond in the fenarimol analogue–TcCYP51 complexes, which is >0.2 Å longer than that in the N-indolyl-oxopyridinyl-4-aminopropanyl-based complexes and >0.3 Å longer than in azoles.67 Attenuated metal-binding is believed to improve selectivity and safety profile of the fenarimol analogues, compared to that of azole antifungal inhibitors, by increasing contributions of specific drug–protein interactions in binding affinity at the expense of nonspecific drug–metal interactions. A similar approach has been used to achieve highly selective CYP17A1 inhibitors, a potential treatment for prostate cancer, via utilizing less avid metal-binding groups.71 Accordingly, in future studies we will explore analogues of our N-indolyl-oxopyridinyl-4-aminopropanyl-based inhibitors with less nucleophilic metal-coordinating heterocycles in an attempt to increase CYP51 selectivity of this class of inhibitors.
Figure 7.
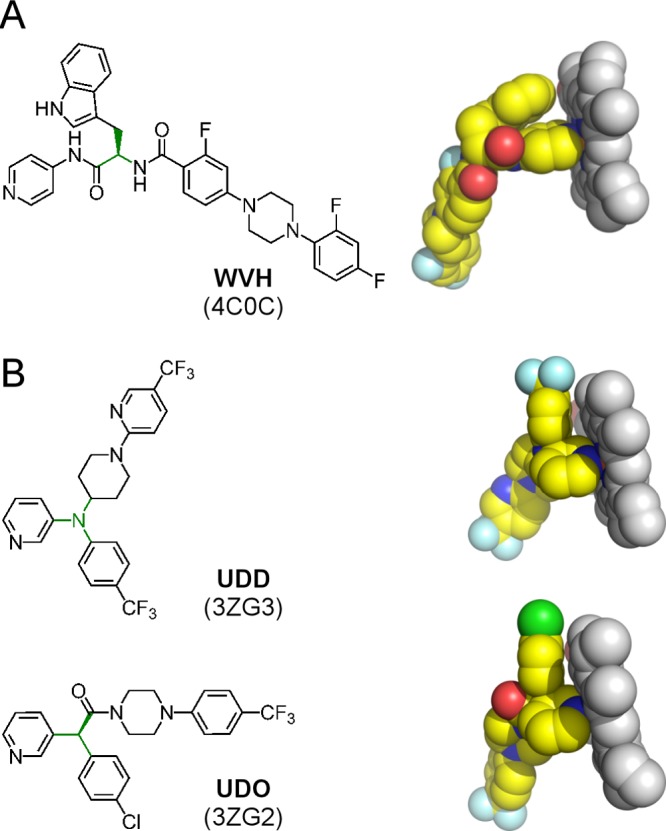
Pyridine-based CYP51 inhibitors. (A) N-Indolyl-oxopyridinyl-4-aminopropanyl-based analogues. (B) Fenarimol analogues. Drug–heme van der Waals interactions are shown as resolved in the X-ray structures of the corresponding drug–target complexes (PDB ID codes are in parentheses). Heme is in gray van der Waals spheres; inhibitors colored by atom types with carbon highlighted in yellow are labeled by the small-molecule codes. The branching points in chemical structures are highlighted in green.
Conclusions
The 4-aminopyridyl-based molecular scaffold possesses features key for translating lead compounds into clinical drug candidates: potency against the therapeutic target, large therapeutic window, oral bioavailability, and high tissue tropism, while terminal half-life and cross-reactivity with human CYPs need further optimization. Only concomitant enhancement of each of these features may allow improvements sufficient to ensure parasitological cure in mice and humans. Over the course of this work, we have assembled drug leads, methodologies, and tools necessary to successfully monitor each aspect of lead improvement. Valuable additions to the toolbox include an application of the T. cruzi Y strain expressing firefly luciferase for quick assessment of bioavailability and potency and inclusion of high-resolution crystal structures as plug-ins for analysis of the pharmacodynamic aspects of lead optimization. The new class of molecules has considerable promise for the development of anti-T. cruzi therapeutics and represents the current trend in the field of drug discovery for Chagas disease toward lead diversification and optimization specifically directed against the T. cruzi molecular target.
Experimental Section
Chemistry
General Procedure for the Synthesis of Inhibitors 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, and 12
To a solution of the appropriate benzoic acid (ca.1.2 equiv), PyBOP (ca. 1.4 equiv), and HOBt (ca. 10 mol %) in dry CH2Cl2 (5 mL) was slowly added triethylamine (ca. 4 equiv) at ambient temperature. The reaction mixture was stirred for 15 min until it became homogeneous. d-Tryptophan derivative 30(45) was added, and the reaction mixture was stirred at room temperature for 1 h. After confirming that the reaction was complete by using TLC analysis, the solvent was removed under reduced pressure. Ethyl acetate (10 mL) was added to the crude product mixture, and this solution was washed then with saturated aqueous NaHCO3 (2 mL × 2) and brine (2 mL × 2). The organic layer was concentrated in vacuo, and the crude product was directly subjected to flash chromatography purification to provide the titled products in ca. 70% yield.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2′,3,5′-trifluoro-[1,1′-biphenyl]-4-carboxamide TFA (1)
The general procedure was followed using carboxylic acid 13 as the acylating agent. The crude product was further purified by HPLC to afford 1 as a white solid (69%): [α]D26 = −70.0 (c = 1.0, MeOH). 1H NMR (400 MHz, DMSO-d6) δ 11.55 (s, 1H), 10.95 (d, J = 2.5 Hz, 1H), 8.83 (dd, J = 6.9, 2.7 Hz, 1H), 8.76–8.58 (m, 2H), 8.15–7.92 (m, 2H), 7.67 (q, J = 7.9 Hz, 2H), 7.59–7.48 (m, 3H), 7.43 (td, J = 9.5, 4.6 Hz, 1H), 7.38–7.26 (m, 3H), 7.06 (ddd, J = 8.1, 7.0, 1.2 Hz, 1H), 7.00–6.92 (m, 1H), 4.98–4.85 (m, 1H), 3.42–3.17 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.77, 163.59, 160.46, 159.53, 157.98, 157.15, 156.44, 154.03, 151.61, 143.78, 138.38, 138.29, 136.09, 130.50, 130.47, 127.08, 124.96, 124.92, 124.88, 124.14, 122.71, 122.57, 121.04, 118.38, 118.33, 118.14, 118.05, 117.89, 117.80, 117.16, 117.12, 116.89, 116.71, 116.68, 116.45, 114.45, 111.41, 109.00, 55.69, 26.99. MS (ESI) m/z 515.2 [M + H]+. HRMS (ESI) m/z for C29H22N4O2F3 [M + H]+ calcd 515.1695, found 515.1696.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-3,3′,4′-trifluoro-[1,1′-biphenyl]-4-carboxamide (2)
The general procedure was followed using 14 as the acylating agent to provide 2 as a light-yellow solid (80%): [α]D26 = −69.3 (c = 1.0, MeOH). 1H NMR (400 MHz, DMSO-d6) δ 10.90 (d, J = 2.5 Hz, 1H), 10.68 (s, 1H), 8.56 (dd, J = 7.4, 3.7 Hz, 1H), 8.51–8.38 (m, 2H), 7.93 (ddd, J = 12.1, 7.7, 2.3 Hz, 1H), 7.77–7.50 (m, 8H), 7.33 (d, J = 8.1 Hz, 1H), 7.25 (d, J = 2.3 Hz, 1H), 7.13–7.02 (m, 1H), 6.97 (t, J = 7.4 Hz, 1H), 4.91 (td, J = 8.2, 5.6 Hz, 1H), 3.43–3.16 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.52, 163.22, 161.08, 158.60, 151.09, 151.05, 150.96, 150.92, 150.32, 148.64, 148.58, 148.52, 148.46, 145.53, 142.34, 142.24, 136.07, 135.30, 130.98, 130.95, 127.19, 123.94, 123.89, 123.86, 122.56, 122.53, 122.09, 121.95, 121.00, 118.50, 118.28, 118.18, 118.01, 116.30, 116.12, 114.46, 114.21, 113.45, 111.35, 109.35, 55.14, 27.43. MS (ESI) m/z 515.2 [M + H]+. HRMS (ESI) m/z for C29H22N4O2F3 [M + H]+ calcd 515.1695, found 515.1695.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-3-fluoro-3′-(trifluoromethyl)-[1,1′-biphenyl]-4-carboxamide hydrochloride (3)
The general procedure was followed using 15 as the acylating agent to provide 3 as a light-yellow solid (73%), which was further purified by HPLC. The HCl salt of 3 was obtained by adding 2 N HCl (ca. 1 mL) to 3 in aqueous solution during evaporation: [α]D26 = −73.1 (c = 1.0, MeOH). 1H NMR (400 MHz, DMSO-d6) δ 15.44 (s, 1H), 12.24 (s, 1H), 11.00 (d, J = 2.4 Hz, 1H), 8.84–8.65 (m, 3H), 8.27–8.16 (m, 2H), 8.13–8.03 (m, 2H), 7.87–7.65 (m, 6H), 7.39–7.29 (m, 2H), 7.10–7.00 (m, 1H), 6.95 (t, J = 7.4 Hz, 1H), 4.98 (ddd, J = 8.7, 6.8, 5.4 Hz, 1H), 3.50–3.22 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 173.04, 163.45, 161.16, 158.68, 152.96, 143.09, 143.00, 142.16, 138.82, 136.08, 131.12, 131.06, 131.03, 130.23, 130.10, 129.78, 127.14, 125.46, 125.22, 124.23, 123.57, 123.54, 123.50, 123.46, 122.91, 122.88, 122.75, 122.14, 122.00, 120.99, 118.55, 118.32, 114.82, 114.60, 111.38, 108.99, 55.94, 26.97. MS (ESI) m/z 547.2 [M + H]+. HRMS (ESI) m/z for C30H23N4O2F4 [M + H]+ calcd 547.1757, found 547.1756.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-3,3′-difluoro-4′-((4-fluorobenzyl)oxy)-[1,1′-biphenyl]-4-carboxamide (4)
The general procedure was followed using 22b as the acylating agent to provide 4 as a light-yellow solid (85%): [α]D26 = −54.6 (c = 1.0, MeOH). 1H NMR (400 MHz, DMSO-d6) δ 10.90 (d, J = 2.5 Hz, 1H), 10.67 (s, 1H), 8.55–8.39 (m, 3H), 7.73 (dd, J = 12.8, 2.3 Hz, 1H), 7.70–7.56 (m, 7H), 7.56–7.51 (m, 2H), 7.35 (t, J = 8.6 Hz, 2H), 7.29–7.19 (m, 3H), 7.06 (ddd, J = 8.0, 6.9, 1.1 Hz, 1H), 7.01–6.86 (m, 1H), 5.23 (s, 2H), 4.91 (td, J = 8.1, 5.6 Hz, 1H), 3.34–3.19 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.53, 163.25, 163.13, 161.21, 160.71, 158.74, 153.26, 150.84, 150.39, 146.52, 146.42, 145.47, 143.12, 143.04, 136.08, 132.65, 132.62, 130.95, 130.92, 130.86, 130.18, 130.10, 127.19, 123.94, 123.18, 123.15, 122.01, 121.98, 121.22, 121.08, 121.00, 118.50, 118.28, 115.76, 115.48, 115.27, 114.68, 114.49, 113.79, 113.55, 113.45, 111.36, 109.34, 69.55, 55.12, 27.45. MS (ESI) m/z 621.3 [M + H]+. HRMS (ESI) m/z for C36H28N4O3F3 [M + H]+ calcd 621.2114, found 621.2115.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2′,3-difluoro-5′-(trifluoromethyl)-[1,1′-biphenyl]-4-carboxamide (5)
The general procedure was followed using 16 as the acylating agent to provide 5 as a light-yellow solid (85%): [α]D27 = −57.6 (c = 0.54, MeOH). 1H NMR (400 MHz, DMSO-d6) δ 10.90 (d, J = 2.5 Hz, 1H), 10.68 (s, 1H), 8.67 (dd, J = 7.4, 3.2 Hz, 1H), 8.51–8.40 (m, 2H), 7.98 (dd, J = 7.1, 2.4 Hz, 1H), 7.89 (ddd, J = 8.5, 4.4, 2.4 Hz, 1H), 7.76–7.66 (m, 2H), 7.66–7.57 (m, 4H), 7.54 (dt, J = 8.0, 1.7 Hz, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.26 (d, J = 2.4 Hz, 1H), 7.07 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.02–6.92 (m, 1H), 4.99–4.83 (m, 1H), 3.39–3.16 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.48, 163.26, 162.31, 160.47, 159.78, 157.98, 150.38, 145.48, 137.75, 137.66, 136.07, 130.59, 130.56, 128.21, 128.18, 128.14, 127.77, 127.38, 127.25, 127.19, 126.19, 126.16, 125.87, 125.83, 125.14, 125.11, 125.06, 123.94, 123.08, 122.93, 122.35, 120.99, 118.50, 118.28, 117.78, 117.54, 116.93, 116.70, 116.67, 113.45, 111.35, 109.36, 55.16, 27.44. MS (ESI) m/z 565.2 [M + H]+. HRMS (ESI) m/z for C30H22N4O2F5 [M + H]+ calcd 565.1663, found 565.1658.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-3,4′-difluoro-3′-(trifluoromethyl)-[1,1′-biphenyl]-4-carboxamide (6)
The general procedure was followed using 17 as the acylating agent to provide 6 as a light-yellow solid (68%): [α]D27 = −69.6 (c = 0.56, MeOH). 1H NMR (400 MHz, DMSO-d6) δ 10.90 (d, J = 2.5 Hz, 1H), 10.73 (s, 1H), 8.59 (dd, J = 7.3, 3.7 Hz, 1H), 8.50–8.42 (m, 2H), 8.22–8.06 (m, 2H), 7.82–7.75 (m, 1H), 7.74–7.66 (m, 4H), 7.66–7.60 (m, 3H), 7.34 (d, J = 8.1 Hz, 1H), 7.26 (d, J = 2.4 Hz, 1H), 7.06 (ddd, J = 8.0, 6.9, 1.2 Hz, 1H), 7.01–6.93 (m, 1H), 4.92 (td, J = 7.9, 5.5 Hz, 1H), 3.39–3.17 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.57, 163.23, 161.09, 160.30, 158.60, 150.05, 145.79, 142.06, 141.98, 136.09, 134.87, 133.88, 133.79, 131.04, 131.01, 127.20, 125.82, 125.77, 123.95, 123.89, 122.87, 122.84, 122.29, 122.15, 121.17, 121.00, 118.49, 118.29, 118.08, 117.87, 114.79, 114.55, 113.50, 111.36, 109.34, 55.19, 27.43. MS (ESI) m/z 565.2 [M + H]+. MS (ESI) m/z 565.2 [M + H]+. HRMS (ESI) m/z for C30H22N4O2F5 [M + H]+ calcd 565.1663, found 565.1664.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-3′-chloro-3,4′-difluoro-[1,1′-biphenyl]-4-carboxamide hydrochloride (7)
The general procedure was followed using 18 as the acylating agent to provide 7 as a light-yellow solid, which was further purified by HPLC. The HCl salt of 7 (58%) was obtained by adding 2 N HCl (ca. 1 mL) to 7 in aqueous solution during evaporation: [α]D27 = −54.9 (c = 0.57, MeOH). 1H NMR (400 MHz, DMSO-d6) δ 12.29 (s, 1H), 11.00 (d, J = 2.5 Hz, 1H), 8.79–8.67 (m, 3H), 8.27–8.17 (m, 2H), 8.03 (dd, J = 7.1, 2.4 Hz, 1H), 7.79 (ddd, J = 8.7, 4.7, 2.4 Hz, 1H), 7.76–7.67 (m, 3H), 7.64 (dd, J = 8.1, 1.7 Hz, 1H), 7.53 (t, J = 9.0 Hz, 1H), 7.47 (t, J = 100.0 Hz, 1H), 7.39–7.29 (m, 2H), 7.04 (ddd, J = 8.1, 6.9, 1.1 Hz, 1H), 6.99–6.88 (m, 1H), 4.98 (ddd, J = 8.8, 6.8, 5.4 Hz, 1H), 3.51–3.23 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 173.02, 163.40, 161.12, 158.72, 158.64, 156.25, 152.96, 142.32, 142.24, 142.12, 136.07, 135.54, 130.99, 130.96, 129.10, 127.79, 127.71, 127.14, 124.22, 122.66, 122.63, 121.82, 121.69, 120.98, 120.42, 120.24, 118.56, 118.31, 117.57, 117.36, 114.59, 114.33, 111.37, 108.98, 55.93, 26.97. MS (ESI) m/z 531.1 [M + H]+. HRMS (ESI) m/z for C29H22N4O2F2Cl [M + H]+ calcd 531.1399, found 531.1391.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-2-fluoro-4-(4-(3-fluorophenyl)piperazin-1-yl)benzamide (8)
The general procedure was followed using 26b as the acylating agent to provide 8 as a light-yellow solid (84%): [α]D26 = −61.2 (c = 1.0, MeOH). 1H NMR (400 MHz, DMSO-d6) δ 10.89 (d, J = 2.4 Hz, 1H), 10.65 (s, 1H), 8.49–8.38 (m, 2H), 7.81 (t, J = 7.5 Hz, 1H), 7.67–7.53 (m, 4H), 7.33 (d, J = 8.1 Hz, 1H), 7.28–7.18 (m, 2H), 7.05 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 6.99–6.91 (m, 1H), 6.90–6.73 (m, 4H), 6.57 (td, J = 8.4, 1.8 Hz, 1H), 4.95–4.83 (m, 1H), 3.43 (dd, J = 6.9, 3.6 Hz, 4H), 3.39–3.19 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 171.69, 164.48, 162.95, 162.92, 162.70, 162.09, 160.26, 154.15, 154.03, 152.48, 152.38, 150.37, 145.49, 136.10, 131.74, 131.70, 130.44, 130.34, 127.21, 123.92, 121.01, 118.45, 118.29, 113.42, 111.36, 111.00, 110.18, 110.05, 109.84, 109.22, 105.08, 104.87, 102.09, 101.84, 100.85, 100.57, 54.91, 47.21, 46.42, 27.57. MS (ESI) m/z 581.5 [M + H]+. HRMS (ESI) m/z for C33H31N6O2F2 [M + H]+ calcd 581.2477, found 581.2480.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4-(4-acetylpiperazin-1-yl)-2-fluorobenzamide Hydrochloride (9)
The general procedure was followed using 24b as the acylating agent to provide 9, which was further purified by HPLC to afford the product as a white solid. The HCl salt of 9 was obtained by adding 2 N HCl (ca. 1 mL) to 9 in aqueous solution during evaporation (67%): [α]D26 = −57.3 (c = 1.0, MeOH). 1H NMR (400 MHz, DMSO-d6) δ 15.42 (s, 1H), 12.25 (s, 1H), 10.99 (d, J = 2.5 Hz, 1H), 8.80–8.65 (m, 2H), 8.26–8.14 (m, 2H), 7.95 (dd, J = 8.4, 5.6 Hz, 1H), 7.67 (d, J = 7.9 Hz, 1H), 7.60 (t, J = 9.0 Hz, 1H), 7.32 (d, J = 8.1 Hz, 1H), 7.29 (d, J = 2.3 Hz, 1H), 7.04 (ddd, J = 8.2, 6.9, 1.1 Hz, 1H), 6.95–6.88 (m, 1H), 6.84–6.72 (m, 2H), 4.92 (dt, J = 7.8, 5.8 Hz, 1H), 3.54 (dd, J = 6.7, 3.9 Hz, 4H), 3.45–3.23 (m, 6H), 2.03 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 173.26, 168.43, 163.16, 163.13, 162.73, 160.28, 154.09, 153.98, 153.04, 142.09, 136.11, 131.70, 131.66, 127.12, 124.19, 121.02, 118.50, 118.33, 114.55, 111.40, 109.96, 109.84, 109.80, 108.88, 100.86, 100.57, 55.71, 46.77, 46.45, 44.91, 26.99, 21.21. MS (ESI) m/z 529.3 [M + H]+. HRMS (ESI) m/z for C29H30N6O3F [M + H]+ calcd 529.2363, found 529.2365.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4-(4-(3-chlorophenyl)piperazin-1-yl)-2-fluorobenzamide (10)
The general procedure was followed using 27b as the acylating agent to provide 10 as a light-yellow solid (61%): [α]D27 = −65.7 (c = 0.72, MeOH). 1H NMR (400 MHz, DMSO-d6) δ 10.90 (d, J = 2.5 Hz, 1H), 10.70 (s, 1H), 8.53–8.39 (m, 2H), 7.82 (t, J = 7.5 Hz, 1H), 7.68–7.58 (m, 4H), 7.33 (d, J = 8.1 Hz, 1H), 7.27–7.19 (m, 2H), 7.05 (ddd, J = 8.1, 7.0, 1.2 Hz, 1H), 7.00 (t, J = 2.2 Hz, 1H), 6.97–6.90 (m, 2H), 6.88–6.76 (m, 3H), 4.98–4.82 (m, 1H), 3.43 (dd, J = 6.8, 3.7 Hz, 4H), 3.37–3.19 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 171.77, 162.96, 162.94, 162.70, 160.26, 154.14, 154.03, 151.91, 150.01, 145.82, 136.10, 133.86, 131.74, 131.70, 130.48, 127.20, 123.93, 121.01, 118.45, 118.36, 118.29, 114.74, 113.84, 113.47, 111.37, 110.17, 110.04, 109.83, 109.20, 100.85, 100.57, 54.95, 47.21, 46.42, 27.54. MS (ESI) m/z 597.2 [M + H]+. HRMS (ESI) m/z for C33H31N6O2FCl [M + H]+ calcd 597.2181, found 597.2188.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4-(4-(3,4-difluorophenyl)piperazin-1-yl)-2-fluorobenzamide (11)
The general procedure was followed using 28b as the acylating agent to provide 11 as a light-yellow solid (64%): [α]D27 = −69.5 (c = 0.46, MeOH). 1H NMR (400 MHz, DMSO-d6) δ 10.89 (d, J = 2.5 Hz, 1H), 10.70 (s, 1H), 8.52–8.36 (m, 2H), 7.82 (t, J = 7.5 Hz, 1H), 7.69–7.55 (m, 4H), 7.33 (d, J = 8.1 Hz, 1H), 7.27 (dt, J = 10.7, 9.4 Hz, 1H), 7.21 (d, J = 2.4 Hz, 1H), 7.11–7.01 (m, 2H), 6.93 (t, J = 7.4 Hz, 1H), 6.89–6.75 (m, 3H), 4.99–4.80 (m, 1H), 3.43 (dd, J = 6.8, 3.7 Hz, 4H), 3.37–3.16 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 171.76, 162.96, 162.93, 162.69, 160.25, 154.15, 154.04, 151.01, 150.88, 150.01, 148.60, 148.47, 148.24, 148.16, 145.82, 141.84, 136.10, 131.73, 131.68, 127.20, 123.93, 121.01, 118.45, 118.28, 117.40, 117.23, 113.47, 111.37, 110.23, 110.11, 109.91, 109.20, 104.77, 104.56, 100.94, 100.66, 54.94, 47.89, 46.48, 27.54. MS (ESI) m/z 599.2 [M + H]+. HRMS (ESI) m/z for C33H30N6O2F3 [M + H]+ calcd 599.2382, found 599.2374.
(R)-N-(3-(1H-Indol-3-yl)-1-oxo-1-(pyridin-4-ylamino)propan-2-yl)-4-(4-(2,4-difluorophenyl)piperazin-1-yl)-2-fluorobenzamide (12)
The general procedure was followed using 29b as the acylating agent to provide 12 as a light-yellow solid (74%): [α]D27 = −52.8 (c = 0.61, MeOH). 1H NMR (400 MHz, DMSO-d6) δ 10.89 (d, J = 2.5 Hz, 1H), 10.64 (s, 1H), 8.50–8.38 (m, 2H), 7.82 (t, J = 7.5 Hz, 1H), 7.69–7.56 (m, 4H), 7.33 (dt, J = 8.1, 0.9 Hz, 1H), 7.28–7.18 (m, 2H), 7.16–6.97 (m, 3H), 6.94 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 6.89–6.77 (m, 2H), 4.96–4.81 (m, 1H), 3.44 (dd, J = 6.7, 3.5 Hz, 4H), 3.34–3.19 (m, 2H), 3.16–3.01 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 171.69, 162.95, 162.92, 162.68, 160.24, 158.48, 156.15, 156.09, 156.03, 155.96, 154.30, 154.19, 153.69, 153.56, 150.36, 145.50, 136.48, 136.44, 136.39, 136.35, 136.10, 131.72, 131.67, 127.21, 123.92, 121.01, 120.31, 120.27, 120.22, 120.18, 118.46, 118.29, 113.43, 111.37, 111.21, 111.17, 111.00, 110.96, 110.31, 110.18, 109.92, 109.22, 104.95, 104.69, 104.44, 100.95, 100.67, 54.91, 50.06, 46.92, 27.57. MS (ESI) m/z 599.2 [M + H]+. HRMS (ESI) m/z for C33H30N6O2F3 [M + H]+ calcd 599.2382, found 599.2380.
Compounds were stored at −20 °C as 10 mM stock solutions in DMSO and freshly diluted immediately prior to use.
Hepatic Microsomal Stability
Hepatic microsomal stability was addressed as described previously.44
CYP Inhibition
Cytochrome P450 inhibition was evaluated in human liver microsomes using four selective marker substrates as described previously.44
Binding Affinity by UV–vis Spectroscopy
Binding affinity of compounds was approximated from the spectrophotometry titration curves generated as previously described.46
T. cruzi Maintenance
T. cruzi, Y luc strain, transformed with episomal firefly luciferase gene, was developed as described elsewhere.47 All media components used in this work were purchased from Sigma unless indicated otherwise. Cultured trypomastigotes were obtained by weekly infection of C2C12 myoblasts, with trypomastigotes being released in the supernatant 4–7 days post infection, collected by centrifugation for 15 min at 3300 rpm. Without selective antibiotic pressure, the luciferase expression in the parasite was detectable for about as long as seven passages in mammalian culture. To maintain high titer of luciferase marker in parasite population, the pressure of G418 antibiotic was applied to epimastigote form. For that, the epimastigotes were cultivated in LIT medium,72 supplemented with 10% fetal bovine serum (FBS) and 200 μg/mL of G418, at 28 °C. Once a month, myoblast cultures were infected with 2–4 week old epimastigotes enriched with the metacyclic trypomastigotes. Forty-eight hours postinfection, the epimastigotes were removed from the medium by successive washing of cultures with phosphate buffer saline (PBS). Seven days postinfection, trypomastigote population enriched with the transgenic parasites expressing luciferase was collected from the medium.
Dose–Response in T. cruzi Cell-Based Assay
To estimate compound’s anti-T. cruzi potency, the EC50 values of compounds were determined in the cell-based assay adapted from Engel and coauthors73 and modified as previously described.51 Briefly, mouse C2C12 myoblasts (ATCC no. CRL-1772) used to harbor parasites were cultivated in Dulbecco’s Modified Eagle’s Medium containing 4.5 g/L glucose (DMEM H-21), supplemented with 5% FBS, 25 mM HEPES, 2 mM l-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. T. cruzi CA-I/72 trypomastigotes74 were obtained from infected-culture supernatants after 4–7 days of infection. Cultures were maintained at 37 °C with 5% CO2. Trypomastigotes and C2C12 cells concentration was determined using a Neubauer hemocytometer.
The assay was performed in triplicate. Sterile, black 384-well plates with clear-bottom wells (Greiner Bio-One) were seeded with mouse C2C12 myoblasts (500 cells/well) and then were infected with CA-I/72 trypomastigotes (2500 parasites/well) in 50 μL of culture medium (DMEM H-21)/well. Culture plates were incubated at 37 °C with 5% CO2. Twenty-four hours postinfection, culture medium was removed and test compounds were added in fresh medium. For this, an intermediate 384-well plate was prepared by serial dilution for all the compounds in 100% DMSO. Then, 50 nL of each sample were diluted in 50 μL of media (DMEM H-21) to final concentrations of 10 μM, 2 μM, 400 nM, 80 nM, 16 nM, 3 nM, 128 pM, 25 pM, and 5 pM and added to the experimental plate followed by incubation at 37 °C with 5% CO2 for 72 h. Wells containing noninfected cells were used as a positive control (100% cell survival), while T. cruzi-infected but untreated cells (0% cell survival) were used as a negative control.
Cells were then fixed for 2 h with 4% paraformaldehyde and rinsed with a solution of 150 mM NaCl, 100 mM NH4Cl, 0.1% Triton X-100, and 0.1% NaN3. After that, they were treated for 4 h with 0.2 μg/mL of the DNA fluorescent dye, DAPI (4,6-diamidino-2-phenylindole), diluted in the same solution. Plates were kept at ambient temperature until image acquisition was performed. Images were acquired by an IN Cell Analyzer 2000 (GE Healthcare), and the procedure and analyses were performed according to previously described.51,73
Cytotoxicity Assay
Cytotoxicity of compounds was assessed against two different cell types, cardiomyocytes and hepatocytes, isolated from 18-day-old mouse embryos as previously described.75,76 Briefly, to obtain cardiomyocytes, cardiac embryo fragments were dissociated in PBS, supplemented with 0.025% trypsin and 0.01% collagenase (Worthington Co., Lakewood, NJ), and plated in a density of 5 × 105 cells/well into a white opaque flat bottom μclear 96-well plate (Greiner Bio-One) coated with 0.01% gelatin. Cultures were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS, 2.5 mM CaCl2, 1 mM l-glutamine, and 2% chicken embryo extract. Hepatocytes were isolated through incubation of embryo liver fragments with 0.05% solution of collagenase type III in Hank’s Balanced Salt Solution (HBSS): 0.14 M NaCl, 5.37 M KCl, 0.84 mM MgSO4, 0.44 mM KH2PO4, 1,50 g/L glucose, 0,0005 g/L insulin, 0.02 M HEPES, pH 7.4. The liver cells were dispersed by pipetting and seeded in a density of 1.5 × 105 cells/well into a white opaque flat bottom μclear 96-well plate (Greiner Bio-One) coated with 0.01% gelatin in hepatocyte adhesion medium (70% MEM/30% 199 medium, supplemented with 10% FBS, 5 mM CaCl2). After 6 h, the cultures were washed in HBSS and the media was changed to a mixture of DMEM/199 at 70/30% ratio supplemented with 10% FBS, 2 mM l-glutamin, 10 μg/mL insulin, 1 ng/mL glucagon, 50 ng/mL EGF, 3.5 × 106 U hydrocortisone, 6.5 ng/mL somatotropin, and 0.5 μg/mL linoleic acid. All cultures were kept at 37 °C in an atmosphere of 5% CO2. For the cytotoxicity assay, the compounds were serially diluted freshly from 10 mM DMSO stock solutions with appropriate medium and added to primary culture cells at final concentrations of 90, 30, 10, 3.3, 1.1, 0.37, 0.12, and 0.04 μM. After 72 h incubation, the viability of the cells was assessed using the ATP-detection luminescence-based kit, Cell Titer Glo (Promega). The EC50 values were determined based in the sigmoidal dose–response curves of the cell growth inhibition. Selectivity index (SI) for the compounds was calculated as a ratio between the mammalian cell and T. cruzi amastigote viability expressed as corresponding EC50 values.
Four-Day Dosing Mouse Model of T. cruzi Infection
The four-day dosing mouse model of T. cruzi infection was used to assess in vivo potency of increasingly optimized inhibitors. Eight-week-old female Swiss Webster albino mice (average weight 20 g) were obtained from Simonsen Laboratories (Gilroy, CA). All animal procedures were approved and carried out in accordance with the guidelines established by the Institutional Animal Care and Use Committee from UCSF (approval no. AN087605-01). Mice were housed at a maximum of five per cage and kept in a specific-pathogen free (SPF) room at 20 to 24 °C under a 12 h light/12 h dark cycle and provided with sterilized water and chow ad libitum. To infect the mice, trypomastigotes of T. cruzi Y luc strain were harvested from culture supernatant and injected intraperitoneally, 105 trypomastigotes per mouse. Three days post infection, mice were anesthetized by inhalation of isofluorane (controlled flow of 1.5% of isofluorane in air was administered through a nose cone via gas anesthesia system). Mice were injected ip with 150 mg/kg d-luciferin potassium salt (Gold Biotechnology) dissolved in PBS and imaged after 5 min using IVIS Spectrum preclinical in vivo imaging system (PerkinElmer, Waltham, MA) and the data acquisition and analysis software LivingImage V4.1 (PerkinElmer, Waltham, MA). Only mice with detectable luminescence at day 3 postinfection were used for treatment. Compound potency was evaluated following intaperitoneal (ip) or oral (po) administration in groups of five mice.
Intraperitoneally, treatment with compounds continued at 40 mg/kg, bid, for four consecutive days. The compounds were administered at 8 mg/mL in 100 μL of 60% DMSO in saline (NaCl 0.9%) per dose. Two control groups included untreated mice, which received 60% DMSO in saline (NaCl 0.9%), and the positive control group, which received 20 mg/kg posaconazole, both ip, bid. Orally, compounds were administered as suspension in 20% 2-hydroxypropyl-β-cyclodextrin (HPβCD) (VWR International) or as solution in Kolliphor HS 15 (Sigma no. 42966), also known as solutol. Only one compound, 7, was fully soluble in HPβCD, while all dissolved in 20% solutol upon overnight incubation. Specified doses were administered bid for four consecutive days. Two control groups included untreated mice, which received 20% HPβCD or solutol, and the positive control group, which received benznidazole 50 mg/kg, both bid by oral gavage. After 4 days of treatment, mice were imaged again as described above. The absolute numbers of measured photons/s/cm2 were averaged between all five mice in each group and compared directly between compound-treated mice and the control groups. Two-tailed paired Student t test was used to assess statistical significance between luminescence values from vehicle-treated and compound-treated groups at day 7 postinfection; values are statistically significant when p ≤ 0.05.
Single Dose PK and Tissue Distribution
Compounds were dosed at 50 mg/kg in eight-week-old female Swiss Webster albino mice via oral gavage. All compounds were formulated to a concentration of 10 mg/mL in 20% solutol/80% water. Nine mice were dosed and n = 3 plasma samples were collected at approximately 0.25, 0.5, 1, 2, 4, 6, 8, 12, and 24 h. Tissue samples were collected from three mice two and 8 h post dose. Plasma samples were treated with 5 times v/v acetonitrile to precipitate protein and filtered through a 0.2 μm filter prior to analysis by LC-MS/MS using an ABSciex 5500. Tissue samples were placed in PBS (1:4 w/v) and disrupted using a probe tip sonicator prior to precipitating protein with acetonitrile and LC-MS/MS analysis.
Sterol Profiling of Intracellular T. cruzi Amastigotes
Sterol profiling was performed on whole-cell lipid extracts, prepared as described previously.51 Posaconazole (100 nM) was used as a positive control and benznidazole (5 μM) as a negative control. Compounds 3 and 12 were tested at 100 nM. Briefly, C2C12 cultures infected with T. cruzi (CAI/72 strain) for 96 h and treated with compounds for 24 h were detached and submitted to lipid extraction with chloroform/methanol, chloroform, and acetonitrile, each step followed by several rounds of washes with water to extract polar molecules. The organic layer was then dried under nitrogen gas and subsequently treated with 75 μL of N,N-bis(trimethylsilyl)-2,2,2-trifluoroacetamide (BSTFA) for 2 h at 37 °C to facilitate chemical derivatization with trimethylsilyl (TMS) groups (BSTFA, Sigma-Aldrich). The TMS-derivatized lipid mixture was analyzed by injecting 3 μL directly into an Agilent HP5790 gas chromatography system outfitted with a DB5-MS analytical column (30 m, 0.25 mm i.d., 0.33 μm film thickness, Agilent) coupled to a mass selective detector. The lipids were separated on the analytical column using a temperature profile that begins at 200 °C for 1 min, increases by 15 °C/min up to 300 °C, and then holds at 300 °C for 20 min. The inlet temperatures of the GC and the MSD were held at 250 and 300 °C, respectively. The mass spectrometer scanned from m/z 50–750 during the course of analysis.
X-ray Structure Analysis
To analyze the inhibitor binding mode, recombinant T. cruzi CYP51, modified by replacing the first 31 residues upstream of Pro32 with the fragment MAKKTSSKGKL77 and by inserting a His6-tag at the C-terminus, was expressed and purified as described elsewhere.42 Concentrated purified protein samples were stored at −80 °C and diluted prior to crystallization to 0.1 mM by mixing with 20 mM potassium phosphate pH 7.5, 10% glycerol, 1 mM DTT, 0.5 mM EDTA, and 300 mM NaCl, supplemented with equimolar inhibitor. Crystallization conditions were determined using commercial high-throughput screening kits available in deep-well format (Hampton Research, Aliso Viejo, CA), a nanoliter drop-setting Mosquito robot (TTP LabTech, Cambridge, MA) operating with 96-well plates, and a hanging drop crystallization protocol. Crystals were further optimized in 96-well plates for diffraction data collection and harvested directly from the 200 nL drops. Prior to data collection, crystals were cryoprotected by plunging them into a drop of reservoir solution supplemented with 20% ethylene glycol, then flash frozen in liquid nitrogen.
Diffraction data were collected at 100–110 K at beamline 8.3.1, Advanced Light Source, Lawrence Berkeley National Laboratory, USA. Data indexing, integration, and scaling were conducted using MOSFLM78 and the programs implemented in the ELVES software suite.79 The crystal structures were determined by molecular replacement using diffraction data processed in the corresponding space groups and atomic coordinates of T. cruzi CYP51 (PDB ID code: 2WX2) as a search model. The final model was built using COOT80 and refinement was performed by using REFMAC5 software.81,82 Data collection and refinement statistics are shown in Supporting Information, Table S3.
Acknowledgments
We thank Dr. Barbara Burleigh, Harvard School of Public Health, and Dr. Ana Rodriguez, New York University, for kindly providing T. cruzi Y luc strain, Dr. Julio Urbina for fruitful discussions and expert advice, Potter Wickware for proof reading of the manuscript, the staff members of beamline 8.3.1, James Holton, George Meigs, and Jane Tanamachi, at the Advanced Light Source at Lawrence Berkeley National Laboratory, for assistance with data collection. We also thank the Ortiz de Montellano Laboratory at UCSF for use of the GC-MS instrument and Renata Soares and Liliane Mesquita (IOC, FIOCRUZ, R.J.) for technical assistance with the hepatocyte and cardiomyocyte cultures. This work was supported by NIH RO1 grant AI095437 (to L.M.P.). C.M.C. was supported by Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (CNPq) and FIOCRUZ. J.B.J. was supported by Dr. Miriam & Sheldon G. Adelson Medical Research Foundation (APNRR Proteomics Platform, A120343). The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under contract no. DE-AC02-05CH11231. Molecular graphics images were produced in part using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR001081).
Glossary
Abbreviations Used
- T. cruzi
Trypanosoma cruzi
- CYP51
cytochrome P450 family 51
- CYP1A2, CYP2C9, CYP2D6, CYP3A4
cytochrome P450 isoforms 1A2, 2C9, 2D6, and 3A4
- SAR
structure–activity relationship
- EC50
half-maximal effective concentration
Supporting Information Available
Synthetic procedures and tabulated spectroscopic data for synthetic intermediates, tissue distribution data, dose–response curves, UV−vis titration curves, lipid profiles by GC-MS chromatograms and crystallographic statistics. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The atomic coordinates and structure factors (PDB ID codes 4C0C, 4UQH, and 4BMM) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Chagas C. Nova trypanozomiaze humana: estudos sobre a morfolojia e o ciclo evolutivo do Schizotrypanum cruzi n. gen., n. sp., ajente etiolojico de nova entidade morbida do homem. Mem. Inst. Oswaldo Cruz 1909, 12159–218. [Google Scholar]
- Russi A. J.; Russi A.; Marin-Neto J. A. Chagas disease. Lancet 2010, 375, 1388–1402. [DOI] [PubMed] [Google Scholar]
- WHO Fact Sheet 340: Chagas Disease (American Trypanosomiasis); World Health Organization: Geneva, 2014; http://www.who.int/mediacentre/factsheets/fs340/en/ (accessed April 30, 2014).
- Lee B. Y.; Bacon K. M.; Bottazzi M. E.; Hotez P. J. Global economic burden of Chagas disease: a computational simulation model. Lancet Infect. Dis. 2013, 134342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coura J. R.; Borges-Pereira J. Chronic phase of Chagas disease: why should it be treated? A comprehensive review. Mem. Inst. Oswaldo Cruz. 2011, 1066641–645. [DOI] [PubMed] [Google Scholar]
- Viotti R.; de Noya B. A.; Araujo-Jorge T.; Grijalva M. J.; Guhl F.; Lopez M. C.; Ramsey J. M.; Ribeiro I.; Schijman A. G.; Sosa-Estani S.; Torrico F.; Gascon J. Towards a paradigm shift in the treatment of chronic Chagas disease. Antimicrob. Agents Chemother. 2014, 582635–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro J. A.; de Mecca M. M.; Bartel L. C. Toxic side effects of drugs used to treat Chagas’ disease (American trypanosomiasis). Hum. Exp. Toxicol. 2006, 258471–479. [DOI] [PubMed] [Google Scholar]
- Buckner F. S.; Urbina J. A. Recent developments in sterol 14-demethylase inhibitors for Chagas Disease. Int. J. Parasitol. Drugs Drug. Resist. 2012, 2, 236–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeres J.; Meerpoel L.; Lewi P. Conazoles. Molecules 2010, 1564129–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan D. J.; Hitchcoch C. A.; Sibley C. M. Current and emerging azole antifungal agents. Clin. Microbiol. Rev. 1999, 12140–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paiva J. A.; Pereira J. M. New antifungal antibiotics. Curr. Opin. Infect. Dis. 2013, 262168–174. [DOI] [PubMed] [Google Scholar]
- Baldwin B. C. Inhibitors of ergosterol biosynthesis as crop protecting agents. Biochem. Soc. Trans. 1990, 18161–62. [DOI] [PubMed] [Google Scholar]
- Urbina J. A.; Payares G.; Contreras L. M.; Liendo A.; Sanoja C.; Molina J.; Piras M.; Piras R.; Perez N.; Wincker P.; Loebenberg D. Antiproliferative effects and mechanism of action of SCH 56592 against Trypanosoma (Schizotrypanum) cruzi: in vitro and in vivo studies. Antimicrob. Agents Chemother. 1998, 4271771–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman C. A.; Malani A. N.; Easley C.; Kirkpatrick P. Posaconazole. Nature Rev. Drug Discovery 2007, 63183–184. [DOI] [PubMed] [Google Scholar]
- Katragkou A.; Tsikopoulou F.; Roilides E.; Zaoutis T. E. Posaconazole: when and how? The clinician’s view. Mycoses 2011, 552110–122. [DOI] [PubMed] [Google Scholar]
- Molina J.; Martins-Filho O.; Brener Z.; Romanha A. J.; Loebenberg D.; Urbina J. A. Activities of the triazole derivative SCH 56592 (posaconazole) against drug-resistant strains of the protozoan parasite Trypanosoma (Schizotrypanum) cruzi in immunocompetent and immunosuppressed murine hosts. Antimicrob. Agents Chemother. 2000, 441150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraz M. L.; Gazzinelli R. T.; Alves R. O.; Urbina J. A.; Romanha A. J. The anti-Trypanosoma cruzi activity of posaconazole in a murine model of acute Chagas’ disease is less dependent on gamma interferon than that of benznidazole. Antimicrob. Agents Chemother. 2007, 5141359–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivieri B. P.; Molina J. T.; de Castro S. L.; Pereira M. C.; Calvet C. M.; Urbina J. A.; Araujo-Jorge T. C. A comparative study of posaconazole and benznidazole in the prevention of heart damage and promotion of trypanocidal immune response in a murine model of Chagas disease. Int. J. Antimicrob. Agents 2010, 36179–83. [DOI] [PubMed] [Google Scholar]
- Pinazo M.-J.; Espinosa G.; Gállego M.; López-Chejade P. L.; Urbina J.; Gascón J. Treatment with posaconazole of a patient with systemic lupus erythematosus and Chagas Disease. Am. J. Trop. Med. Hyg. 2010, 824583–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton J. Chagas disease: pushing through the pipeline. Nature 2010, 4657301S12–S15. [DOI] [PubMed] [Google Scholar]
- Nomeir A. A.; Kumari P.; Hilbert M. J.; Gupta S.; Loebenberg D.; Cacciapuoti A.; Hare R.; Miller G. H.; Lin C. C.; Cayen M. N. Pharmacokinetics of SCH 56592, a new azole broad-spectrum antifungal agent, in mice, rats, rabbits, dogs, and cynomolgus monkeys. Antimicrob. Agents Chemother. 2000, 443727–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lignell A.; Lowdin E.; Cars O.; Chryssanthou E.; Sjolin J. Posaconazole in human serum: a greater pharmacodynamic effect than predicted by the non-protein-bound serum concentration. Antimicrob. Agents Chemother. 2011, 5573099–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe R. Failure of ketoconazole to cure chronic murine Chagas’ disease. J. Infect. Dis. 1988, 15861408–1409. [DOI] [PubMed] [Google Scholar]
- Brener Z.; Cancado J. R.; Galvao L. M.; da Luz Z. M.; Filardi Lde S.; Pereira M. E.; Santos L. M.; Cancado C. B. An experimental and clinical assay with ketoconazole in the treatment of Chagas disease. Mem. Inst. Oswaldo Cruz 1993, 881149–153. [DOI] [PubMed] [Google Scholar]
- Moreira A. A.; de Souza H. B.; Amato Neto V.; Matsubara L.; Pinto P. L.; Tolezano J. E.; Nunes E. V.; Okumura M. Evaluation of the therapeutic activity of itraconazole in chronic infections, experimental and human, by Trypanosoma cruzi. Rev. Inst. Med. Trop. Sao Paulo 1992, 342177–180. [PubMed] [Google Scholar]
- Diniz Lde F.; Caldas I. S.; Guedes P. M.; Crepalde G.; de Lana M.; Carneiro C. M.; Talvani A.; Urbina J. A.; Bahia M. T. Effects of ravuconazole treatment on parasite load and immune response in dogs experimentally infected with Trypanosoma cruzi. Antimicrob. Agents Chemother. 2010, 5472979–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drug Trial for Leading Parasitic Killer of the Americas Shows Mixed Results but Provides New Evidence for Improved Therapy; Drugs for Neglected Diseases Initiative: Geneva, November14, 2013; http://www.dndi.org/media-centre/press-releases/1700-e1224.html (accessed April 30, 2014).
- Molina I.; Gomez i Prat J.; Salvador F.; Trevino B.; Sulleiro E.; Serre N.; Pou D.; Roure S.; Cabezos J.; Valerio L.; Blanco-Grau A.; Sanchez-Montalva A.; Vidal X.; Pahissa A. Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. N. Engl. J. Med. 2014, 370201899–1908. [DOI] [PubMed] [Google Scholar]
- Kohl N. E.; Mosser S. D.; deSolms S. J.; Giuliani E. A.; Pompliano D. L.; Graham S. L.; Smith R. L.; Scolnick E. M.; Oliff A.; Gibbs J. B. Selective inhibition of ras-dependent transformation by a farnesyltransferase inhibitor. Science 1993, 26051161934–1937. [DOI] [PubMed] [Google Scholar]
- Karp J. E.; Kaufmann S. H.; Adjei A. A.; Lancet J. E.; Wright J. J.; End D. W. Current status of clinical trials of farnesyltransferase inhibitors. Curr. Opin. Oncol. 2001, 136470–476. [DOI] [PubMed] [Google Scholar]
- Buckner F.; Yokoyama K.; Lockman J.; Aikenhead K.; Ohkanda J.; Sadilek M.; Sebti S.; Van Voorhis W.; Hamilton A.; Gelb M. H. A class of sterol 14-demethylase inhibitors as anti-Trypanosoma cruzi agents. Proc. Natl. Acad. Sci. U. S. A. 2003, 1002515149–15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucke O.; Gelb M. H.; Verlinde C. L.; Buckner F. S. The protein farnesyltransferase inhibitor Tipifarnib as a new lead for the development of drugs against Chagas disease. J. Med. Chem. 2005, 48175415–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chennamaneni N. K.; Arif J.; Buckner F. S.; Gelb M. H. Isoquinoline-based analogs of the cancer drug clinical candidate tipifarnib as anti-Trypanosoma cruzi agents. Bioorg. Med. Chem. Lett. 2009, 19, 6582–6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus J. M.; Tatipaka H. B.; McGuffin S. A.; Chennamaneni N. K.; Karimi M.; Arif J.; Verlinde C. L.; Buckner F. S.; Gelb M. H. Second generation analogues of the cancer drug clinical candidate tipifarnib for anti-Chagas disease drug discovery. J. Med. Chem. 2010, 53103887–3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus J. M.; Verlinde C. L.; Karimi M.; Lepesheva G. I.; Gelb M. H.; Buckner F. S. Rational modification of a candidate cancer drug for use against Chagas disease. J. Med. Chem. 2009, 5261639–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner F. S.; Bahia M. T.; Suryadevara P. K.; White K. L.; Shackleford D. M.; Chennamaneni N. K.; Hulverson M. A.; Laydbak J. U.; Chatelain E.; Scandale I.; Verlinde C. L.; Charman S. A.; Lepesheva G. I.; Gelb M. H. Pharmacological characterization, structural studies, and in vivo activities of anti-chagas disease lead compounds derived from tipifarnib. Antimicrob. Agents Chemother. 2012, 5694914–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan M.; Abbott M. J.; Alexander P. W.; Armstrong T.; Best W. M.; Berven B.; Botero A.; Chaplin J. H.; Charman S. A.; Chatelain E.; von Geldern T. W.; Kerfoot M.; Khong A.; Nguyen T.; McManus J. D.; Morizzi J.; Ryan E.; Scandale I.; Thompson R. A.; Wang S. Z.; White K. L. Analogues of fenarimol are potent inhibitors of Trypanosoma cruzi and are efficacious in a murine model of Chagas disease. J. Med. Chem. 2012, 5594189–4204. [DOI] [PubMed] [Google Scholar]
- Keenan M.; Alexander P. W.; Diao H.; Best W. M.; Khong A.; Kerfoot M.; Thompson R. C.; White K. L.; Shackleford D. M.; Ryan E.; Gregg A. D.; Charman S. A.; von Geldern T. W.; Scandale I.; Chatelain E. Design, structure–activity relationship and in vivo efficacy of piperazine analogues of fenarimol as inhibitors of Trypanosoma cruzi. Bioorg. Med. Chem. 2013, 2171756–1763. [DOI] [PubMed] [Google Scholar]
- Keenan M.; Chaplin J. H.; Alexander P. W.; Abbott M. J.; Best W. M.; Khong A.; Botero A.; Perez C.; Cornwall S.; Thompson R. A.; White K. L.; Shackleford D. M.; Koltun M.; Chiu F. C.; Morizzi J.; Ryan E.; Campbell M.; von Geldern T. W.; Scandale I.; Chatelain E.; Charman S. A. Two analogues of fenarimol show curative activity in an experimental model of Chagas disease. J. Med. Chem. 2013, 562410158–10170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalta F.; Dobish M. C.; Nde P. N.; Kleshchenko Y. Y.; Hargrove T. Y.; Johnson C. A.; Waterman M. R.; Johnston J. N.; Lepesheva G. I. VNI Cures Acute and Chronic Experimental Chagas Disease. J. Infect. Dis. 2013, 2083504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podust L. M.; von Kries J. P.; Nasser Eddine A.; Kim Y.; Yermalitskaya L. V.; Kuehne R.; Ouellet H.; Warrier T.; Altekoster M.; Lee J.-S.; Rademann J.; Oschkinat H.; Kaufmann S. H. E.; Waterman M. R. Small molecule scaffolds for CYP51 inhibitors identified by high-throughput screening and defined by x-ray crystallography. Antimicrob. Agents Chemother. 2007, 51113915–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.-K.; Doyle P. S.; Yermalitskaya L. V.; Mackey Z. B.; Ang K. K. H.; McKerrow J. H.; Podust L. M. Trypanosoma cruzi CYP51 inhibitor derived from a Mycobacterium tuberculosis screen hit. PLoS Negl. Trop. Dis. 2009, 32e372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kries J. P.; Warrier T.; Podust L. M. Identification of small-molecule scaffolds for P450 inhibitors. Curr. Protoc. Microbiol. 2010, 1617.417.4.1–17.4.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. Y.; Calvet C. M.; Gunatilleke S. S.; Ruiz C.; Cameron M. D.; McKerrow J. H.; Podust L. M.; Roush W. R. Rational development of 4-aminopyridyl-based inhibitors targeting Trypanosoma cruzi CYP51 as anti-Chagas agents. J. Med. Chem. 2013, 56197651–7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. Y.; Calvet C. M.; Vieira D. F.; Gunatelleke S. S.; Cameron M. D.; McKerrow J. H.; Podust L. M.; Roush W. R. The R-configuration of 4-aminopyridyl-based inhibitors of CYP51 confers superior efficacy against Trypanosoma cruzi. ACS Med. Chem. Lett. 2014, 5, 434–439 10.1021/ml500010m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira D. F.; Choi J. Y.; Roush W. R.; Podust L. M. Expanding the binding envelope of CYP51 inhibitors targeting Trypanosoma cruzi with 4-aminopyridyl-based sulfonamide derivatives. ChemBioChem 2014, 15, 1111–1120 10.1002/cbic.201402027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andriani G.; Chessler A. D.; Courtemanche G.; Burleigh B. A.; Rodriguez A. Activity in vivo of anti-Trypanosoma cruzi compounds selected from a high throughput screening. PLoS Negl. Trop. Dis. 2011, 58e1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbina J. A.; Payares G.; Molina J.; Sanoja C.; Liendo A.; Lazardi K.; Piras M. M.; Piras R.; Perez N.; Wincker P.; Ryley J. F. Cure of short- and long-term experimental Chagas’ disease using D0870. Science 1996, 2735277969–971. [DOI] [PubMed] [Google Scholar]
- Liendo A.; Lazardi K.; Urbina J. A. In vitro antiproliferative effects and mechanism of action of the bis-triazole D0870 and its S(−) enantiomer against Trypanosoma cruzi. J. Antimicrob. Chemother. 1998, 412197–205. [DOI] [PubMed] [Google Scholar]
- Guengerich F. P. Cytochromes P450, drugs, and diseases. Mol. Interventions 2003, 34194–204. [DOI] [PubMed] [Google Scholar]
- Gunatilleke S. S.; Calvet C. M.; Johnston J. B.; Chen C. K.; Erenburg G.; Gut J.; Engel J. C.; Ang K. K.; Mulvaney J.; Chen S.; Arkin M. R.; McKerrow J. H.; Podust L. M. Diverse inhibitor chemotypes targeting Trypanosoma cruzi CYP51. PLoS Negl. Trop. Dis. 2012, 67e1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle P. S.; Chen C.-K.; Johnston J. B.; Hopkins S. D.; Leung S. S. F.; Jacobson M. P.; Engel J. C.; McKerrow J. H.; Podust L. M. A nonazole CYP51 inhibitor cures Chagas’ Disease in a mouse model of acute infection. Antimicrob. Agents Chemother. 2010, 5462480–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKerrow J. H.; Doyle P. S.; Engel J. C.; Podust L. M.; Robertson S. A.; Ferreira R.; Saxton T.; Arkin M.; Kerr I. D.; Brinen L. S.; Craik C. S. Two approaches to discovering and developing new drugs for Chagas disease. Mem. Inst. Oswaldo Cruz 2009, 1044263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftsson T.; Brewster M. E. Cyclodextrins as functional excipients: methods to enhance complexation efficiency. J. Pharm. Sci. 2012, 10193019–3032. [DOI] [PubMed] [Google Scholar]
- Stokes A. H.; Kemp D. C.; Faiola B.; Jordan H. L.; Merrill C. L.; Hailey J. R.; Brown R. E.; Bailey D. W. Effects of solutol (Kolliphor) and cremophor in polyethylene glycol 400 vehicle formulations in Sprague–Dawley rats and beagle dogs. Int. J. Toxicol. 2013, 323189–197. [DOI] [PubMed] [Google Scholar]
- Chen C.-K.; Leung S. S. F.; Guilbert C.; Jacobson M. P.; McKerrow J. H.; Podust L. M. Structural characterization of CYP51 from Trypanosoma cruzi and Trypanosoma brucei bound to the antifungal drugs posaconazole and fluconazole. PLoS Negl. Trop. Dis. 2010, 4, e651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Poulos T. L. Crystallization of cytochromes P450 and substrate–enzyme interactions. Curr. Top. Med. Chem. 2004, 4161789–1802. [DOI] [PubMed] [Google Scholar]
- Pochapsky T. C.; Kazanis S.; Dang M. Conformational plasticity and structure/function relationships in cytochromes P450. Antioxid. Redox Signaling 2010, 1381273–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulos T. L.; Finzel B. C.; Howard A. J. High-resolution crystal structure of cytochrome P450cam. J. Mol. Biol. 1987, 195, 687–700. [DOI] [PubMed] [Google Scholar]
- Papadopoulou M. V.; Bloomer W. D.; Rosenzweig H. S.; Ashworth R.; Wilkinson S. R.; Kaiser M.; Andriani G.; Rodriguez A. Novel 3-nitro-1H-1,2,4-triazole-based compounds as potential anti-Chagasic drugs: in vivo studies. Future Med. Chem. 2013, 5151763–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeiro M. N.; Werbovetz K.; Boykin D. W.; Wilson W. D.; Wang M. Z.; Hemphill A. Novel amidines and analogues as promising agents against intracellular parasites: a systematic review. Parasitology 2013, 1408929–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyland K. V.; Asfaw S. H.; Olson C. L.; Daniels M. D.; Engman D. M. Bioluminescent imaging of Trypanosoma cruzi infection. Int. J. Parasitol. 2008, 38121391–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A.; Tarleton R. L. Transgenic parasites accelerate drug discovery. Trends Parasitol. 2012, 28390–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques C.; Henriques-Pons A.; Meuser-Batista M.; Ribeiro A. S.; de Souza W. In vivo imaging of mice infected with bioluminescent Trypanosoma cruzi unveils novel sites of infection. Parasites Vectors 2014, 7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbina J. A.; Payares G.; Sanoja C.; Lira R.; Romanha A. J. In vitro and in vivo activities of ravuconazole on Trypanosoma cruzi, the causative agent of Chagas disease. Int. J. Antimicrob. Agents 2003, 21127–38. [DOI] [PubMed] [Google Scholar]
- Li Y.; Theuretzbacher U.; Clancy C. J.; Nguyen M. H.; Derendorf H. Pharmacokinetic/pharmacodynamic profile of posaconazole. Clin. Pharmacokinet. 2010, 496379–396. [DOI] [PubMed] [Google Scholar]
- Hargrove T. Y.; Wawrzak Z.; Alexander P. W.; Chaplin J. H.; Keenan M.; Charman S. A.; Perez C. J.; Waterman M. R.; Chatelain E.; Lepesheva G. I. Complexes of Trypanosoma cruzi sterol 14alpha-demethylase (CYP51) with two pyridine-based drug candidates for Chagas disease: structural basis for pathogen selectivity. J. Biol. Chem. 2013, 2884431602–31615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich F. P. Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 1–17. [DOI] [PubMed] [Google Scholar]
- Wester M. R.; Yano J. K.; Schoch G. A.; Yang C.; Griffin K. J.; Stout C. D.; Johnson E. F. The structure of human cytochrome P450 2C9 complexed with flurbiprofen at 2.0-A resolution. J. Biol. Chem. 2004, 2793435630–35637. [DOI] [PubMed] [Google Scholar]
- Williams P. A.; Cosme J.; Ward A.; Angove H. C.; Matak Vinkovic D.; Jhoti H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature 2003, 4246947464–468. [DOI] [PubMed] [Google Scholar]
- Rafferty S. W.; Eisner J. R.; Moore W. R.; Schotzinger R. J.; Hoekstra W. J. Highly-selective 4-(1,2,3-triazole)-based P450c17a17,20-lyase inhibitors. Bioorg. Med. Chem. Lett. 2014, 24112444–2447. [DOI] [PubMed] [Google Scholar]
- Camargo E. P. Growth and differentiation in Trypanosoma cruzi. I. Origin of metacyclic trypanosomes in liquid media. Rev. Inst. Med. Trop. Sao Paulo 1964, 6, 93–100. [PubMed] [Google Scholar]
- Engel J. C.; Ang K. K. H.; Chen S.; Arkin M. R.; McKerrow J. H.; Doyle P. S. Image-based high-throughput drug screening targeting the intracellular stage of Trypanosoma cruzi, the agent of Chagas’ Disease. Antimicrob. Agents Chemother. 2010, 5483326–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel J. C.; Doyle P. S.; Dvorak J. A. Trypanosoma cruzi: biological characterization of clones derived from chronic chagasic patients. II. Quantitative analysis of the intracellular cycle. J. Protozool. 1985, 32180–83. [DOI] [PubMed] [Google Scholar]
- Meirelles M. N.; de Araujo-Jorge T. C.; Miranda C. F.; de Souza W.; Barbosa H. S. Interaction of Trypanosoma cruzi with heart muscle cells: ultrastructural and cytochemical analysis of endocytic vacuole formation and effect upon myogenesis in vitro. Eur. J. Cell Biol. 1986, 412198–206. [PubMed] [Google Scholar]
- Porrozzi R.; Soares R.; Meuser M.; Guguen-Guillouzo C.; Meirelles M. N. Invasion and development of Trypanosoma cruzi in primary cultures of mouse embryo hepatocytes. Mem. Inst. Oswaldo Cruz 1997, 921117–120. [DOI] [PubMed] [Google Scholar]
- von Wachenfeldt C.; Richardson T. H.; Cosme J.; Johnson E. F. Microsomal P450 2C3 is expressed as a soluble dimer in Escherichia coli following modification of its N-terminus. Arch. Biochem. Biophys. 1997, 3391107–114. [DOI] [PubMed] [Google Scholar]
- Leslie A. G. W., Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 ESF-EAMCB Newslett. Protein Crystallogr. 1992, 26. [Google Scholar]
- Holton J.; Alber T. Automated protein crystal structure determination using ELVES. Proc. Natl. Acad. Sci. U. S. A. 2004, 10161537–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994, 50, 760–763.15299374 [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25131605–1612. [DOI] [PubMed] [Google Scholar]
- DeLano W. L.The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.