

Abstract
Antagonist and partial agonist modulators of the dopamine D3 receptor (D3R) have emerged as promising therapeutics for the treatment of substance abuse and neuropsychiatric disorders. However, development of druglike lead compounds with selectivity for the D3 receptor has been challenging because of the high sequence homology between the D3R and the dopamine D2 receptor (D2R). In this effort, we synthesized a series of acylaminobutylpiperazines incorporating aza-aromatic units and evaluated their binding and functional activities at the D3 and D2 receptors. Docking studies and results from evaluations against a set of chimeric and mutant receptors suggest that interactions at the extracellular end of TM7 contribute to the D3R versus D2R selectivity of these ligands. Molecular insights from this study could potentially enable rational design of potent and selective D3R ligands.
Introduction
Dysregulation of the dopaminergic system is implicated in several pathological conditions including schizophrenia, Parkinson’s disease, depression, and addiction. Dopaminergic signaling is mediated through two types of receptors termed D1-like (D1, D5) and D2-like (D2, D3, D4) receptors. Among the various approaches, targeting the dopamine D3 receptor (D3R) with antagonist or partial agonist ligands has emerged as a promising area for the development of medications for the treatment of substance abuse and neuropsychiatric disorders.1,2 The D3 dopamine receptor subtype is expressed primarily in mesolimbic regions of the brain including the nucleus accumbens and has been implicated in the pathophysiology of drug addiction.3 Studies in animal models have demonstrated that D3R activation is involved in the reinforcing and motivational effects of cocaine.4−9 Long-term exposure to cocaine results in up-regulation of D3 receptors as demonstrated in post-mortem studies of cocaine-overdose fatalities.10,11 Positron emission tomography (PET) studies have also shown an up-regulation of D3R over D2R in methamphetamine polydrug abusers.12 Preclinical studies with a number of D3R antagonist or partial agonist ligands, such as those shown in Figure 1 (1–5), have demonstrated that D3R ligands can effectively suppress motivation to self-administer drugs and prevent drug-associated cue-induced craving and relapse to drug taking.13−20
Figure 1.

Structures of dopamine D3 receptor selective ligands.
In addition, several lines of evidence indicate that D3 receptors play an important role in the pathophysiology of schizophrenia.21 Elevated levels of D3R expression in the mesolimbic regions of the brain of schizophrenic patients have been demonstrated.22 Overexpression of D3R has been proposed to be responsible for the sensitization to dopamine agonists. Inhibition of D3R function may, therefore, attenuate positive symptoms associated with schizophrenia without causing the extrapyramidal side effects associated with classical D2R antagonists. Moreover, D3R antagonists have been shown to enhance D3 receptor mediated release of acetylcholine in the frontal cortex and, therefore, may have beneficial effects on attention and memory loss associated with schizophrenia.21 Indeed, studies with D3R selective or D3R preferring antagonists have confirmed their efficacy as antipsychotic and procognitive agents.23−27
In the design and development of novel D3R ligands, a primary challenge is achieving a high degree of selectivity for D3R over the highly homologous D2R for ligands with druglike characteristics. These issues, as well as the progress made in the development of D3R selective ligands, have been the subject of several reviews.28−34 In the search for novel D3R selective ligands, compounds possessing a 4-phenylpiperazine tethered to an amide via a four-carbon linker such as that found in structures 1 and 3 have emerged as a particularly promising group of ligands. Previous structure–activity relationship (SAR) studies on this class of compounds, generically represented in Figure 2, have elucidated the importance of the length and composition of the linker, the carboxamide function, the substituent group on the piperazine (referred to as the “head group”), and the substituent group on the amide moiety (referred to as the “tail group”) in modulating the affinity and intrinsic activity of this class of compounds.35−38
Figure 2.
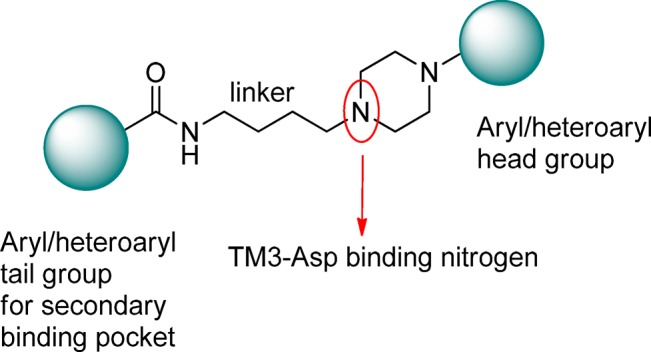
Schematic representation of the generic pharmacophore for the acylaminobutylpiperazine class of ligands.
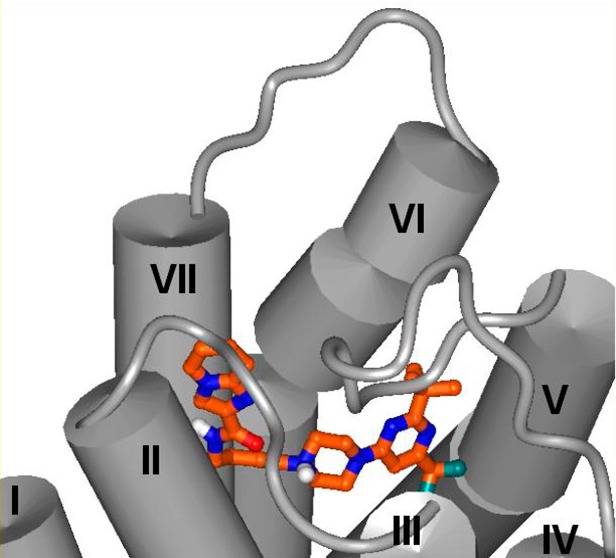
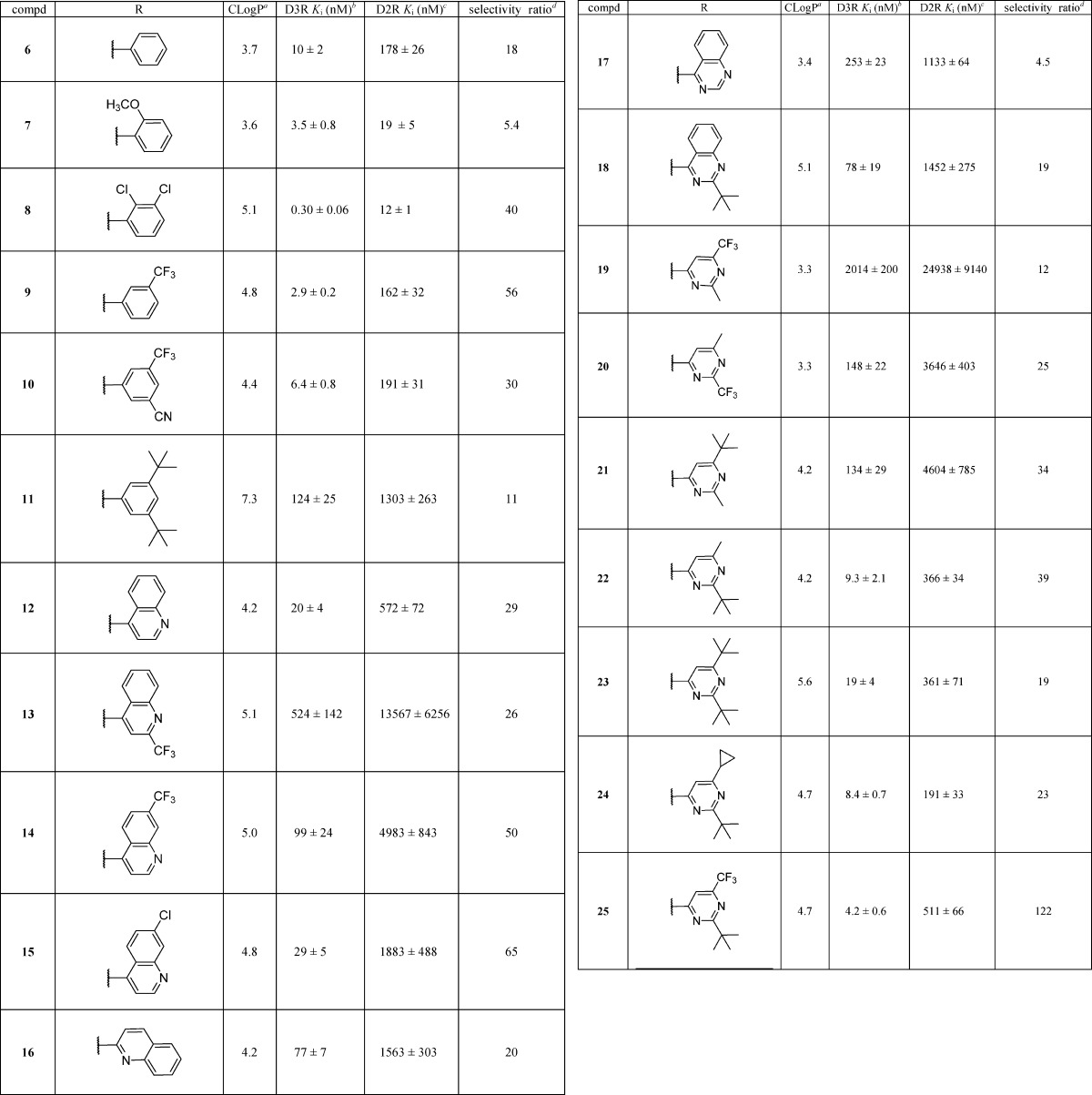
Structural comparisons of the D3R crystal structure39 and D2R homology model as well as docking studies suggest that a putative orthosteric binding site near transmembrane helices (TM) 5 and 6 and part of extracellular loop II (ELII) may contribute to D3R selectivity of the ligands that occupy this site.40,41 The head group of arylpiperazine class of ligands is accommodated in this region. Hydrophobic substituents on the head groups could potentially explore differences in the residues Val350 (D3R)/Ile365 (D2R) and Thr353 (D3R)/Ile368 (D2R) at this orthosteric binding site. Therefore, we pursued a series of ligands possessing various aryl or heteroaryl head groups with nonpolar aliphatic substituents that may explore this region (Table 1). In an effort to keep the overall lipophilicity low, in this series of ligands (6–25) we incorporated imidazo[1,2-a]pyridine as the tail group.
Table 1. Binding Affinity of Imidazo[1,2-a]pyridinecarboxamides with Head Group Variations.
Partition coefficients (CLogP) was calculated using ChemBioOffice Ultra 2010.
Displacement of [125I]IABN from HEK cell membranes stably expressing human D3R.
Displacement of [125I]IABN from HEK cell membranes stably expressing human D2LR. Ki values are the mean ± SEM from three or more independent experiments.
D2R Ki/D3R Ki.
The use of a heteroaryl functionality such as 2-indolyl-35,36,42−44 and 2-benzofuranyl16,35,42−47 as tail groups has been shown to yield ligands with selectivity for D3 versus D2 receptors. Recent studies have implicated interaction of these substituents at a secondary binding pocket (SBP) near the transmembrane helices 1, 2, and 7 and ELI and ELII as contributing to the D3R selectivity of such ligands.38,39 To gain insight into such selectivity conferring tail group interactions, we pursued another series of compounds possessing isosteric heterocyclic systems in place of imidazo[1,2-a]pyridine (26–31). Additional analogues were designed to investigate potential salt bridge/polar interactions (32–36) and hydrophobic interactions (37–39) of the tail group (Table 2). The results and insights gained from the synthesis, receptor binding, and molecular modeling studies are presented herein.
Table 2. Binding Affinity of 2-tert-Butyl-6-trifluoromethylpyrimidines with Tail Group Variations.
Partition coefficients (CLogP) was calculated using ChemBioOffice Ultra 2010.
Displacement of [125I]IABN from HEK cell membranes stably expressing human D3R.
Displacement of [125I]IABN from HEK cell membranes stably expressing human D2LR. Ki values are the mean ± SEM from three or more independent experiments.
D2R Ki/D3R Ki.
Results and Discussion
Synthesis
The target compounds 6–25 listed in Table 1 and 26–34 and 37–39 listed in Table 2 were synthesized using a general procedure involving the coupling of a carboxylic acid with the appropriate aminobutylpiperazine, as depicted in Scheme 1. The coupling of the amine with the acids was performed using either BOP-Cl or HATU as the coupling agent. The desired aminobutylpiperazine intermediates 43 were prepared from the corresponding piperazines 41 by alkylation with bromobutylphthalimide 40 followed by deprotection of the resulting intermediate 42 with hydrazine hydrate.
Scheme 1. General Synthesis of Target Compounds.

Reagents and conditions: (a) K2CO3, CH3CN, reflux; (b) N2H4·H2O, MeOH, reflux; (c) R′CO2H, BOP-Cl, Et3N, CH2Cl2, rt or R′CO2H, HATU, Et3N, CH3CN, rt.
The 5-hydroxymethyl and 5-(dimethylaminomethyl) compounds 35 and 36 were synthesized as shown in Scheme 2 starting with 2-amino-6-hydroxymethylpyridine (44).
Scheme 2. Synthesis of Target Compounds 35 and 36.

Reagents and conditions: (a) BrCH2COCO2Et, EtOH, reflux, 3 h (70%); (b) TIPSCl, imidazole, DMF, 70 °C, 2 h (72%); (c) (i) NaOH, MeOH–THF–H2O, 2 h, (ii) AcOH, 81%; (d) amine [43, R = 2-(tert-butyl)-6-(trifluoromethyl)pyrimidin-4-yl], HATU, Et3N, CH3CN, rt, 16 h (22%); (e) Et4NF, THF, rt, 16h (62%); (f) (i) MeSO2Cl, Et3N, CH2Cl2, 1 h, (ii) Me2NH, THF, rt, 6 h (23%).
Binding Affinities of Head Group Variants
The affinity of the compounds at D3 and D2 receptors were determined using previously established displacement binding assays using [125I]IABN as the radioligand and membrane preparations from HEK cells stably expressing human D3 or D2L receptors.43,48 The data for compounds containing head group variations are shown in Table 1. Compounds 6–10, possessing phenyl and substituted phenyl groups, displayed Ki values of <10 nM at D3R with D2R/D3R selectivity ratio in the range of 5.4- to 56-fold. Compared to the unsubstituted phenylpiperazine compound 6, the 2-methoxyphenylpiperazine compound 7 displayed ∼3-fold enhancement in affinity at the D3R. However, it also displayed nearly 10-fold higher affinity at D2R, thus resulting in a reduction of D2R/D3R selectivity. The highest D3R affinity (in the subnanomolar range) was displayed by compound 8, the 2,3-dichlorophenylpiperazine analogue. Earlier studies have shown that the effect of 2-methoxyphenyl and 2,3-dichlorophenyl substituents on binding affinity and selectivity varies depending upon the linker and the nature of the arylamide moiety.47,49 The improved affinity and selectivity of compound 8 compared to compound 7, however, are associated with an increase in lipophilicity (CLogP of 3.6 for 7 vs 5.1 for 8). Interestingly, the introduction of a trifluoromethyl group at the 3-position of the phenyl ring of compound 6 led to an increase in affinity at D3R without significantly affecting affinity at D2R, thus improving the binding selectivity (compound 9 D2R Ki/D3R Ki = 56). In addition, the introduction of an electron withdrawing cyano substituent at the 5-position (compound 10) led to a modest decrease in affinity at D3R and D2R as well as a decrease in D3R selectivity. The introduction of a tert-butyl group at the 3- and 5-position gave compound 11, which displayed significant reductions in affinity at both the D3R and the D2R. Incorporation of a bicyclic heterocyclic functionality, such as 4-quinolinyl, 2-quinolinyl, and 4-quinazolinyl rings, led to compounds (12–18) with lower affinities at both D3R and D2R. Whereas the unsubstituted 4-quinolinyl compound 12 and its 7-chloro analogue 15 displayed moderate affinity at D3R, the 2-trifluoromethyl and 7-trifluoromethyl analogues 13 and 14 had much lower affinity at D3R.
Further, a series of compounds were prepared in which various substituents were introduced at the 2- and 6-position of the pyrimidine ring. The isomeric methyl-trifluoromethyl pyrimidines 19 and 20 displayed low affinities at the D3R. However, the isomeric methyl-tert-butyl compounds 21 and 22 displayed interesting differences in their binding profiles at D3R and D2R. For example, the 2-methyl-6-tert-butyl isomer 21 had greatly reduced affinity at both receptors, while the 2-tert-butyl-6-methyl isomer 22 had Ki < 10 nM at D3R with nearly a 40-fold selectivity over D2R. Incorporation of a bulky tert-butyl group at the 2- and 6-position resulted in compound 23 with lower affinity (D3R Ki = 19 nM) compared to the compound 24 (D3R Ki = 8.4 nM) possessing a tert-butyl and cyclopropyl group at the 2- and 6-position, respectively. Among the pyrimidinyl variants containing a tert-butyl group at the 2-position (compounds 22–25), the most dramatic effect was observed with the 6-trifluoromethyl compound 25, which was the most potent (D3R Ki = 4.2 nM) and D3R-selective (D2R Ki/D3R Ki ratio =122) compound among the 4-pyrimidinylpiperazines. The improved affinity and selectivity of the 2-tert-butyl compound 25 as compared to the 2-methyl compound 19 highlight the importance of the tert-butyl substituent at the 2-position of the pyrimidine ring for D3R affinity and selectivity. To gain insight into the role of tert-butyl substituents in influencing binding affinity and D2R/D3R selectivity, we performed a retrospective analysis of the binding modes of compounds 19–25.
The docked poses of these compounds display a preference for two opposite orientations of the pyrimidine ring. The docked poses of compounds that exemplify these opposite orientations are shown in Figure 3 for compounds 21 and 22 and in Figure S1 (Supporting Information) for compounds 19 and 20. In the binding mode of compound 21, the nitrogen at the 3-position of the pyrimidine ring is buried facing the intracellular side which we defined as the “b” (buried) orientation as opposed to the “e” (exposed) orientation shown for compound 22. Analogously, compound 19 has “b” as opposed to “e” orientation in compound 20. The “e” orientation exposes a more polar surface of the pyrimidine ring to the aqueous environment than “b”, and it is expected to be the more favorable orientation. Binding affinity differences between the isomers 21 and 22 or between isomers 19 and 20 support this notion that the “e” orientation is the more favorable of the two. The docked pose of compound 25 (Figure 4 and Figure S2 in Supporting Information) is very similar to that obtained for compound 22 with the pyrimidine ring having the “e” orientation. In this orientation, the trifluoromethyl group forms polar interactions with Thr115 and Ser196 while the tert-butyl group participates in favorable hydrophobic interactions with Ile183 (ELII), Val189 (TM5), Val350 (TM6) in D3R corresponding to Ile184, Val190, and Ile365 in D2R. This interaction may contribute to the binding potency and D3R selectivity of compound 25 for the following reasons. (1) The D2R side chain of residue Ile365 exposed in this pocket is bulkier than the corresponding V350 residue in the D3R, contributing to a smaller available space in D2R than in the D3R. (2) Val190 of D2R participates in an interhelical interaction with Ile368. Such an interaction is absent in D3R where the residue corresponding to Ile368 is Thr353 which hydrogen-bonds to the helical backbone of TM6. This difference likely affects the interhelical packing between the extracellular ends of TM5 and TM6 and may contribute to D3R/D2R subtype selectivity of ligands.
Figure 3.
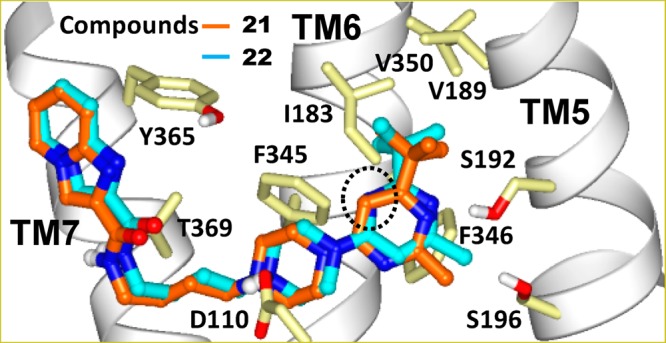
Docked poses of compounds 21 and 22 at the D3 receptor are illustrated. For clarity, only the side chain orientations of the refined D3 crystal structure used for docking are shown. Atoms are colored by atom type (C, light yellow; O, red; N, blue) except for ligand carbon atoms as shown. The region of the pyrimidine ring relevant to the “e” and “b” orientations is circled.
Figure 4.
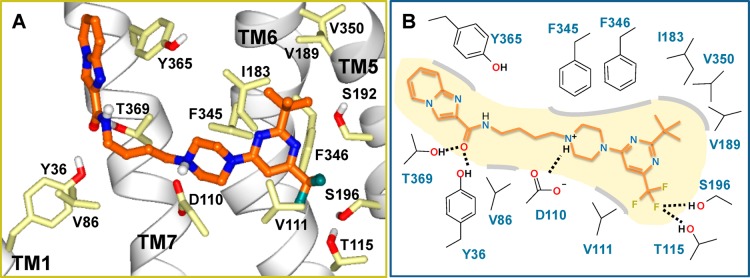
(A) Docked pose of compound 25 in the D3R binding site. Residues forming favorable interactions with the ligand are shown. Atoms are colored by atom type (C, light yellow; O, red; N, blue) except ligand carbon atoms are colored orange. TM1 and TM5–7 helices are displayed as ribbons. (B) Schematic representation of the interactions between compound 25 and D3R residues. Polar interactions are indicated with dashed lines and nonpolar/steric interactions with gray contour lines.
Binding Affinities of Tail Group Variants
The binding affinities of this series of compounds are listed in Table 2. Replacement of the imidazo[1,2-a]pyridine group in compound 25 with a 2-indolyl moiety gave compound 26 that had lower binding potency at both D3R and D2R (2.6-fold and 1.8-fold, respectively) and was less selective for D3R (122-fold vs 83-fold). The 2-benzimidazole compound 27 had D3R binding potency comparable to that of compound 25 but with reduced D3R selectivity. The incorporation of a 2-pyridothienyl group gave compound 28 with reduced binding affinity at the D3R. Introduction of an additional nitrogen atom in the imidazopyridine system gave the imidazopyrimidine (29), imidazopyrazine (30), and imidazopyridazine (31) analogues, all of which showed slightly reduced D3R binding affinity and D3R selectivity. The amino, dimethylaminomethyl, and hydroxymethyl compounds 32–36, designed to explore potential polar and salt bridge interactions by these substituents, displayed moderate binding affinity at D3R with Ki values in the range of 25–45 nM and diminished (<21-fold) D3R selectivity.
Previous studies have suggested that the occupation of a secondary binding pocket formed by transmembrane helices TM1, -2, -7 and extracellular loops ELI and ELII by tail groups of aminobutylpiperazines contributes to the D3R selectivity.38 Prior to the determination of the D3R crystal structure, Geneste and co-workers docked a ligand series related to compound 25 into a D3R model and predicted the positioning of the amide and tail groups near TM7 to form interactions with Thr368.50−52 While our docking results predicted a single preferred orientation for the head group region of compound 25 and its analogues, we found that the arylamide tail group may adopt three distinct possible orientations within the secondary pocket formed by transmembrane helices TM1, -2, -7 and extracellular loops ELI and ELII at D2R and D3R. In one of these possible orientations the tail group is in proximity to Glu90 (D3R)/Glu95 (D2R), while a second orientation places the tail group near the ELII loop residues Val180 (D3R)/Glu181 (D2R) and Ser182 (D3R)/Ile183 (D2R). In the third possible orientation the tail group forms aromatic interactions with Tyr365 (D3R)/Tyr379 (D2R) while its amide group hydrogen-bonds with Thr369 (D3R)/Thr383 (D2R). In order to gain insight as to which might be the most likely placement of the imidazo[1,2-a]pyridine tail group of compound 25 and its analogues in the secondary binding pocket, we designed the two analogues 34 and 36 containing a charged amine substituent introduced at distinct positions of the imidazo[1,2-a]pyridine, which could potentially interact with Glu90 (D3R)/Glu95 (D2R) or Glu181 (D2R). Binding affinities of these analogues and lead compound 25 were evaluated at the wild type receptors and a set of chimeric and single-point mutant receptors to test for interactions predicted by these alternative binding modes.
Binding affinities of compound 25 and the designed analogues 34 and 36 at chimeric receptors D3/D2 ELII (human D3 receptor with the D2 ELII loop), D2/D3 ELII (human D2 receptor with the D3 ELII loop) and the single point D3 receptor mutants Glu90Ala, Glu90Gln, Ser182Ile are listed in Table 3. All three compounds displayed only a moderate change in affinities between the wild type D3R and D3R-D2R ELII, suggesting the lack of significant stabilizing interactions between the positively charged tail groups of compounds 34 and 36 with Glu181 in D3/D2 ELII and, by extension, in the D2R. This is further corroborated by the observation that, compared to affinity at wild type D2R, these compounds show slight improvement in affinity at the D2/D3 ELII chimeric receptor that has a valine substitution for Glu181. Furthermore, compounds 25, 34, and 36 did not display a marked change in their binding affinities at Glu90Ala, Glu90Gln, Ser182Ile compared to the wild type D3R, indicating the lack of significantly stabilizing salt bridge or polar interactions between the positively charged amine substituent of compounds 34 and 36 with Glu90 or Ser182 of D3R.
Table 3. Affinity of Selected Compounds at Human Dopamine D2R, D3R, Chimeric, and Mutant Receptors.
|
Ki ± SEM (nM)a |
|||||||
|---|---|---|---|---|---|---|---|
| compd | D3R wild type | D2R wild type | D3R-D2R ELIIb | D2R-D3R ELIIc | D3R (S182I)d | D3R (E90A)e | D3R (E90Q)f |
| 25 | 8.4 ± 0.8 (3) | 702 ± 167 (3) | 40 ± 9 (3) | 148 ± 10 (3) | 12 ± 2 (3) | 13 ± 3 (3) | 20 ± 4 (3) |
| 34 | 9.1 ± 3.0 (4) | 191 ± 5 (3) | 27 ± 6 (3) | 72 ± 8 (3) | 16 ± 2 (3) | 16 ± 5 (4) | 19 ± 3 (3) |
| 36 | 16 ± 5 (4) | 245 ± 30 (3) | 44 ± 7 (3) | 176 ± 25 (3) | 24 ± 5 (3) | 12 ± 4 (4) | 12 ± 2 (3) |
Binding affinities derived from competition binding experiments using [125I]IABN as the radioligand. The number of independent experiments is shown in parentheses.
Chimeric D3 receptor possessing extracellular loop II of D2 receptor.
Chimeric D2 receptor possessing extracellular loop II of D3 receptor.
D3 receptor with serine 182 mutated to isoleucine.
D3 receptor with glutamate 90 mutated to alanine.
D3 receptor with glutamate 90 mutated to glutamine.
These results suggest that the side chains of Glu90 (D3)/Glu95 (D2) or Glu181 (D2)/Val180 (D3) do not significantly contribute to the binding of the arylamide tail groups of compounds 25, 34, and 36. In docked poses, the tail group orientation that is consistent with these results is shown in Figure 5. The protonated dimethylamino nitrogen in the docked poses of compounds 34 and 36 are too distant (6.4 and 8.7 Å, respectively) to engage in salt bridge interactions with the carboxylate group of Glu90 of the D3R. Binding interactions of compounds 34 and 36 include hydrogen bonding between the amide and Thr369 in TM7. The distance between the tail group’s amide NH of compounds 34 and 36 and the Thr369 side chain oxygen are 2.4 and 2.9 Å, respectively. Thr369 also hydrogen-bonds with a ring nitrogen of the imidazo[1,2-a]pyridine in compounds 34 and 36 (heavy atom distances of 3.2 and 3.1 Å, respectively). These hydrogen bonds may play a role in the positioning of the imidazopyridine ring for favorable aromatic interactions with Tyr365 in D3R (Tyr379 in D2R) in TM7.
Figure 5.
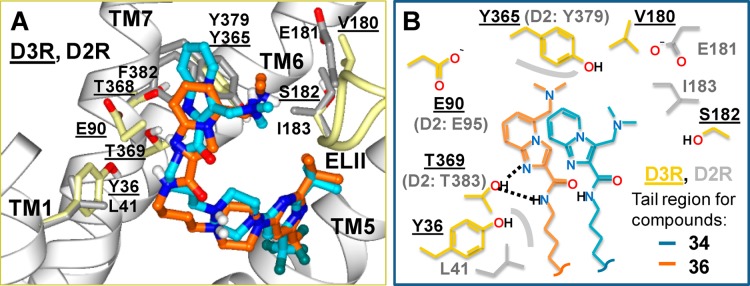
(A) Docked poses of compounds 34 and 36 at the D3R crystal structure. D3R residues are shown with light yellow colored carbons and underlined residue numbers. D2R residues with sequence differences from D3R are shown with carbons colored gray. Amino acids included in mutation experiments and discussed in text are also shown. Ligand carbon atoms are in cyan for 34 and orange for 36. Fluorine atoms are colored dark green. All other atoms are colored by atom type. (B) Schematic representation of the tail region of the poses shown in (A). Polar interactions are illustrated with dashed lines, nonpolar/steric interactions with gray contour lines.
In order to test the hypothesis that tail groups engage in aromatic interactions as predicted by our docked poses, we designed and evaluated binding affinities of compounds 37–39 (Table 2). Whereas the piperidine-4-carboxamide analogues 37 and 38 displayed only moderate affinity, the cyclohexanecarboxamide analogue 39 displayed single digit nanomolar affinity at D3R. Interestingly, its affinity at D2R was markedly diminished, thus providing a ligand with >150-fold selectivity for D3R over D2R. Docking results of compound 39 at D3R and D2R predict aromatic–aromatic interactions between the phenyl ring of the 4-phenylcyclohexanecarboxamide group of compound 39 and the Tyr365 D3R side chain (Figure 6) corresponding to Tyr379 in D2R. Sequence and structural comparison between the D3R crystal structure and D2R model suggests that interactions with this tyrosine in TM7 may contribute to D3R selectivity of ligands as follows. The nonconservative amino acid difference Thr368 (D3R)/Phe382 (D2R) within the interhelical region of TM6—TM7 likely affects interhelical packing near the extracellular end of these helices leading to structural differences between D2R and D3R. Another sequence difference at the extracellular end of TM7, at its TM1 interface, is Tyr36 in D3R corresponding to Leu41 in D2R. Furthermore, the region of the TM7—TM1 interface is rich in sequence difference amino acids. TM1 is the least conserved helix, only 65% identical between D2R, D3R sequences as compared to overall 78% identity within the seven transmembrane helical region. These differences may position the extracellular end of TM7 closer to TM1 and more distant from TM6 in the case of the D2R compared to D3R. The D3R selectivity of ligands, therefore, may arise from direct interactions with residues near the extracellular end of TM7 such as Tyr365 (D3R)/Tyr379 (D2R). Indeed, it has been suggested that subtle differences in relative positioning of TM1 and TM7 helices in D2R and D3R may contribute to selectivity even if the ligands interact with the same amino acid side chains in D2R and D3R.39 Consistent with this notion, docked poses of ligands with high D3R selectivity (e.g., 25–27 and 39) display interactions between their pendent aryl group and the tyrosine residue at the extracellular end of TM7 in both D3R and D2R. In a recent study, mutagenesis results coupled with docking and molecular dynamics simulations led to the identification of Gly94 (D3R) in the ELI loop as an important contributor to D3R/D2R selectivity of ligands through its ability to modulate the size and shape of the secondary binding pocket.53 The ELI loop in D3R contains two glycine residues (Gly93, Gly94), while this loop in D2R is one residue shorter containing one glycine residue (Gly98). Interestingly, in the docked poses of our ligands, Gly94 is in proximity to tail groups, for example, in the case of compound 25 (Figure S2 in Supporting Information), the α-carbon of Gly94 is at 3.4 Å distance from the closest heavy atom in the imidazopyridine ring of this ligand docked at D3R.
Figure 6.
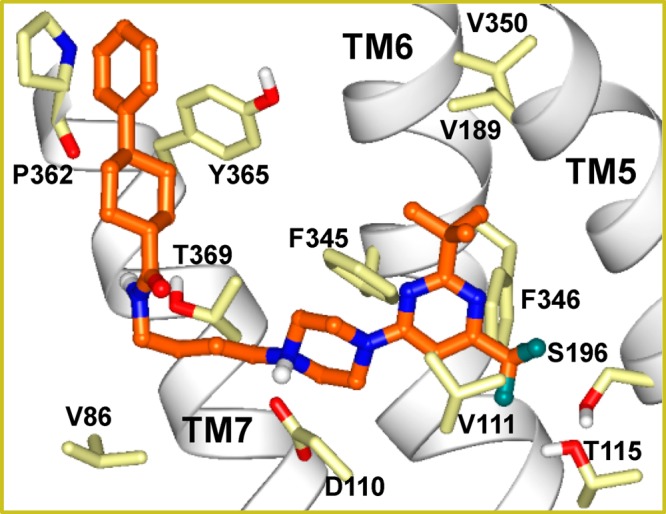
Docked pose of compound 39 in the D3R binding site. Residues forming favorable interactions with the ligand are shown. Atoms are colored by atom type. Fluorine atoms are colored dark green. Ligand carbon atoms are shown in orange.
Functional Activity
In an effort to assess the intrinsic activity profile of the compounds, functional activity evaluations were performed using assays that measure various end points such as the mitogenesis assay, the cyclase assay, β-arrestin recruitment assay, and the GTPγS binding assay. In the mitogenesis assay using CHOp cells expressing human D3 and D2 dopamine receptors, the evaluated compounds did not display appreciable intrinsic efficacy at D3 or D2 receptors (Table 4).54 The maximum stimulation of [3H]thymidine incorporation at a ligand concentration of 10 μM was <20% of the full agonist quinpirole. These compounds displayed varying antagonist potencies in inhibiting mitogenesis induced by the agonist quinpirole at the D3R and D2R. With the exception of compound 7 which displayed D3R antagonist IC50 value (3.0 nM) similar to its D3R binding Ki value (3.5 nM), the D3R antagonist potencies of all of the evaluated compounds were weaker than their binding affinities at the D3R. The D2R/D3R antagonist selectivity of the compounds in the mitogenesis assay was also much lower than the selectivity in the binding assay. The pyrimidinyl compound 25 displayed moderate antagonist potency with IC50 of 157 nM at D3R. However, it displayed 15-fold D3R antagonist selectivity due to its much weaker potency at D2R.
Table 4. Efficacy of Selected Ligands in Mitogenesis Assay.
| agonist
activity |
antagonist activity |
||||
|---|---|---|---|---|---|
| compd | D3R % stimulationa | D2R % stimulationa | D3R IC50 (nM)b | D2R IC50 (nM)b | selectivity ratioc |
| 7 | 6.4 | <20 | 3.0 ± 1.3 | 26 ± 8 | 8.7 |
| 8 | <20 | <20 | 7.2 ± 3.3 | 55 ± 16 | 7.6 |
| 12 | <20 | ndd | 120 ± 22 | ndd | nae |
| 17 | <20 | ndd | 940 ± 150 | ndd | nae |
| 25 | 0 | 0 | 157 ± 34 | 2300 ± 1100 | 15 |
| 27 | <20 | <20 | 208 ± 74 | 490 ± 150 | 2.4 |
| 29 | <20 | <1 | 51 ± 21 | 205 ± 48 | 4.0 |
| 30 | <20 | <20 | 245 ± 33 | 420 ± 120 | 1.7 |
| 31 | <1 | <1 | 637 ± 99 | 1160 ± 110 | 1.8 |
Percent stimulation at 10 μM normalized to the maximal stimulation by quinpirole. Results are from two independent experiments.
Inhibition potency against stimulation of mitogenesis by standard agonist quinpirole (30 nM). IC50 values are mean ± SEM from at least three independent experiments, each conducted with duplicate determinations.
(D2R IC50)/(D3R IC50).
Not determined because of weak binding affinity at D2R (Ki > 500 nM).
Not applicable.
Selected compounds were evaluated in the adenylyl cyclase assay which measures the ability of the compounds to inhibit forskolin-dependent stimulation of adenylyl cyclase activity.43,48 The evaluations were carried out using HEK 293 cells expressing human D3 and D2 dopamine receptors (Table 5).
Table 5. Efficacy of Selected Ligands in Adenylyl Cyclase Assay.
| compd | D3R % inhibitiona | D2R % inhibitiona |
|---|---|---|
| 7 | 44 ± 6 | 25 ± 5 |
| 8 | 66 ± 13 | 45 ± 1 |
| 9 | 70 ± 4 | 32 ± 4 |
| 12 | 50 ± 6 | 1.4 ± 6.7 |
| 17 | 70 ± 18 | 19 ± 6 |
| 25 | 28 ± 7 | –1.9 ± 13 |
| 26 | 38 ± 4 | ndb |
| 27 | 12 ± 11 | 5.6 ± 4.1 |
| 29 | 51 ± 10 | 9.2 ± 7.3 |
| 30 | 38 ± 5 | ndb |
| 31 | 32 ± 7 | ndb |
| 34 | 47 ± 4 | 8.9 ± 5.0 |
| 39 | 52 ± 13 | ndb |
Percent inhibition values were normalized to the percent inhibition of the full agonist quinpirole at D3R (100 nM) and D2R (1 μM). For D3 receptors the maximum inhibition by quinpirole ranged from 38% to 53%, and for D2 receptors the maximum inhibition was >90%. The test compounds were used at a concentration equal to approximately 10 times the Ki value in the radioligand binding assays. The values are the mean ± SEM from three or more independent experiments.
Not determined because of insolubility of the compounds at 10 times their binding Ki values at D2R.
In the adenylyl cyclase inhibition assay, all of the evaluated compounds displayed a partial agonist profile at D3R with intrinsic efficacies ranging from 12% (compound 27) to 70% (compounds 9 and 17). Interestingly, among the compounds possessing identical arylamide groups (7–9, 12, 17, and 25), compound 25, which has a 2-tert-butyl-6-trifluoromethylpyrimidinyl head group, displayed lower efficacy at the D3R (28%) than compounds possessing substituted phenyl, 4-quinolinyl, and 4-quinazolinyl head groups. Among the compounds possessing a 2-tert-butyl-6-trifluoromethylpyrimidinyl head group (25–27, 29–31, 34, and 39), the benzimidazole-2-carboxamide 27 displayed the lowest efficacy at the D3R (12%).
Binding of agonists at D3R and D2R is known to lead to the recruitment of β-arrestin to the receptor. Selected compounds were evaluated for their activity on β-arrestin-2 recruitment using a cell-based receptor/β-arrestin interaction assay (DiscoveRx PathHunter).55 In this assay, the interaction of β-arrestin with a GPCR is monitored using β-galactosidase (β-gal) enzyme fragment complementation. Activation of the receptor by an agonist results in the translocation of β-arrestin to active receptor, which leads to the formation of an active β-galactosidase enzyme. The activity of β-galactosidase is then measured by addition of chemiluminescent detection reagents. These assays were performed according to manufacturer’s protocol using CHO-K1 cells expressing human D2L receptors and U2OS cells expressing human D3 receptors. Intrinsic agonist or inverse agonist activity of the compounds was determined by evaluating the ability of the compounds to stimulate or inhibit basal activity. In these evaluations, all of the tested compounds (Table 6) were found to be devoid of agonist activity and displayed very weak inverse agonist activity at both D3R and D2R. The maximum inhibition of the basal activity observed was <21% (compound 33) at D3R and <30% (compound 34) at D2R at 500 nM concentration of the test compounds. The antagonist potency of the compounds was assessed by determining the ability of these compounds to inhibit the agonist activity of ligands PD128907 and pergolide at D3R and D2R, respectively. Haloperidol was included as a standard antagonist ligand. The results are presented in Table 6.
Table 6. Antagonist Potencies of Selected Ligands in the β-Arrestin-2 Recruitment Assay.
| compd | D3R IC50 ± SEM, nMa | D2R IC50 ± SEM, nMb | selectivity ratioc |
|---|---|---|---|
| 7 | 0.7 ± 0.1 | 6.6 ± 5.1 | 9.4 |
| 8 | 0.2 ± 0.1 | 91 ± 7 | 455 |
| 9 | 1.8 ± 0.8 | 60 ± 12 | 33 |
| 12 | 52 ± 7 | 161 ± 78 | 3.1 |
| 17 | 409 ± 25 | 248 ± 63 | 0.6 |
| 25 | 0.6 ± 0.1 | 261 ± 54 | 435 |
| 26 | 5.6 ± 1.8 | 418 ± 80 | 75 |
| 27 | 8.5 ± 2.1 | 301 ± 50 | 35 |
| 29 | 0.4 ± 0.2 | 37 ± 17 | 93 |
| 30 | 0.3 ± 0.1 | 49 ± 12 | 163 |
| 31 | 0.3 ± 0.1 | 22 ± 15 | 73 |
| 33 | 14 ± 3 | 311 ± 53 | 22 |
| 34 | 7.2 ± 3.7 | 217 ± 10 | 30 |
| 36 | 29 ± 3 | 281 ± 43 | 9.7 |
| 39 | 6.8 ± 1.8 | >500 | >73 |
| haloperidol | 0.06 ± 0.02 | 0.16 ± 0.02 | 2.7 |
IC50 values for inhibition of (+)-PD128907-induced arrestin translocation in U2OS cells expressing D3R.
IC50 values for inhibition of pergolide-induced arrestin translocation in CHO-K1 cells expressing D2LR.
(D2R IC50)/(D3R IC50).
Most of the evaluated compounds displayed potent (<1.0 nM) to moderately potent (>1.0 nM) antagonist activity in inhibiting agonist stimulated recruitment of β-arrestin-2 by D3 receptors. Compounds that displayed subnanomolar antagonist potency at D3R include 7, 8, 25, 29–31. The antagonist potency of the compounds at the D3R, in general, correlates with their binding affinities (Pearson’s correlation coefficient between binding pKi and antagonist pIC50, r = 0.77; P = 0.008). Similar to their binding affinity at D2R, the antagonist potency of these ligands at D2R was weaker than at D3R. The binding affinity and antagonist potency at D2R, however, was not as well correlated, as they were at D3R. In this assay, compounds 8 and 25, both possessing the imidazo[1,2-a]pyridine tail group, with the 2,3-dichlorophenyl (8) or 2-tert-butyl-6-trifluoromethyl-4-pyrimidinyl (25) head group emerged as high potency ligands with very high (>400-fold) D3R selectivity. A comparison of the profile of compound 25 with compound 26 indicates that incorporation of the imidazo[1,2-a]pyridine-2-carboxamide group (25) provides a more favorable D3R binding and D3R antagonist functional selectivity than the indole-2-carboxamide group (26).
On the basis of the relatively high D3 over D2 antagonist selectivity in the β-arrestin-2 recruitment assay, compounds 8, 25, 30, and 39 were selected for evaluation in the [35S]GTPγS binding functional assays using CHO cell lines expressing human D3R and D2R.56−58 These compounds did not stimulate [35S]GTPγS binding, and up to 1 μM concentration these compounds caused less than 30% inhibition of basal binding at D2R and D3R. Compounds 25, 30, and 39 at 10 μM caused inhibition of 40–60% of D3R basal [35S]GTPγS binding, indicating some inverse agonist activity which did not appear pharmacologically relevant compared with their binding affinity assessed with [125I]IABN. The potencies of these compounds as antagonists were then determined using the “shift” experiments59 to obtain antagonist Ke values based on their ability to reduce the apparent potency of dopamine in stimulating [35S]GTPγS binding in CHO-D2R and CHO-D3R cells (Table 7). For compound 39, the antagonist potency at D2R could not be determined in shift experiments, as drug concentrations substantially higher than 1 μM were required to shift the dopamine curve; such high concentrations had inverse agonist activity invalidating the shift method.
Table 7. Antagonist Potencies of Selected Compounds in [35S]GTPγS Binding Assay.
| compd | CHO-hD3 Ke (nM)a | CHO-hD2 Ke (nM)a | selectivity ratiob |
|---|---|---|---|
| 8 | 0.0038 ± 0.0013 | 1.1 ± 0.2 | 289 |
| 25 | 1.3 ± 0.4 | 111 ± 25 | 85 |
| 30 | 0.33 ± 0.05 | 25 ± 6 | 76 |
| 39 | 19 ± 4 | nac | nac |
The antagonist Ke value was calculated from “shift” experiments as described in Experimental Section. Dopamine dose–response curves were generated in the absence or presence of compound 8 (10 nM for D2R; 0.1 nM for D3R), compound 25 or 39 (1 μM for D2R; 100 nM for D3R), or compound 30 (50 nM for D2R; 1 nM for D3R). The fixed concentrations of drug were chosen as being high enough to shift the dopamine stimulation curves to the right but low enough to not appreciably inhibit [35S]GTPγS binding below baseline based on the inverse agonist activity. The Ke was calculated from the increase in ED50 observed with test compound (see Experimental Section). Results are the mean ± SEM for three to four independent experiments assayed in triplicate.
(CHO-hD2 Ke)/(CHO-hD3 Ke).
Measurement could not be made with shift protocol: Concentrations of drug that by themselves did not appreciably inhibit binding below baseline did not substantially shift the dopamine curve to the right.
Among the compounds possessing the 2-tert-butyl-6-trifluoromethylpyrimidinylpiperazine head group (compounds 25, 30, and 39), compounds 25 and 39 displayed D3R antagonist potency somewhat similar to their binding potency. The antagonist potency of imidazopyrazine compound 30 was moderately higher at D3R and D2R compared to its binding affinities at these receptors. Interestingly, compound 8 which contains the 2,3-dichlorphenylpiperazinyl head group displayed extraordinary potency at D3R in the picomolar range.
Increasing evidence indicates that GPCR ligands including D3R and D2R ligands can display differing intrinsic efficacy and/or potency for different signaling pathways linked to the same receptor.48,60−65 These types of effects have been referred to by various terms which include stimulus trafficking, functional selectivity, collateral efficacy, and biased agonism. The manifestation of functional selectivity in our series of compounds is exemplified by the functional activity profile of compounds 8, 25, and 30. At D3R, these compounds behave as antagonists in the [35S]GTPγS binding, β-arrestin-2 recruitment, and mitogenesis assays but function as partial agonists in the cyclase assay.
Among the compounds studied, compound 25 displayed favorable D3R binding affinity (Ki < 5 nM) and D2R/D3R binding selectivity (>100-fold) with antagonist–partial agonist functional activity profile and was chosen for pharmacological evaluations in vivo. This compound, coded as SR 21502, was evaluated in rats and was found to produce significant decreases in cocaine reward, cocaine seeking, preference for cocaine-associated environments, and cocaine induced locomotor activity at doses that had no effect on food reward or spontaneous locomotor activity, indicating that it selectively inhibits cocaine’s rewarding and stimulant effects.66,67
Summary and Conclusions
In an effort to identify dopamine D3 receptor ligands with selectivity for D3R versus D2R, we designed a series of ligands based on the acylaminobutylarylpiperazine pharmacophore incorporating, primarily, aza-aromatic systems on the acyl and piperazine moieties. Docking of these ligands at the binding sites of the human D3R crystal structure and our D2R homology model provide insights into molecular features contributing to binding affinity and binding selectivity of this panel of ligands. Among the ligands possessing a 4-pyrimidinylpiperazine group, the affinity differences between isomeric compounds at the D3 receptor could be traced to a favorable orientation of the pyrimidine ring nitrogen positioned toward the solvent-exposed extracellular side of the receptor with simultaneous occupation of a small pocket near helices TM5 and TM6 formed by Val189, Ile183, and Val350 by a hydrophobic substituent from the 2-position of the pyrimidine ring. The results from binding experiments with chimeric and mutant receptors support the hypothesis that D3 receptor selectivity over D2 receptor conferred by tail groups such as the imidazo[1,2-a]pyridine arises through interaction with the tyrosine residue (D3R Tyr365, D2R Tyr379) at the extracellular end of TM7, which is a region with significant structural differences between D2 and D3 receptors. These structural insights provide a basis for the rational design of future ligands with improved binding affinity and selectivity for the dopamine D3 receptors.
Results from functional assays characterized the investigated class of compounds as antagonists or partial agonists with varying potencies depending upon the functional end point of the assay. From this series, compound 25 was selected as a lead compound and evaluated in rat models of cocaine self-administration and conditioned place preference. Results from these studies showed that compound 25 was effective at significantly attenuating cocaine self-administration, cocaine reward, cue-induced reinstatement of cocaine-seeking and blocking cocaine-induced place preference.66,67 Further studies on the development of dopamine D3R selective ligands are in progress and will be the subject of future publications.
Experimental Section
General Methods
Melting points were determined in open capillary tubes with a Mel-Temp melting point apparatus and are uncorrected. 1H NMR spectra were recorded on a Nicolet 300NB spectrometer operating at 300.635 MHz. Chemical shifts are expressed in parts per million downfield from tetramethylsilane. Spectral assignments were supported by proton decoupling. Mass spectra were recorded on a Varian MAT 311A double-focusing mass spectrometer in the fast atom bombardment (FAB) mode or on a Bruker BIOTOF II in electrospray ionization (ESI) mode. Elemental analyses were performed by Atlantic Microlab, Inc. (Atlanta, GA) or by the Spectroscopic and Analytical Laboratory of Southern Research Institute. Analytical results indicated by elemental symbols were within ±0.4% of the theoretical values. Thin layer chromatography (TLC) was performed on Analtech silica gel GF 0.25 mm plates. Flash column chromatography was performed with E. Merck silica gel 60 (230–400 mesh). Yields are of purified compounds and were not optimized. On the basis of NMR and combustion analysis data, all final compounds reported in the manuscript are >95% pure.
General Amidation Procedures
Procedure A
To a solution of the carboxylic acid (1 equiv, 1 mmol) in anhydrous CH2Cl2 (10 mL) was added BOP-Cl (1 equiv), and the mixture was stirred at room temperature for 2.5 h under nitrogen atmosphere. Triethylamine (3 equiv) and the appropriate amine (1 equiv) were added, and the mixture was stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure, and the residue was partitioned between CHCl3 and water. The organic layer was separated, dried over anhydrous sodium sulfate, filtered, and the filtrate was evaporated under reduced pressure. The residue obtained was purified by column chromatography over silica gel to obtain the desired product.
Procedure B
To a solution of the carboxylic acid (1 equiv, 1 mmol) in acetonitrile (12 mL) was added HATU (1 equiv), and the mixture was stirred at room temperature for 15 min. Triethylamine (3 equiv) and the appropriate amine (1 equiv) were added, and the mixture was stirred at room temperature overnight. The mixture was concentrated under reduced pressure, and the residue obtained was partitioned between CHCl3 and saturated aqueous sodium bicarbonate. The organic layer was separated, dried over anhydrous sodium sulfate, filtered, and the solvent was removed under reduced pressure. The crude product thus obtained was purified by chromatography over a column of silica gel to obtain the desired product.
N-(4-(4-Phenylpiperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (6)
To a solution of imidazo[1,2-a]pyridine-2-carboxylic acid (0.162 g, 1.0 mmol) in anhydrous CH2Cl2 (10 mL) was added 0.254 g (1.0 mmol) of BOP-Cl, and the mixture was stirred at room temperature for 2.5 h under nitrogen atmosphere. Triethylamine (0.42 mL, 3.0 mmol) and 4-(4-phenylpiperazin-1-yl)butan-1-amine (0.233 g, 1.0 mmol) were added, and the mixture was stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure, and the residue was partitioned between CHCl3 and water. The organic layer was separated, dried over anhydrous sodium sulfate, filtered, and the filtrate was evaporated under reduced pressure. The residue obtained was purified by column chromatography over silica gel (CHCl3–MeOH, 95:5) to obtain 0.164 g (44%) of the desired product 6 as a colorless solid. Mp 169–171 °C. TLC Rf = 0.39 (CHCl3–MeOH, 92.5:7.5). 1H NMR (DMSO-d6) δ 1.47–1.63 (m, 4H), 2.34 (t, 2H) 2.44–2.52 (m, 4H), 3.11 (t, 4H), 3.22–3.35 (m, 2H), 6.76 (td, 1H), 6.89 (dd, J = 8.6, 9.0 Hz, 2H), 6.97 (td, 1H), 7.19 (td, 2H), 7.32 (td, 1H), 7.55 (dd, J = 9.0, 9.0 Hz, 1H), 8.31–8.39 (m, 2H), 8.56 (dt, 1H). ESI MS m/z 378 (M + H)+. Anal. (C22H27N5O) C, H, N.
N-(4-(4-(2-Methoxyphenyl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (7)
Imidazo[1,2-a]pyridine-2-carboxylic acid (0.243 g, 1.5 mmol) was reacted with 4-(4-(2-methoxyphenyl)piperazin-1-yl)butan-1-amine (0.40 g, 1.5 mmol) in the presence of BOP-Cl (0.42 g, 1.65 mmol) and triethylamine (3.0 mL, 21.6 mmol) in CH2Cl2 (15 mL) according to general procedure A. The crude product was purified by column chromatography over silica gel (EtOAc–MeOH, 10:1) to obtain compound 7 (0.141 g, 23%) as a colorless solid. Mp 169–171 °C. TLC Rf = 0.49 (CHCl3–MeOH, 95:5). 1H NMR (DMSO-d6) δ 1.42–1.58 (m, 4H), 2.31 (t, J = 6.58 Hz, 2H), 2.49 (t, 4H), 2.94 (s, 4H), 3.27–3.32 (m, 2H), 3.75 (s, 3H), 6.80–6.99 (m, 5H), 7.28–7.38 (m, 1H), 7.56 (d, 1H), 8.34–8.42 (m, 2H), 8.56–8.60 (m, 1H). ESI MS m/z 408 (M + H)+. Anal. (C23H29N5O2·H2O) C, H, N.
N-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (8)
Imidazo[1,2-a]pyridine-2-carboxylic acid (0.649 g, 4.0 mmol) was reacted with 4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-1-amine (1.21 g, 4.0 mmol) in the presence of BOP-Cl (1.02 g, 4.0 mmol) and triethylamine (2.0 mL, 14.4 mmol) in CH2Cl2 (30 mL) according to general procedure A. The crude product was purified by column chromatography over silica gel (EtOAc–MeOH, 7:1) to obtain compound 8 (0.268 g, 15%) as a colorless solid. Mp 122–124 °C. TLC Rf = 0.12 (CHCl3–MeOH, 95:5). 1H NMR (DMSO-d6) δ 1.39–1.57 (m, 4H), 2.24 (t, J = 6.6 Hz, 2H), 2.44 (bs, 4H), 2.98 (s, 4H), 3.21 (m, 2H), 6.95 (t, 1H), 7.09–7.16 (m, 1H), 7.27–7.37 (m, 3H), 7.56 (dd, J = 9.12, 9.23 Hz, 1H), 8.34 (d, J = 0.66 Hz, 1H), 8.37 (t, 1H), 8.56 (m, 1H). ESI MS m/z 446 (M + H)+. Anal. (C22H25Cl2N5O·0.25H2O) C, H, N.
N-(4-(4-(3-(Trifluoromethyl)phenyl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (9)
To a solution of imidazo[1,2-a]pyridine-2-carboxylic acid (0.08 g, 0.5 mmol) in acetonitrile (6 mL) was added HATU (0. 190 g, 0.5 mmol), and the mixture was stirred at room temperature for 15 min. Triethylamine (0.21 mL, 1.5 mmol) and 4-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)butan-1-amine (0.15 g, 0.5 mmol) were then added, and the mixture was stirred at room temperature overnight. The mixture was concentrated under reduced pressure, and the residue obtained was partitioned between CHCl3 and saturated aqueous sodium bicarbonate. The organic layer was separated, dried over anhydrous sodium sulfate, filtered, and the solvent was removed under reduced pressure. The crude product thus obtained was purified by chromatography over a column of silica gel (CHCl3–MeOH, 96:4) to obtain 0.065 g (29%) of the desired product 9 as a colorless solid. Mp 144–146 °C. TLC Rf = 0.33 (CHCl3–MeOH, 92.5:7.5). 1H NMR (DMSO-d6) δ 1.45–1.62 (m, 4H), 2.35 (t, 2H), 2.47–2.51 (m, 4H), 3.22 (t, 4H), 3.27–3.34 (m, 2H), 6.96–6.99 (m, 1H), 7.04 (d, J = 7.9 Hz, 1H), 7.14 (bs, 1H), 7.19 (dd, J = 8.2, 8.6 Hz, 1H), 7.31–7.34 (m, 1H), 7.37–7.42 (m, 1H), 7.56 (dd, J = 9.0, 9.0 Hz, 1H), 8.30–8.40 (m, 2H), 8.57 (dt, 1H). ESI MS m/z 446 (M + H)+. Anal. (C23H26F3N5O·0.25H2O) C, H, N.
N-(4-(4-(3-Cyano-5-(trifluoromethyl)phenyl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (10)
This compound was prepared from imidazo[1,2-a]pyridine-2-carboxylic acid and 3-(4-(4-aminobutyl)piperazin-1-yl)-5-(trifluoromethyl)benzonitrile according to general procedure A. The crude product was purified by column chromatography over silica gel (CHCl3–MeOH, 97:3) to obtain compound 10 as a colorless solid in 29% yield. Mp 124–126 °C. TLC Rf = 0.40 (CHCl3–MeOH, 92.5:5). 1H NMR (DMSO-d6) δ 1.45–1.66 (m, 4H), 2.35 (t, 2H), 2.44–2.56 (m, 4H), 3.18–3.35 (m, 6H), 6.93–7.02 (m, 1H), 7.31–7.32 (m, 1H), 7.46 (s, 1H), 7.50 (s, 1H), 7.55–7.62 (m, 1H), 7.64 (s, 1H), 8.34 (s, 1H), 8.43 (t, 1H), 8.54–8.62 (m, 1H). ESI MS m/z 471 (M + H)+. Anal. (C24H25F3N6O·0.25H2O) C, H, N.
N-(4-(4-(3,5-Di-tert-butylphenyl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (11)
This compound was prepared from imidazo[1,2-a]pyridine-2-carboxylic acid and 4-(4-(3,5-di-tert-butylphenyl)piperazin-1-yl)butan-1-amine according to general procedure B. The crude product was purified by column chromatography over silica gel (CHCl3–MeOH, 96:4) to obtain compound 11 as a colorless solid in 28% yield. TLC Rf = 0.37 (CHCl3–MeOH, 95:5). 1H NMR (DMSO-d6) δ 1.25 (s, 18H), 1.45–1.64 (bs, 4H), 2.32–2.72 (bs, 6H), 3.10–3.22 (bs, 4H), 3.28–3.38 (m, 2H), 6.72 (d, J = 1.5 Hz, 2H), 6.86 (t, 1H), 6.97 (td, 1H), 7.32 (td, 1H), 7.55 (dt, 1H), 8.34 (d, J = 0.8 Hz, 1H), 8.39 (t, 1H), 8.52 (dt, 1H). ESI MS m/z 490 (M + H)+. Anal. (C30H43N5O·H2O) C, H, N.
N-(4-(4-(Quinolin-4-yl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (12)
This compound was prepared from imidazo[1,2-a]pyridine-2-carboxylic acid (0.648 g, 4.0 mmol) and 4-(4-(quinolin-4-yl)piperazin-1-yl)butan-1-amine (1.14 g, 4.0 mmol) in the presence of BOP-Cl (1.38 g, 5.4 mmol) and triethylamine (1.67 mL 12.0 mmol) in CH2Cl2 (20 mL) according to general procedure A. The crude product was purified by column chromatography over silica gel (CHCl3–MeOH, 95:5) to obtain 0.19 g (11%) of compound 12 as a colorless solid. Mp 126–128 °C. TLC Rf = 0.09 (CHCl3–MeOH, 95:5). 1H NMR (DMSO-d6) δ 1.46–1.65 (m, 4H), 2.38–2.47 (m, 2H), 2.65 (bs, 4H), 3.18 (bs, 4H), 3.26–3.39 (m, 2H), 6.95 (d, J = 5.27 Hz, 1H), 6.99 (d, J = 1.99 Hz, 1H), 7.28–7.39 (m, 1H), 7.50–7.60 (m, 2H), 7.62–7.72 (m, 1H), 7.92 (dd, J = 8.35, 8.34 Hz, 1H), 7.99 (d, J = 7.42 Hz, 1H), 8.35 (s, 1H), 8.35 (t, J = 5.93 Hz, 1H), 8.57 (d, J = 6.92 Hz, 1H), 8.66 (d, J = 4.94 Hz, 1H). ESI MS m/z 429 (M + H)+. Anal. (C25H28 N6O·0.25H2O) C, H, N.
N-(4-(4-(2-(Trifluoromethyl)quinolin-4-yl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (13)
This compound was prepared from imidazo[1,2-a]pyridine-2-carboxylic acid and 4-(4-(2-(trifluoromethyl)quinolin-4-yl)piperazin-1-yl)butan-1-amine according to general procedure A. The crude product was purified by column chromatography over silica gel (CHCl3–MeOH, 97:3) to afford compound 13 as a colorless solid in 16% yield. Mp 122–124 °C. TLC Rf = 0.56 (CHCl3–MeOH, 90:10). 1H NMR (DMSO-d6) δ 1.51–1.68 (m, 4H), 2.44 (t, 2H), 2.67 (bs, 4H), 3.26–3.44 (m, 6H), 6.97 (ddd, J = 1.2, 1.2, 1.0 Hz, 1H), 7.24 (s, 1H), 7.31–7.65 (m, 1H), 7.56 (dd, J = 9.2, 9.2 Hz, 1H), 7.68–7.74 (m, 1H), 7.78–7.88 (m, 1H), 8.03–8.12 (m, 2H), 8.35 (s, 1H), 8.40 (t, 1H), 8.56–8.60 (m, 1H). ESI MS m/z 497 (M + H)+. Anal. (C26H27F3N6O·H2O) C, H, N.
N-(4-(4-(7-(Trifluoromethyl)quinolin-4-yl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (14)
This compound was prepared from imidazo[1,2-a]pyridine-2-carboxylic acid and 4-(4-(7-(trifluoromethyl)quinolin-4-yl)piperazin-1-yl)butan-1-amine according to general procedure A. The crude product was purified by column chromatography over silica gel (CHCl3–MeOH, 97:3) to obtain compound 14 as an off-white solid in 34% yield. Mp 55–58 °C. TLC Rf = 0.35 (CHCl3–MeOH, 92.5:7.5). 1H NMR (DMSO-d6) δ 1.61–1.73 (bs, 2H), 1.87–1.94 (bs, 2H), 3.18–3.30 (bs, 2H), 3.33–3.48 (m, 2H), 3.55–4.38 (m, 8H), 7.29 (s, 1H), 7.44 (d, J = 6.4 Hz, 1H), 7.74 (bs, 2H), 7.95 (d, J = 8.6 Hz, 1H), 8.41 (d, J = 8.8 Hz, 1H), 8.58 (s, 1H), 8.74 (bs, 1H), 8.88 (bs, 1H), 8.96 (d, J = 6.1 Hz, 1H), 9.16 (bs, 1H). ESI MS m/z 497 (M + H)+. Anal. (C26H27F3N6O·H2O) C, H, N.
N-(4-(4-(7-Chloroquinolin-4-yl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (15)
This compound was prepared from imidazo[1,2-a]pyridine-2-carboxylic acid and 4-(4-(7-chloroquinolin-4-yl)piperazin-1-yl)butan-1-amine according to general procedure A. The crude product was purified by column chromatography over silica gel (EtOAc–MeOH, 80:20) to afford compound 15 as an off-white solid in 28% yield. Mp 155–157 °C. TLC Rf = 0.67 (CHCl3–MeOH, 85:15). 1H NMR (DMSO-d6) δ 1.47–1.65 (m, 4H), 2.42 (t, 2H), 2.62–2.72 (bs, 4H), 3.14–3.23 (bs, 2H), 3.27–3.46 (m, 4H), 6.96 (d, J = 3.1 Hz, 1H), 6.98–7.18 (m, 1H), 7.28–7.34 (m, 1H), 7.50–7.56 (m, 2H), 7.96 (d, J = 2.2 Hz, 1H), 8.00 (d, J = 9.0 Hz, 1H), 8.34 (s, 1H), 8.38 (t, 1H), 8.58–8.62 (m, 1H), 8.69 (d, J = 4.9 Hz, 1H). ESI MS m/z 463 (M + H)+. Anal. (C25H27ClN6O·0.25H2O) C, H, N.
N-(4-(4-(Quinolin-2-yl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (16)
This compound was prepared from imidazo[1,2-a]pyridine-2-carboxylic acid and 4-(4-(quinolin-2-yl)piperazin-1-yl)butan-1-amine according to general procedure A. The crude product was purified by column chromatography over silica gel (EtOAc–MeOH, 92.5:7.5) to obtain compound 16 as a colorless solid in 33% yield. Mp 137–139 °C. TLC Rf = 0.63 (CHCl3–MeOH, 92.5:7.5). 1H NMR (DMSO-d6) δ 1.47–1.63 (m, 4H), 2.35 (bs, 2H), 2.44–2.58 (bs, 4H), 3.31–3.49 (m, 2H), 3.68 (bs 4H), 6.97 (dt, 1H), 7.17–7.25 (m, 2H), 7.31–7.36 (m, 1H), 7.52 (dt, 1H), 7.55–7.78 (m, 2H), 7.68 (d, J = 7.9 Hz, 1H), 8.02 (d, J = 9.2 Hz, 1H), 8.34 (s, 1H), 8.38 (t, 1H), 8.56 (dd, J = 6.8, 6.9 Hz, 1H). ESI MS m/z 429 (M + H)+. Anal. (C25H28N6O·0.25H2O) C, H, N.
N-(4-(4-(Quinazolin-4-yl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (17)
This compound was prepared from imidazo[1,2-a]pyridine-2-carboxylic acid (0.65 g, 4.0 mmol) and 4-(4-(quinazolin-4-yl)piperazin-1-yl)butan-1-amine (1.136 g, 4.0 mmol) in the presence of BOP-Cl (1.38 g, 5.42 mmol) and triethylamine (1.68 mL, 12 mmol) in CH2Cl2 (20 mL) according to general procedure A. The crude product was purified by column chromatography over silica gel (EtOAc–MeOH, 8:1) to obtain 0.12 g (7%) of compound 17 as a pale yellow solid. Mp 130–134 °C. TLC Rf = 0.10 (CHCl3–MeOH, 95:5). 1H NMR (DMSO-d6) δ 1.66–1.78 (m, 4H), 2.45–2.54 (m, 2H), 2.64–2.74 (m, 4H), 3.42–3.55 (m, 2H), 3.82–3.94 (m, 4H), 6.91–7.00 (m, 1H), 7.32–7.40 (m, 1H), 7.50–7.61 (m, 2H), 7.76–7.86 (m, 2H), 7.97–8.05 (m, 2H), 8.27 (d, J = 0.76 Hz, 1H), 8.43–8.50 (m, 1H), 8.56 (s, 1H). ESI MS m/z 430 (M + H)+. Anal. (C24H27N7O) C, H, N.
N-(4-(4-(2-(tert-Butyl)quinazolin-4-yl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (18)
This compound was prepared from imidazo[1,2-a]pyridine-2-carboxylic acid and 4-(4-(2-(tert-butyl)quinazolin-4-yl)piperazin-1-yl)butan-1-amine according to general procedure A. The crude product was purified by chromatography over a column of silica gel using CHCl3–MeOH, 92.5:7.5, as the eluent to obtain compound 18 as a pale yellow solid in 18% yield. Mp 59–61 °C. TLC Rf = 0.44 (CHCl3–MeOH, 92.5:7.5). 1H NMR (DMSO-d6) δ 1.50 (s, 9H), 1.52–1.68 (m, 2H), 1.77–1.88 (m, 2H), 3.17 (bs, 2H), 3.24–3.41 (m, 4H), 3.64 (d, J = 11.1 Hz, 2H), 4.11 (bs, 2H), 4.85 (bs, 2H), 7.32 (t, 1H), 7.64–7.82 (m, 3H), 8.04 (t, 1H), 8.20 (d, J = 8.4 Hz, 1H), 8.45 (d, J = 8.2 Hz, 1H), 8.75 (s, 1H), 8.86 (d, J = 6.8 Hz, 1H), 9.10 (s, 1H). ESI MS m/z 486 (M + H)+. Anal. (C28H35N7O·0.25H2O) C, H, N.
N-(4-(4-(2-Methyl-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (19)
This compound was prepared using general procedure B by reacting imidazo[1,2-a]pyridine-2-carboxylic acid (0.081 g, 0.5 mmol) with 4-(4-(2-methyl-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butan-1-amine (0.159 g, 0.5 mmol) in the presence of HATU (0.19 g, 0.5 mmol) and triethylamine (0.13 mL, 0.75 mmol) in acetonitrile (7.0 mL). The crude product was purified by column chromatography over silica gel (CHCl3–MeOH, 96:4) to obtain 0.096 g (42%) of compound 19 as a crystalline white solid. Mp 156–158 °C. TLC Rf = 0.67 (CHCl3–MeOH, 90:10). 1H NMR (DMSO-d6) δ 1.44–1.61 (m, 4H), 2.34 (t, 2H), 2.42 (s, 3H), 2.44–2.50 (m, 4H), 3.25–3.34 (m, 2H), 3.71 (bs, 4H), 6.95–6.99 (m, 1H), 7.05 (s, 1H), 7.31–7.36 (m, 1H), 7.56 (d, J = 9.0 Hz, 1H), 8.31 (s, 1H), 8.37 (t, 1H), 8.57 (d, J = 6.6 Hz, 1H). ESI MS m/z 462 (M + H)+. Anal. (C22H26 F3N7O·0.25H2O) C, H, N.
N-(4-(4-(6-Methyl-2-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (20)
This compound was prepared from imidazo[1,2-a]pyridine-2-carboxylic acid (0.102 g, 0.63 mmol) and 4-(4-(6-methyl-2-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butan-1-amine (0.20 g, 0.63 mmol) in the presence of HATU (0.24 g, 0.63 mmol) and triethylamine (0.26 mL, 0.189 mmol) in acetonitrile (7.0 mL) according to general procedure B. The crude product was purified by column chromatography over silica gel (CHCl3–MeOH, 95:5) to obtain 0.076 g (26%) of compound 20 as a colorless solid. Mp 118–120 °C. TLC Rf = 0.60 (CHCl3–MeOH, 90:10). 1H NMR (DMSO-d6) δ 1.43–1.61 (m, 4H), 2.33 (t, 2H), 2.37 (s, 3H), 2.42 (t, 4H), 3.26–3.34 (m, 2H), 3.65 (bs, 4H), 6.96 (s, 1H), 6.97–6.99 (m, 1H), 7.31–7.36 (m, 1H), 7.56 (m, 1H), 8.31 (d, J = 0.8 Hz, 1H), 8.37 (t, 1H), 8.55–8.59 (m, 1H). ESI MS m/z 462 (M + H)+. Anal. (C22H26 F3N7O·0.25H2O) C, H, N.
N-(4-(4-(6-(tert-Butyl)-2-methylpyrimidin-4-yl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (21)
This compound was prepared from imidazo[1,2-a]pyridine-2-carboxylic acid and 4-(4-(6-(tert-butyl)-2-methylpyrimidin-4-yl)piperazin-1-yl)butan-1-amine (0.305 g, 1.0 mmol) according to general procedure B. The crude product was purified by column chromatography over silica gel (CHCl3–MeOH, 96:4) to obtain compound 21 as a colorless viscous oil in 35% yield. TLC Rf = 0.32 (CHCl3–MeOH, 95:5). 1H NMR (DMSO-d6) δ 1.21 (s, 9H), 1.45–1.64 (m, 4H), 2.22–2.35 (m, 2H), 2.34 (s, 3H), 2.46 (t, 4H), 3.28–3.34 (m, 2H), 3.58 (t, 4H), 6.47 (s, 1H), 6.96 (td, 1H), 7.33 (td, 1H), 7.58 (dd, J = 8.6, 8.2 Hz, 1H), 8.37 (t, 2H), 8.56 (dt, 1H). ESI MS m/z 450 (M + H)+. Anal. (C25H35N7O·75H2O) C, H, N.
N-(4-(4-(2-(tert-Butyl)-6-methylpyrimidin-4-yl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (22)
This compound was prepared from imidazo[1,2-a]pyridine-2-carboxylic acid and 4-(4-(2-(tert-butyl)-6-methylpyrimidin-4-yl)piperazin-1-yl)butan-1-amine according to general procedure B. The crude product was purified by column chromatography over silica gel (CHCl3–MeOH, 96:4) to obtain compound 22 as a light brown viscous oil in 20% yield. TLC Rf = 0.17 (CHCl3–MeOH, 95:5). 1H NMR (DMSO-d6) δ 1.25 (s, 9H), 1.44–1.60 (m, 4H), 2.23 (s, 3H), 2.23–2.46 (m, 6H), 3.24–3.44 (m, 2H), 3.58 (bs, 4H), 6.47 (s, 1H), 6.97 (td, 1H), 7.37 (td, 1H), 7.56 (dd, J = 9.4, 9.0 Hz, 1H), 8.34 (s, 1H), 8.37 (t, 1H), 8.56 (dt, 1H). ESI MS m/z 450 (M + H)+. Anal. (C25H35N7O·H2O) C, H, N.
N-(4-(4-(2,6-Di-tert-butylpyrimidin-4-yl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (23)
This compound was prepared from imidazo[1,2-a]pyridine-2-carboxylic acid and 4-(4-(2,6-di-tert-butylpyrimidin-4-yl)piperazin-1-yl)butan-1-amine according to general procedure B. The crude product was purified by column chromatography over silica gel (CHCl3–MeOH, 92:8) to obtain compound 23 as an off-white solid in 30% yield. Mp 90–94 °C. TLC Rf = 0.54 (CHCl3–MeOH, 95:5). 1H NMR (DMSO-d6) δ 1.24 (s, 9H), 1.27 (s, 9H), 1.45–1.60 (m, 4H), 2.33 (t, 2H), 2.41 (t, 4H), 3.28–3.34 (m, 2H), 3.60 (t, 4H), 6.45 (s, 1H), 6.96 (td, 1H), 7.33 (td, 1H), 7.58 (dd, J = 9.4, 9.0 Hz, 1H), 8.34 (d, J = 0.8 Hz, 1H), 8.38 (t, 1H) 8.57 (dt, 1H). ESI MS m/z 492 (M + H)+. Anal. (C28H41N7O·0.75H2O) C, H, N.
N-(4-(4-(2-(tert-Butyl)-6-cyclopropylpyrimidin-4-yl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (24)
This compound was prepared from imidazo[1,2-a]pyridine-2-carboxylic acid and 4-(4-(2-(tert-butyl)-6-cyclopropylpyrimidin-4-yl)piperazin-1-yl)butan-1-amine according to general procedure B. The crude product was purified by column chromatography over silica gel (CHCl3–MeOH, 92:8) to obtain compound 24 as a colorless solid in 36% yield. Mp 34–37 °C. TLC Rf = 0.19 (CHCl3–MeOH, 95:5). 1H NMR (DMSO-d6) δ 0.81–0.96 (2m, 4H), 1.28 (s, 9H), 1.45–1.60 (m, 4H), 1.84–1.90 (m, 1H), 2.32 (t, 2H), 2.40 (t, 4H), 3.27–3.33 (m, 2H), 3.58 (t, 4H), 6.49 (s, 1H), 6.97 (td, 1H), 7.32 (td, 1H), 7.56 (dd, J = 9.4, 9.0 Hz, 1H), 8.37 (t, 2H), 8.56 (dt, 1H). ESI MS m/z 476 (M + H)+. Anal. (C27H37N7O·H2O) C, H, N.
N-(4-(4-(2-(tert-Butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butyl)imidazo[1,2-a]pyridine-2-carboxamide (25)
Imidazo[1,2-a]pyridine-2-carboxylic acid (5.89 g, 36.6 mmol) was reacted with 4-(4-(2-(tert-butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butan-1-amine (13.14 g, 36.6 mmol) in the presence of BOP-Cl (9.3 g, 36.6 mmol) and triethylamine (15.0 mL) in CH2Cl2 (350 mL) as described in general procedure A. The crude product was purified by column chromatography over silica gel (EtOAc–hexane, 2:1) to yield 6.0 g (32%) of the desired product. A solution of the free base in ether was treated with 1.0 M solution of HCl in Et2O to obtain the dihydrochloride salt as a light brown solid. Mp 253–255 °C. TLC Rf = 0.29 (CHCl3–MeOH, 90:10). 1H NMR (DMSO-d6) δ 1.31 (s, 9H), 1.62–1.69 (m, 2H), 1.80–1.88 (m, 2H), 2.88–3.19 (broad hump, 1H), 3.10–3.17 (m, 2H), 3.37–3.33 (q, 2H), 3.52–3.60 (m, 4H), 4.32–4.86 (b, 2H), 7.11 (s, 1H), 7.13–7.18 (m, 1H), 7.50–7.57 (t, 1H), 7.65 (dd, J = 9.2, 9.0 Hz, 1H), 8.53 (s, 1H), 8.57 (bs, 1H), 8.68 (d, J = 6.9 Hz, 1H), 11.0–11.8 (b, 1H). ESI MS m/z 504 (M + H)+. Anal. (C25H32F3N7O·2HCl·0.25H2O) Calcd: C, 51.68; H, 5.98; N, 16.87.Cl, 12.20. Found: C, 51.64; H, 6.00; N, 16.89, Cl, 11.97.
N-(4-(4-(2-(tert-Butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butyl)-1H-indole-2-carboxamide (26)
This compound was prepared from indole-2-carboxylic acid and 4-(4-(2-(tert-butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butan-1-amine according to general procedure B. The crude product was purified by column chromatography over silica gel (CHCl3–MeOH, 92:8) to obtain compound 26 as a colorless solid in 35% yield. Mp 70–72 °C. TLC Rf = 0.54 (CHCl3–MeOH, 92.5:7.5). 1H NMR (DMSO-d6) δ 1.28 (s, 9H), 1.43–1.63 (m, 4H), 2.35 (t, 2H), 2.42 (t, 4H), 3.26–3.34 (m, 2H), 3.71 (bs, 4H), 7.00–7.02 (m, 2H), 7.16–7.19 (m, 2H), 7.41 (d, J = 8.2 Hz, 1H), 7.59 (d, J = 8.3 Hz, 1H), 8.44 (t, 1H), 7.78 (t, 1H). ESI MS m/z 503 (M + H)+. Anal. (C26H33 F3N6O·0.25H2O) C, H, N.
N-(4-(4-(2-(tert-Butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butyl)-1H-benzo[d]imidazole-2-carboxamide (27)
This compound was prepared from benzimidazole-2-carboxylic acid and 4-(4-(2-(tert-butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butan-1-amine according to general procedure A. The crude product was purified by column chromatography over silica gel (CHCl3–MeOH, 92:8) to obtain compound 27 as a colorless solid in 18% yield. Mp 80–72 °C. TLC Rf = 0.56 (CHCl3–MeOH, 90:10). 1H NMR (DMSO-d6) δ 1.28 (s, 9H), 1.44–1.66 (m, 4H), 2.36 (t, 2H), 2.43 (t, 4H), 3.28–3.48 (m, 2H), 3.64–3.68 (m, 4H), 7.03 (s, 1H), 7.22–7.36 (m, 2H), 7.52 (d, J = 2.0 Hz, 1H), 7.70 (d, J = 7.5 Hz, 1H), 8.97 (t, 1H), 13.12 (s, 1H). ESI MS m/z 504 (M + H)+. Anal. (C25H32F3N7O) C, H, N.
N-(4-(4-(2-(tert-Butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butyl)thieno[2,3-b]pyridine-2-carboxamide (28)
This compound was prepared from thieno[2,3-b]pyridine-2-carboxylic acid (0.10 g, 0.56 mmol) and 4-(4-(2-(tert-butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butan-1-amine (0.20 g, 0.56 mmol) in the presence of HATU (0.211 g, 0.56 mmol) and triethylamine (0.23 mL, 1.68 mmol) in acetonitrile (7.0 mL) according to general procedure B. The crude product was purified by column chromatography over silica gel (CHCl3–MeOH, 92:8) to yield 0.125 g (43%) of compound 28 as a colorless solid. Mp 122–124 °C. TLC Rf = 0.49 (CHCl3–MeOH, 92.5:7.5). 1H NMR (DMSO-d6) δ 1.28 (s, 9H), 1.44–1.62 (m, 4H), 2.34 (t, 2H), 2.45 (t, 4H), 3.33 (q, 2H), 3.71 (bs, 4H), 7.03 (s, 1H), 7.49 (q, 1H), 8.08 (s, 1H), 8.40 (dd, J = 1.6, 1.6 Hz, 1H), 8.65 (dd, J = 1.5, 1.5 Hz, 1H), 8.84 (t, 1H). ESI MS m/z 521 (M + H)+. Anal. (C25H31F3N6OS·H2O) C, H, N.
N-(4-(4-(2-(tert-Butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butyl)imidazo[1,2-a]pyrimidine-2-carboxamide (29)
This compound was prepared from imidazo[1,2-a]pyrimidine-2-carboxylic acid (0.50 g, 3.07 mmol) and 4-(4-(2-(tert-butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butan-1-amine (1.105 g, 3.07 mmol) in the presence of BOP-Cl (0.783 g, 3.08 mmol) and triethylamine (1.25 mL, 9.0 mmol) in CH2Cl2 (15 mL) according to general procedure A. The crude product was purified by column chromatography over silica gel (EtOAc–MeOH, 6:1) to obtain 0.776 g (50%) of compound 29 as a colorless solid. Mp 136–138 °C. TLC Rf = 0.47 (CHCl3–MeOH, 95:5). 1H NMR (DMSO-d6) δ 1.80 (s, 9H), 1.37–1.57 (m, 4H), 2.24 (t, J = 5.96 Hz, 2H), 2.44–2.48 (m, 4H), 2.98–3.06 (m, 2H), 3.21–3.26 (m, 2H), 3.62 (bs, 2H), 7.03 (s, 1H), 7.15 (dd, J = 6.92, 6.91 Hz, 1H), 8.29 (s, 1H), 8.57 (t, 1H), 8.63 (dd, J = 4.05 and 4.18 Hz, 1H), 8.98 (dd, J = 6.91, 6.81 Hz, 1H). ESI MS m/z 505 (M + H)+. Anal. (C24H31F3N8O·H2O) C, H, N.
N-(4-(4-(2-(tert-Butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butyl)imidazo[1,2-a]pyrazine-2-carboxamide (30)
This compound was prepared from imidazo[1,2-a]pyrazine-2-carboxylic acid (0.50 g, 3.07 mmol) and 4-(4-(2-(tert-butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butan-1-amine (0.75 g, 2.09 mmol) in the presence of BOP-Cl (1.1 g, 4.32 mmol) and triethylamine (3.0 mL, 21.52 mmol) in CH2Cl2 (20 mL) according to general procedure A. The crude product was purified by column chromatography over silica gel (CHCl3–MeOH, 92:8) to obtain 0.412 g (39%) of compound 30 as an off-white solid. Mp 200–202 °C. TLC Rf = 0.25 (CHCl3–MeOH, 95:5). 1H NMR (DMSO-d6) δ 1.28 (s, 9H), 1.47–1.67 (m, 4H), 2.32 (t, J = 5.96 Hz, 2H), 2.44–2.48 (m, 4H), 3.21–3.36 (m, 2H), 3.71 (bs, 4H), 7.04 (s, 1H), 7.95 (d, J = 4.66 Hz, 1H), 8.50 (s, 1H), 8.61–8.66 (m, 2H), 9.12 (d, J = 0.77 Hz, 1H). ESI MS m/z 505 (M + H)+. Anal. (C24H31F3N8O·2HCl·0.75H2O) C, H, N.
N-(4-(4-(2-(tert-Butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butyl)-6-chloroimidazo[1,2-b]pyridazine-2-carboxamide (31)
This compound was prepared from 6-chloroimidazo[1,2-b]pyridazine-2-carboxylic acid (0.591 g, 3.0 mmol) and 4-(4-(2-(tert-butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butan-1-amine (1.078 g, 3.0 mmol) in the presence of BOP-Cl (0.764 g, 3.0 mmol) and triethylamine (1.25 mL, 9.0 mmol) in CH2Cl2 (20 mL) using general procedure A. The crude product was purified by column chromatography over silica gel (EtOAc–MeOH, 10:1) to obtain 0.242 g (15%) of compound 31 as a colorless solid. Mp 119–121 °C. TLC Rf = 0.59 (CHCl3–MeOH, 95:5). 1H NMR (DMSO-d6) δ 1.27 (s, 9H), 1.43–1.62 (m, 4H), 2.29 (t, J = 4.63 Hz, 2H), 2.45 (t, J = 1.81 Hz, 4H), 3.34 (bs, 2H), 3.62 (bs, 4H), 7.03 (s, 1H), 7.46 (d, J = 9.56 Hz, 1H), 8.23 (dd, J = 9.61, 9.56 Hz, 1H), 8.54 (t, J = 5.96 Hz, 1H), 8.67 (d, J = 0.65 Hz, 1H). ESI MS m/z 539 (M + H)+. Anal. (C24H30ClF3N8O·0.5H2O) C, H, N.
3-Amino-N-(4-(4-(2-(tert-butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butyl)thieno[2,3-b]pyridine-2-carboxamide (32)
This compound was prepared from 3-aminothieno[2,3-b]pyridine-2-carboxylic acid (0.109 g, 0.56 mmol) and 4-(4-(2-(tert-butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butan-1-amine (0.20 g, 0.56 mmol) in the presence of HATU (0.213 g, 0.56 mmol) and triethylamine (0.23 mL, 1.68 mmol) in acetonitrile (10 mL) according to general procedure B. The crude product was purified by column chromatography over silica gel (CHCl3–MeOH, 96:4) to obtain 0.09 g (30%) of compound 32 as an off-white solid. Mp 138–140 °C. TLC Rf = 0.46 (CHCl3–MeOH, 92.5:7.5). 1H NMR (DMSO-d6) δ 1.28 (s, 9H), 1.42–1.62 (m, 4H), 2.33 (t, 2H), 2.43 (t, 4H), 3.24 (q, 2H), 3.71 (s, 4H), 7.03 (s, 1H), 7.14 (s, 2H), 7.44 (q, 1H), 7.71 (t, 1H), 8.4 (dd, J = 1.6, 1.6 Hz, 1H), 8.61 (dd, J = 1.5, 1.5 Hz, 1H). ESI MS m/z 536 (M + H)+. Anal. (C25H32F3N7OS·0.25H2O) C, H, N.
N-(4-(4-(2-(tert-Butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butyl)-3-((dimethylamino)methyl)-1H-indole-2-carboxamide (33)
Step 1
To an ice cold solution of dimethylamine (15 mL, 2 M solution in THF, 30.0 mmol) was added acetic acid (3.65 mL) followed by 2.20 mL of 37% aqueous formaldehyde solution (30.0 mmol). Ethyl indole-2-carboxylate (3.0 g, 15.8 mmol) in methanol (150 mL) was then added, and the resulting solution was heated under reflux for 4 h. The mixture was concentrated to 20% of its volume in vacuo and diluted with water (50 mL). The aqueous solution was washed with CHCl3 (2 × 50 mL). The aqueous layer was separated, chilled, and basified to pH 12 by addition of 20% aqueous NaOH. The mixture was extracted with CH2Cl2 (3 × 50 mL). The organic extract was dried with sodium sulfate and the solvent was removed under reduced pressure to obtain ethyl 3-((dimethylamino)methyl)-1H-indole-2-carboxylate. Yield 2.8 g (72%). 1H NMR (DMSO-d6) δ 1.34–1.38 (m, 3H), 2.16 (s, 6H), 3.89 (s, 2H), 4.31–4.37 (m, 2H), 7.05–7.42 (m, 4H), 11.59 (s, 1H). ESI MS m/z 247 (M + H)+.
Step 2
The above ester (1.0 g, 4.06 mmol) was dissolved in 20 mL of dioxane–water (90:10), treated with NaOH (0.8 g, 20.0 mmol), and the mixture was stirred at room temperature for 4 h. The reaction mixture was diluted with EtOAc (75 mL), and the organic layer was separated, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to obtain 3-((dimethylamino)methyl)-1H-indole-2-carboxylic acid. Yield (0.58 g, 65%). ESI MS m/z 219 (M + H)+ The acid thus obtained was used in the next step without further purification.
Step 3
The acid (0.091 g, 0.42 mmol) obtained above was coupled with 4-(4-(2-(tert-butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butan-1-amine (0.151 g, 0.42 mmol) in the presence of HATU (0.16 g, 0.42 mmol) and triethylamine (0.18 mL, 1.26 mmol) in acetonitrile (10 mL) according to general procedure B. The crude product was purified by column chromatography over silica gel (CHCl3–MeOH, 97:3) to obtain 0.10 g (43%) of compound 33 as an off-white solid. Mp 174–176 °C. TLC Rf = 0.43 (CHCl3–MeOH, 92.5:7.5). 1H NMR (DMSO-d6) δ 1.28 (s, 9H), 1.56–1.66 (m, 4H), 2.23 (s, 6H), 2.37 (t, 2H), 2.44 (t, 4H), 3.28–3.38 (m, 2H), 3.70 (bs, 4H), 7.03 (s, 1H), 7.04–7.08 (m, 2H), 7.17 (td, 1H), 7.40 (d, 1H, J = 8.2 Hz), 7.65 (d, J = 8.2 Hz, 2H), 10.40 (s, 1H), 11.58 (s, 1H). ESI MS m/z 560 (M + H)+. Anal. (C29H40F3N7O) C, H, N.
N-(4-(4-(2-(tert-Butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butyl)-3-((dimethylamino)methyl)imidazo[1,2-a]pyridine-2-carboxamide (34)
This compound was prepared from 3-((dimethylamino)methyl)imidazo[1,2-a]pyridine-2-carboxylic acid dihydrochloride (0.164 g, 0.56 mmol) and 4-(4-(2-(tert-butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butan-1-amine (0.20 g, 0.56 mmol) in the presence of HATU (0.213 g, 0.56 mmol) and triethylamine (0.235 mL, 1.68 mmol) in acetonitrile (7.0 mL) according to general procedure B. The crude product was purified by column chromatography over silica gel (CHCl3–MeOH, 98:2) to obtain 0.119 g (38%) of compound 34 as a colorless oil. TLC Rf = 0.38 (CHCl3–MeOH, 95:5). 1H NMR (DMSO-d6) δ 1.28 (s, 9H), 1.41–1.64 (m, 4H), 2.16 (s, 6H), 2.34 (t, 2H), 2.43 (t, 4H), 3.22–3.36 (m, 2H), 3.72 (bs, 4H), 4.17 (s, 2H), 6.92–7.05 (m, 2H), 7.38 (t, 1H), 7.57 (d, J = 9.0 Hz, 1H), 8.38 (t, 1H), 8.44 (d, J = 6.7 Hz, 1H). ESI MS m/z 561 (M + H)+. Anal. (C28H39F3N8O·0.5H2O) C, H, N.
N-(4-(4-(2-(tert-Butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butyl)-5-(hydroxymethyl)imidazo[1,2-a]pyridine-2-carboxamide (35)
Step 1
A solution of 2-amino-6-hydroxymethylpyridine (5.0 g, 40.28 mmol) and ethyl 3-bromopyruvate (1.57 g, 80.56 mmol) in ethanol (150 mL) was stirred with heating under reflux for 3 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue obtained was purified by chromatography over a column of silica gel using hexane–EtOAc (75:25) to yield 6.2 g (70%) of ethyl 5-(hydroxymethyl)imidazo[1,2-a]pyridine-2-carboxylate (45). ESI MS m/z 221 (M + H)+.
Step 2
To a stirred solution of the above compound (3.89 g, 17.66 mmol) and imidazole (3.5 g, 51.4 mmol) in DMF (12 mL) was added chlorotriisopropylsilane (6.82 g, 35.4 mmol) dropwise, and the reaction mixture was heated at 70 °C for 2 h. After cooling to room temperature, the reaction mixture was diluted with EtOAc (200 mL) and washed with saturated aqueous sodium carbonate (150 mL). The organic phase was washed with brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The residue thus obtained was purified by silica gel column using CH2Cl2–EtOAc (1:1) as the eluent to obtain 4.8 g (72%) of ethyl 5-(((triisopropylsilyl)oxy)methyl)imidazo[1,2-a]pyridine-2-carboxylate (46). ESI MS m/z 377 (M + H)+.
Step 3
To a solution of the above silyl ether 4.8 g (12.7 mmol) in 50 mL of methanol/THF/water (2:2:1) was added NaOH (2.1 g). The mixture was stirred at room temperature for 2 h. The mixture was neturalized by the addition of acetic acid, and the solvents were removed under reduced pressure. The residue obtained was purified over a column of silica using CH2Cl2–MeOH (7:1) to give 3.6 g (81%) of 5-(((triisopropylsilyl)oxy)methyl)imidazo[1,2-a]pyridine-2-carboxylic acid (47). ESI MS m/z 349 (M + H)+.
Step 4
To a solution of the above acid (1.02 g, 2.92 mmol) in acetonitrile (15 mL) was added HATU (1.11 g, 2.92 mmol) and Et3N (0.6 mL, 4.31 mmol). The mixture was stirred for 10 min, and 4-(4-(2-(tert-butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butan-1-amine (1.05 g, 2.92 mmol) was added. The stirring was continued for 16 h at room temperature. The reaction mixture was concentrated under reduced pressure, and the residue was dissolved in CHCl3 and washed with aqueous sodium bicarbonate. The CHCl3 extract was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The crude product thus obtained was purified by chromatography over a column of silica gel (CHCl3–MeOH 97:3) to obtain 0.45 g (22%) of N-(4-(4-(2-(tert-butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butyl)-5-(((triisopropylsilyl)oxy)methyl)imidazo[1,2-a]pyridine-2-carboxamide (48).
Step 5
To a solution of the above compound 0.45 g (0.65 mmol) in THF (20 mL) was added tetraethylammonium fluoride (0.146 g, 0.98 mmol), and the mixture was stirred at room temperature for 16 h. The mixture was then concentrated and partitioned between CHCl3 and water. The organic layer was separated, dried over anhydrous sodium sulfate, filtered, and the solvent was removed under reduced pressure. The residue thus obtained was purified by chromatography over a column of silica gel (CHCl3–MeOH, 92:8) to obtain 0.216 g (62%) of the desired product as a colorless oil. TLC Rf = 0.46 (CHCl3–MeOH, 92.5:7.5). 1H NMR (DMSO-d6) δ 1.28 (s, 9H), 1.41–1.60 (m, 4H), 2.34 (t, 2H), 2.44 (t, 4H), 3.01–3.28 (m, 2H), 3.70 (bs, 4H), 4.78 (d, J = 7.8 Hz, 2H), 5.71–5.78 (m, 1H), 6.94 (dd, J = 6.6, 6.6 Hz, 1H), 7.03 (s, 1H), 7.30–7.40 (m, 1H), 7.53 (d, J = 9.0 Hz, 1H), 8.28 (s, 1H), 8.40 (t, 1H). ESI MS m/z 534 (M + H)+. Anal. (C26H34F3N7O2·75H2O) C, H, N.
N-(4-(4-(2-(tert-Butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butyl)-5-((dimethylamino)methyl)imidazo[1,2-a]pyridine-2-carboxamide (36)
A solution of hydroxymethyl compound 35 (0.125 g, 0.235 mmol) and triethylamine (0.1 mL, 0.702 mmol) in dichloromethane (5 mL) was cooled in ice bath and treated dropwise with methanesulfonyl chloride (0.15 g, 1.3 mmol). The mixture was allowed to warm to room temperature and stirred at room temperature for 1 h. To the mixture was then added dimethylamine (0.25 mL, 1 M solution in THF), and the mixture was stirred at room temperature for 6 h. Volatiles were removed under reduced pressure, and the residue was partitioned between CHCl3 and saturated aqueous sodium bicarbonate. The organic layers were separated and dried over anhydrous sodium sulfate. Filtration, removal of the solvent under reduced pressure, and purification by silica gel column chromatography (CHCl3–MeOH 97:3) afforded 0.03 g (23%) of the desired product 36 as a colorless solid. Mp 62–64 °C. TLC Rf = 0.64 (CHCl3–MeOH, 92.5:7.5). 1H NMR (DMSO-d6) δ 1.28 (s, 9H), 1.44–1.61 (m, 4H), 2.21 (s, 6H), 2.34 (t, 2H), 2.43 (t, 4H), 3.09–3.35 (m, 2H), 3.72 (bs, 4H), 3.76 (s, 2H), 6.93 (d, J = 6.7 Hz, 1H), 7.03 (s, 1H), 7.26–7.34 (m, 1H), 7.55 (d, J = 9.0 Hz, 1H), 8.31 (s, 1H), 8.39 (t, 1H). ESI MS m/z 561 (M + H)+. Anal. (C28H39F3N8O·H2O) C, H, N.
N-(4-(4-(2-(tert-Butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butyl)-1-phenylpiperidine-4-carboxamide (37)
This compound was prepared from 1-phenylpiperidine-4-carboxylic acid and 4-(4-(2-(tert-butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butan-1-amine according to general procedure A. The crude product was purified by column chromatography over silica gel (EtOAc–MeOH, 10:1) to obtain compound 37 as a colorless solid in 26% yield. Mp 178–180 °C. TLC Rf = 0.30 (CHCl3–MeOH, 92.5:7.5). 1H NMR (DMSO-d6) δ 1.28 (s, 9H), 1.42–1.46 (m, 4H), 1.58–1.77 (m, 4H), 2.22–2.35 (m, 3H), 2.42 (t, 4H), 2.61–2.69 (m, 2H), 3.06 (dd, J = 11.7, 11.7 Hz, 2H), 3.66–3.72 (m, 6H), 6.71–6.77 (m, 1H), 6.91–6.94 (m, 2H), 7.04 (s, 1H), 7.16–7.22 (m, 2H), 7.79 (t, 1H). ESI MS m/z 547 (M + H)+. Anal. (C29H41F3N6O·25H2O) C, H, N.
1-Benzyl-N-(4-(4-(2-(tert-butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butyl)piperidine-4-carboxamide (38)
This compound was prepared from 1-benzylpiperidine-4-carboxylic acid and 4-(4-(2-(tert-butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butan-1-amine according to general procedure A. The crude product was purified by column chromatography over silica gel (EtOAc–MeOH, 95:5) to obtain compound 38 as a pale yellow solid in 41% yield. Mp 36–37 °C. TLC Rf = 0.49 (CHCl3–MeOH, 92.5:7.5). 1H NMR (DMSO-d6) δ 1.28 (s, 9H), 1.36–1.47 (m, 4H), 1.54–1.68 (m, 2H), 1.84–1.93 (m, 2H), 2.07–2.11 (m, 2H), 2.29 (t, 2H), 2.40 (t, 2H), 2.78 (d, J = 11.6 Hz, 4H), 3.02 (dd, J = 11.8, 11.6 Hz, 2H), 3.42 (s, 2H), 3.60–3.88 (bs, 4H), 7.03 (s, 1H), 7.22–7.33 (m, 5H), 7.69 (t, 1H). ESI MS m/z 561 (M + H)+. Anal. (C30H43F3N6O·H2O) C, H, N.
trans-N-(4-(4-(2-(tert-Butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butyl)-4-phenylcyclohexanecarboxamide (39)
This compound was prepared from trans-4-phenylcyclohexanecarboxylic acid and 4-(4-(2-(tert-butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butan-1-amine (0.359 g, 1.0 mmol) according to general procedure A. The crude product was purified by column chromatography over silica gel (EtOAc–MeOH, 80:20) to obtain compound 39 as a colorless solid in 30% yield. Mp 137–139 °C. TLC Rf = 0.69 (CHCl3–MeOH, 92.5:7.5). 1H NMR (DMSO-d6) δ 1.28 (s, 9H), 1.37–1.55 (m, 8H), 1.78–1.86 (m, 4H), 2.11–2.19 (m, 1H), 2.31 (t, 2H), 2.42 (t, 4H), 2.49–2.51 (m, 1H), 3.06 (dd, J = 11.9, 11.7 Hz, 2H), 3.71 (bs, 4H), 7.04 (s, 1H), 7.13–7.31 (m, 5H), 7.71 (t, 1H). ESI MS m/z 546 (M + H)+. Anal. (C30H42F3N5O·25H2O) C, H, N.
4-(4-(6-(tert-Butyl)-2-methylpyrimidin-4-yl)piperazin-1-yl)butan-1-amine (43a)
Step 1
A solution of 4-(tert-butyl)-6-chloro-2-methylpyrimidine (7.98 g, 43.2 mmol) in absolute ethanol (100 mL) was added dropwise to a boiling solution of piperazine (18.46 g, 215 mmol) in ethanol (175 mL) over a period of 2 h. The reaction mixture was refluxed for 12 h, cooled to room temperature, and the solvent was removed under reduced pressure. The residue was treated with ice-cold water (1 L) and stirred for 15 min. After the mixture was allowed to stand for an hour, the solid crystalline product obtained was collected by filtration and dried under reduced pressure over P2O5 to obtain 9.09 g (90%) of 4-(tert-butyl)-2-methyl-6-(piperazin-1-yl)pyrimidine. ESI MS m/z 235 (M + H)+.
Step 2
A mixture of the above piperazine (7.5 g, 32.0 mmol), N-(4-bromobutyl)phthalimide (9.03 g, 32.0 mmol), and potassium carbonate (5.30 g, 38.35 mmol) in acetonitrile (200 mL) was stirred at room temperature overnight. The reaction mixture was filtered, and the filtrate was concentrated under reduced pressure. The residue thus obtained was purified by flash chromatography over a column of silica gel using CHCl3–MeOH 98:2 as the eluent to obtain 11.5 g (82%) of 2-(4-(4-(6-(tert-butyl)-2-methylpyrimidin-4-yl)piperazin-1-yl)butyl)isoindoline-1,3-dione. ESI MS m/z 436 (M + H)+.
Step 3
A mixture of the above (11.5 g, 32.05 mmol) and hydrazine hydrate (64%, 5.01 g, 64.10 mmol) in methanol (150 mL) was heated under reflux for 2 h. The reaction mixture was cooled to room temperature, and the volatiles were removed under reduced pressure. The residue was dissolved in ether (150 mL), filtered, and the filtrate was concentrated under reduced pressure. The residue obtained was purified by chromatography over a column of silica using CHCl3–MeOH–NH4OH (85:13:2) as the eluent to yield 7.6 g (94%) of the desired product. 1H NMR (DMSO-d6) δ 1.22 (s, 9H), 1.37–1.38 (m, 2H), 1.41–1.50 (m, 2H), 2.28 (t, J = 7.0 Hz, 2H), 2.39 (t, J = 5.1 Hz, 2H), 2.54 (t, J = 6.8 Hz, 2H), 3.57 (s, 4H), 6.47 (s, 1H). ESI MS m/z 306 (M + H)+.
4-(4-(2-(tert-Butyl)-6-methylpyrimidin-4-yl)piperazin-1-yl)butan-1-amine (43b)
This compound was prepared from 2-(tert-butyl)-4-chloro-6-methylpyrimidine using procedures analogous to those described for compound 43a. Overall yield: 48%. 1H NMR (DMSO-d6) δ 1.25 (s, 9H), 1.26–1.38 (m, 2H), 1.40–1.50 (m, 2H), 2.23 (s, 3H), 2.28 (t, J = 3.7 Hz, 2H), 2.39 (t, J = 5.1 Hz, 2H), 2.51 (t, J = 6.9 Hz, 2H), 3.57 (t, J = 4.9 Hz, 4H), 6.46 (s, 1H). ESI MS m/z 306 (M + H)+.
4-(4-(2-(tert-Butyl)-6-cyclopropylpyrimidin-4-yl)piperazin-1-yl)butan-1-amine (43c)
This compound was prepared from 2-(tert-butyl)-4-chloro-6-cyclopropylpyrimidine using procedures analogous to those described for compound 43a. Overall yield: 28%. 1H NMR (DMSO-d6) δ 0.83–0.88 (m, 2H), 0.92–0.97 (m, 2H), 1.22 (s, 9H), 1.34–1.38 (m, 2H), 1.44–1.49 (m, 2H), 2.28 (t, J = 7.2 Hz, 2H), 2.39 (t, J = 4.9 Hz, 2H), 2.56 (t, J = 6.8 Hz, 2H), 3.57 (t, J = 4.9 Hz, 4H), 6.49 (s, 1H). ESI MS m/z 332 (M + H)+.
4-(4-(2-(tert-Butyl)-6-(trifluoromethyl)pyrimidin-4-yl)piperazin-1-yl)butan-1-amine (43d)
This compound was prepared from 2-(tert-butyl)-4-chloro-6-(trifluoromethyl)pyrimidine using procedures analogous to those described for compound 43a. Overall yield: 33%. 1H NMR (DMSO-d6) δ 1.36 (s, 9H), 1.48–1.61 (m, 4H), 2.38 (t, J = 7.25 Hz, 2H), 2.51 (t, J = 5.15 Hz, 2H), 2.72 (t, J = 6.71 Hz, 2H), 3.72 (s, 4H), 6.59 (s, 1H). ESI MS m/z 360 (M + H)+.
4-(4-(2,6-Di-tert-butylpyrimidin-4-yl)piperazin-1-yl)butan-1-amine (43e)
This compound was prepared from 2,4-di-tert-butyl-6-chloropyrimidine using procedures analogous to those described for compound 43a. Overall yield: 46%. 1H NMR (DMSO-d6) δ 1.24 (s, 9H), 1.27 (s, 9H), 1.34–1.49 (m, 4H), 2.29 (t, J = 7.5 Hz, 2H), 2.40 (t, J = 5.0 Hz, 2H), 2.53 (t, J = 6.71 Hz, 2H), 3.59 (t, J = 4.8 Hz, 4H), 6.47 (s, 1H). ESI MS m/z 348 (M + H)+.
4-(4-(2-(tert-Butyl)quinazolin-4-yl)piperazin-1-yl)butan-1-amine (43f)
This compound was prepared from 2-(tert-butyl)-4-chloroquinazoline using procedures analogous to those described for compound 43a. Overall yield: 66%. ESI MS m/z 342 (M + H)+.
4-(4-(7-Chloroquinolin-4-yl)piperazin-1-yl)butan-1-amine (43g)
This compound was prepared from 2-(tert-butyl)-4-chloroquinazoline using procedures analogous to those described for compound 43a. Overall yield: 66%. ESI MS m/z 319 (M + H)+.
Binding Affinity
A filtration binding assay was used to characterize the binding properties of the D2, D3, chimeric D2/D3, and mutant dopamine receptors. Direct binding and competition curves were performed using [125I]IABN with dopamine receptors stably expressed in HEK 293 cells. Stably transfected cells were harvested by centrifugation. The cell pellet was resuspended in cold (4 °C) homogenization buffer (50 mM Tris-HCl, pH 7.4, with 10 mM EDTA, 150 mM NaCl) by vortexing and then homogenizing with a Polytron (Brinkmann Instruments, Westbury, NY). The homogenate was centrifuged at 12 000g at 4 °C and the membrane pellet resuspended in buffer and kept at −80 °C. Tissue homogenates (50 μL) were suspended in 50 mM Tris-HCl/150 mM NaCl/10 mM EDTA buffer, pH 7.5, and incubated with 50 μL of [125I]IABN and 50 μL of test ligand at 37 °C for 60 min. Nonspecific binding was defined using 2 μM (+)-butaclamol. For competition experiments, the radioligand concentration was generally equal to the Kd value and the concentration of the competitive inhibitor ranged over 5 orders of magnitude. Binding was terminated by addition of cold wash buffer (10 mM Tris-HCl/150 mM NaCl, pH 7.5) and filtration over a glass-fiber filter (Whatman no. 32, Piscataway, NJ). Filters were washed, and the radioactivity was measured using a Packard γ counter with an efficiency of 75%. The protein concentration of the membranes was determined using a BCA reagent (Pierce, Rockford, IL) and BSA as the protein standard.
Estimates of the Kd and maximum binding sites (Bmax)50 were obtained using unweighted linear regression analysis of data. Data from competitive inhibition experiments were modeled using nonlinear regression analysis to determine the concentration of inhibitor that inhibits 50% of the specific binding of the radioligand (IC50). Since transfected cells expressing receptor were used for this study, competition curves were modeled for a single site using
where B is the amount of ligand bound to tissue, B0 is the amount of ligand bound in the absence of competitive inhibitor, L is the concentration of the competitive inhibitor, Bns is the nonspecific binding of the radioligand (defined using a high concentration of a structurally dissimilar competitive inhibitor), and IC50 is the concentration of competitive inhibitor that inhibits 50% of the total specific binding. Data from competition dose–response curves were analyzed using Tablecurve program (Jandel/Systat Software Inc., San Jose, CA). IC50 values were converted to equilibrium dissociation constants (Ki values).
Generation of Chimeric and Mutant Receptors
Four methods were utilized to construct the D2/D3 receptor chimeras used in these studies. The methods for the preparation of these chimeric receptor genes have been previously described.37,68,69 The first method exploits a unique Pst1 restriction site in the first intracellular loop (I1) that is common to both the human D2 and human D3 receptor cDNAs. The second method involved making chimeric D2/D3 receptors. A human D3 receptor cDNA and a human D2 receptor cDNA were cloned in tandem into the pIRESneo2 vector with a unique restriction site (Hpa1) located between the two receptor cDNAs specifically within the junctions at the helical TMS regions TMS IV and TMS V, using site directed mutagenesis techniques (Quick-Change site-directed mutagenesis kit, Stratagene/Agilent Technologies, Santa Clara, CA). Finally, the D2/D3E2 and D3/D2E2 receptor loop chimeras were prepared using the Quick Change kit strategy with synthetic oligonucleotides encoding the E2 loop with the appropriate 5′ and 3′ flanking region. The authenticity of the chimeric receptor was verified by DNA sequencing, and the expression of the receptor construct in HEK 293 cells was verified by radioligand binding using [125I]IABN.
Mitogenesis Assays
Agonist stimulation of D2 or D3 dopamine receptors leads to an increase in mitogenic activity.54 These assays are based on [3H]thymidine uptake by cells that are proliferating. CHO cells expressing human D2 or D3 receptors (CHOp-D2 or CHOp-D3) were maintained in α-MEM with 10% FBS, 0.05% pen–strep, and 200 μg/mL of G418. To measure stimulation of mitogenesis (agonist assay) or inhibition of quinpirole stimulation of mitogenesis (antagonist assay), CHOp-D2 or CHOp-D3 cells were seeded in a 96-well plate at a concentration of 5000 cells/well. The cells were incubated at 37 °C in α-MEM with 10% FBS. After 48–72 h, the cells were rinsed twice with serum-free α-MEM and incubated for 24 h at 37 °C. Serial dilutions of test compounds were made by the Biomek robotics system in serum-free α-MEM. In the functional assay for agonists, the medium was removed and replaced with 100 μL of test compound in serum-free α-MEM. In the antagonist assay, the serial dilution of the putative antagonist test compound was added in 90 μL (1.1× of final concentration) and 300 nM quinpirole (30 nM final) was added in 10 μL. After another 24 h incubation at 37 °C of CHOp-D2 cells or 16 h incubation of CHOp-D3 cells, 0.25 μCi [3H]thymidine in α-MEM supplemented with 10% FBS was added to each well and the plates were further incubated for 2 h at 37 °C. The cells were trypsinized by addition of 10× trypsin solution (1% trypsin in calcium–magnesium-free phosphate-buffered saline), and the plates were filtered and counted as usual. Quinpirole was run as an internal control, and dopamine was included for comparative purposes. Butaclamol was run as standard antagonist.
Data Analysis
Data were analyzed using GraphPad Prism, and agonist potency was expressed as EC50 values or % stimulation. Antagonist potency was expressed as IC50 values. In these assays, based on the % maximum stimulation as compared to the standard agonist quinpirole (100%), the compounds were classified into following categories: ≥90% full agonist; 70–90% partial to full agonist; <70% partial agonist; 0–20% potential antagonist.
Whole Cell Adenylyl Cyclase Assay
Whole cell cyclic AMP accumulation was measured by an adaptation of the method of Shimizu and co-workers.70 Transfected HEK 293 cells were treated with serum-free medium containing [2,8-3H]adenine, and cells were incubated at 37 °C for 75 min. The medium was replaced with serum-free medium containing 0.1 mM 3-isobutyl-1-methylxanthine (Sigma-Aldrich, St. Louis, MO). Forskolin (100 μM final) was added in the presence or absence of test drug to a total volume of 500 μL and incubated at 37 °C for 20 min. The reaction was stopped by addition of 500 μL of 10% trichloroacetic acid and 1 mM cyclic AMP. After centrifugation, the supernatants were fractionated using Dowex AG1-X8 and neutral alumina to separate the [3H]ATP and the [3H]cAMP. Individual samples were corrected for column recovery by monitoring the recovery of the cyclic AMP using spectrophotometric analysis at OD 259 nm. The percent conversion of [3H]ATP into [3H]cAMP was then calculated as the percent of inhibition of the [3H]cAMP accumulation relative to the assay performed in the absence of a test compound, minus basal activity. The classic D2-like dopamine receptor antagonist and agonist (haloperidol and quinpirole, respectively) were included in the assay design as reference compounds. The percent maximum response is the value for the inhibition normalized to the value obtained for the full agonist quinpirole. Values are reported as the mean values ± SEM. The concentrations for all test compounds was ≥10× the Ki value of the compound at human D2 or D3 dopamine receptors, obtained from competitive radioligand binding experiments.69
β-Arrestin Recruitment Assays
Effect of compounds on agonist induced recruitment of β-arrestin by D3R and D2LR was measured using DiscoverRx PathHunter eXpress kits. Briefly, U2OS DRD3 (DiscoveRx no. 93-0591E3) and CHO-K1 DRD2L (DiscoveRx no. 93-0579E2) cells were seeded at a density of 2500 cells/well in 384-well white, clear-bottom plates (Corning no. 3707) with PathHunter Cell Plating Reagents, CP0 (DiscoveRx no. 93-0563R0) and CP2 (DiscoveRx no. 93-0563R2), respectively, and incubated at 37 °C for 48 h. For the determination of EC80 values of standard agonists, the cells were treated with multiple concentrations (starting from 190 nM, 1:3 dilution, 12 dose points) of (+)-PD128907 (DiscoveRx no. 92-1163) for D3R and Pergolide (DiscoveRx no. 92-1162) for D2LR. After incubation at 37 °C for 90 min, the DiscoveRx detection reagent was added, the plates were incubated at room temperature for 60 min, and the luminescence was measured on a Synergy 4 plate reader (BioTek). The data were plotted, and the agonist EC80 values were calculated using GraphPad Prism software. For antagonist activity, the cells were plated as described above and treated with multiple concentrations (starting from 500 nM, half log dilution, 10 dose points) of the test compounds in plating reagent with 0.1% DMSO. Following a 30 min preincubation at 37 °C, the cells were treated with an EC80 dose (8 nM PD128907 for D3R and 6 nM pergolide for D2R) of the agonist and incubated at 37 °C for an additional 90 min. The DiscoveRx detection reagent was then added, and the plates were incubated at room temperature for 60 min. Luminescence was measured, and the IC50 values were calculated. For determination of agonist or inverse agonist activity, the cells were plated and incubated for 48 h as described above and treated with multiple concentrations (starting from 5 μM, half log dilution, 11 dose points) of the test compounds, incubated at 37 °C for 120 min, treated with detection reagents, and the luminescence was measured. The activity was calculated as percent stimulation or inhibition of the basal luminescence in the absence of the inhibitors, as the EC50 or IC50 values of the compounds were greater than the highest tested concentration of 5 μM. All of the compounds were tested in the counterscreen for inhibition of β-galactosidase enzyme using the EFC BlockDetect kit (DiscoveRx no. 92-0004) in dose response mode and were found to be devoid of any significant inhibitory activity at the highest tested concentration of 5 μM.
[35S]GTPγS Binding Assays
CHO cells expressing human D2L and D3 receptors served as the source for membrane fractions which were washed and resuspended with assay buffer containing Mg2+, Na+, EGTA, and bovine serum albumin as in our previous work.55−57 The GTPγS binding assays were performed in a final volume of 0.5 mL for CHO D2 and 1 mL for CHO D3 containing test compound or dopamine (1 mM for D2 cells, and 100 μM for D3 cells) as indicator of binding plateau, [35S]GTPγS (0.11 nM for CHO D2 and 0.07 nM for CHO D3, 1250 Ci/mmol, PerkinElmer, Boston, MA 02118), and cell membrane suspension (in assay buffer and GDP for final concentration of 3 μM for CHO D2 and 6 μM for CHO D3). After preincubation with test compounds, cell membranes and GDP were placed for 15 min at 30 °C in a shaking water bath. [35S]GTPγS was added, and incubation proceeded for an additional 45 min. Cell membranes were harvested on Brandel GF/B filtermats with a 24-pin Brandel harvester (Biomedical Research & Development Laboratories, Inc., Gaithersburg, MD). Nonspecific binding of [35S]GTPγS measured in the presence of 10 μM GTPγS was a very small fraction (5% or less) of basal binding in the absence of test compounds and does not impact the EC50 (concentration producing half-maximal stimulation) of the test compound estimated by nonlinear logarithmic fitting (logistics model) with OriginPro 7.0. The plateau binding (maximal binding stimulation) with test compound was expressed as percent of maximal binding observed with the full agonist dopamine (% Emax). The test compounds 8, 25, 30, and 39 did not stimulate GTPγS binding in three independent experiments. The last three compounds at high micromolar concentrations showed weak inverse agonist activity at D3R resulting in [35S]GTPγS binding below baseline, but this was not further studied in the present work. Antagonist activity was assessed by testing a fixed concentration that by itself had little or no inverse agonist effect for its ability to shift the concentration curve of an agonist stimulating [35S]GTPγS binding as described for opioid receptors by Sally and co-workers.59 The fixed concentrations of test compound were as listed in the Results and Discussion, and the agonist used was dopamine (0.001 μM to 100 uM for CHO D2 and 0.1 nM to 10 μM for CHO D3). Ke is the functional Ki (equilibrium dissociation constant) of an antagonist and is calculated according to the following equation: [test compound]/(EC50-2/EC50-1 – 1), where EC50-2 is the EC50 value in the presence of the test compound and EC50-1 is the value in the absence of the test compound.
Molecular Modeling and Ligand Docking
Refinement of the Dopamine D3 Receptor Crystal Structure
The human D3R crystal structure in complex with the antagonist eticlopride at 2.89 Å resolution39 (PDB code 3PBL) was refined using Prime preparation and refinement tools of the Protein Preparation Wizard implemented in the Schrödinger software package. After the addition of hydrogens and detection of disulfide bonds the structure was optimized by applying default parameters of the Impref utility using the OPLS2001 force field. Ligand structures were prepared using the LigPrep utility at neutral pH.
D2R Homology Model Development
A model of the D2 receptor was developed based on the human D2 receptor sequence (UniProtKB accession code P14416) and the D3 receptor crystal structure (PDB code 3PBL) as the homology modeling template. The generated model includes transmembrane and loop regions except for intracellular loop 3, which is missing in the D3 crystal structure where it is replaced by T4-lysozyme. InsightII/Homology modeling tools were used for initial homology model building. D2 loop conformations in loop regions containing gap(s) within the D3/D2 sequence alignment were generated in sets of 10. From these, D2 loop structures that closely overlap with the corresponding D3 loops in the crystal structure were selected. Loop side chains were adjusted using a built-in rotamer library to avoid steric clashes with the rest of the structure. Disulfide bridges involving any cysteine residues in loops were reproduced in the D2 model structure except for a disulfide bond that is within extracellular loop III, D3:Cys355-Cys358, which could not be reproduced in D2 because the loop is shorter in the D2R than in the D3R. The sequence identity between our D2 model and the D3 crystal structure is 71.5% (including loops). In order to relax the obtained D2 homology model structure, refinement tools of the Protein Preparation Wizard were applied. The structure was optimized in the OPLS2001 force field using default parameters of the Impref utility, analogous to the refinement of the D3 receptor crystal structure. The root-mean-square deviation of α-carbon atoms between the refined D2R model and D3R crystal structure was 0.7.
InducedFit Ligand Docking
InducedFit docking combines Glide docking with Prime structural refinement tools to account for side chain flexibility in the ligand binding site.71 The center of the docking grid was defined as the centroid of the cocrystallized eticlopride; the enclosing box size was determined automatically. InducedFit settings were at default values except for removing the side chain of a Tyr (D3:Tyr373, D2:387) in the initial step of the protein preparation constrained refinement in order to facilitate the initial docking of ligands longer than eticlopride. In the InducedFit workflow this step was followed by an initial Glide docking run using softened potentials (van der Waals scaling of 0.70 for the receptor and 0.50 for ligands). The obtained structures were then refined using Prime side chain optimization of residues within 5 Å from docked ligand poses. The derived “induced-fit” receptor structures were then utilized for the final step of Glide re-docking with default parameters, applied to structures within 30 kcal/mol of the lowest energy structures. The same InducedFit protocols were applied for ligand docking at both D3 and D2 receptor structures.
Acknowledgments
This investigation was supported by NIH Grant R01DA024675 and Contract N01-DA-1-8827 from the National Institute on Drug Abuse (NIDA). We thank the NIDA Addiction Treatment Discovery Program for providing mitogenesis functional assay data through Inter-Agency Agreement Y1-DA-5007-05. We thank Dr. James M. Riordan, Mark D. Richardson, Joan C. Bearden, and Jackie W. Truss for analytical and spectral data. We are grateful to Dr. John A. Secrist III for his encouragement, valuable comments, and suggestions throughout the course of this work.
Glossary
Abbreviations Used
- BOP-Cl
bis(2-oxo-3-oxazolidinyl)phosphinic chloride
- CHO
Chinese hamster ovary
- CLogP
calculated log of partition coefficient
- D2R
dopamine D2 receptor
- D3R
dopamine D3 receptor
- EL
extracellular loop
- GPCR
G-protein-coupled receptor
- HATU
O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HEK
human embryonic kidney
- IABN
2,3-dimethoxy-5-iodo-N-(9-benzyl-9-azabicyclo[3.3.1]nonan-3-yl)benzamide
- TM
transmembrane domain
Supporting Information Available
Figures showing docked poses; Cartesian coordinates of D3R, D2R residues, and docked compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
# G.Z.: Department of Basic Science, Touro University California, Vallejo, California, 94592.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Joyce J. N.; Millan M. J. Dopamine D3 receptor antagonists as therapeutic agents. Drug Discovery Today 2005, 10, 917–925. [DOI] [PubMed] [Google Scholar]
- Sokoloff P.; Diaz J.; Le Foll B.; Guillin O.; Leriche L.; Bezard E.; Gross C. The dopamine D3 receptor: a therapeutic target for the treatment of neuropsychiatric disorders. CNS Neurol. Disord.: Drug Targets 2006, 5, 25–43. [DOI] [PubMed] [Google Scholar]
- Heidbreder C. A.; Gardner E. L.; Xi Z. X.; Thanos P. K.; Mugnaini M.; Hagan J. J.; Ashby C. R. Jr. The role of central dopamine D3 receptors in drug addiction: a review of pharmacological evidence. Brain Res. Rev. 2005, 49, 77–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acri J. B.; Carter S. R.; Alling K.; Geter-Douglass B.; Dijkstra D.; Wikstrom H.; Katz J. L.; Witkin J. M. Assessment of cocaine-like discriminative stimulus effects of dopamine D3 receptor ligands. Eur. J. Pharmacol. 1995, 281, R7–R9. [DOI] [PubMed] [Google Scholar]
- Spealman R. D. Dopamine D3 receptor agonists partially reproduce the discriminative stimulus effects of cocaine in squirrel monkeys. J. Pharmacol. Exp. Ther. 1996, 278, 1128–1137. [PubMed] [Google Scholar]
- Nader M. A.; Mach R. H. Self-administration of the dopamine D3 agonist 7-OH-DPAT in rhesus monkeys is modified by prior cocaine exposure. Psychopharmacology (Berlin, Ger.) 1996, 125, 13–22. [DOI] [PubMed] [Google Scholar]
- Caine S. B.; Koob G. F. Modulation of cocaine self-administration in the rat through D-3 dopamine receptors. Science (New York, N.Y.) 1993, 260, 1814–1816. [DOI] [PubMed] [Google Scholar]
- Self D. W.; Barnhart W. J.; Lehman D. A.; Nestler E. J. Opposite modulation of cocaine-seeking behavior by D1- and D2-like dopamine receptor agonists. Science (New York, N.Y.) 1996, 271, 1586–1589. [DOI] [PubMed] [Google Scholar]
- Cheung T. H.; Loriaux A. L.; Weber S. M.; Chandler K. N.; Lenz J. D.; Schaan R. F.; Mach R. H.; Luedtke R. R.; Neisewander J. L. Reduction of cocaine self-administration and D3 receptor-mediated behavior by two novel dopamine D3 receptor-selective partial agonists, OS-3-106 and WW-III-55. J. Pharmacol. Exp. Ther. 2013, 347, 410–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley J. K.; Mash D. C. Adaptive increase in D3 dopamine receptors in the brain reward circuits of human cocaine fatalities. J. Neurosci. 1996, 16, 6100–6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal D. M.; Moraes C. T.; Mash D. C. Up-regulation of D3 dopamine receptor mRNA in the nucleus accumbens of human cocaine fatalities. Mol. Brain Res. 1997, 45, 335–339. [DOI] [PubMed] [Google Scholar]
- Boileau I.; Payer D.; Houle S.; Behzadi A.; Rusjan P. M.; Tong J.; Wilkins D.; Selby P.; George T. P.; Zack M.; Furukawa Y.; McCluskey T.; Wilson A. A.; Kish S. J. Higher binding of the dopamine D3 receptor-preferring ligand [11C]-(+)-propyl-hexahydro-naphtho-oxazin in methamphetamine polydrug users: a positron emission tomography study. J. Neurosci. 2012, 32, 1353–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilla M.; Perachon S.; Sautel F.; Garrido F.; Mann A.; Wermuth C. G.; Schwartz J. C.; Everitt B. J.; Sokoloff P. Selective inhibition of cocaine-seeking behaviour by a partial dopamine D3 receptor agonist. Nature 1999, 400, 371–375. [DOI] [PubMed] [Google Scholar]
- Stemp G.; Ashmeade T.; Branch C. L.; Hadley M. S.; Hunter A. J.; Johnson C. N.; Nash D. J.; Thewlis K. M.; Vong A. K.; Austin N. E.; Jeffrey P.; Avenell K. Y.; Boyfield I.; Hagan J. J.; Middlemiss D. N.; Reavill C.; Riley G. J.; Routledge C.; Wood M. Design and synthesis of trans-N-[4-[2-(6-cyano-1,2,3, 4-tetrahydroisoquinolin-2-yl)ethyl]cyclohexyl]-4-quinolinecarboxamide (SB-277011): a potent and selective dopamine D(3) receptor antagonist with high oral bioavailability and CNS penetration in the rat. J. Med. Chem. 2000, 43, 1878–1885. [DOI] [PubMed] [Google Scholar]
- Vorel S. R.; Ashby C. R. Jr.; Paul M.; Liu X.; Hayes R.; Hagan J. J.; Middlemiss D. N.; Stemp G.; Gardner E. L. Dopamine D3 receptor antagonism inhibits cocaine-seeking and cocaine-enhanced brain reward in rats. J. Neurosci. 2002, 22, 9595–9603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundt P.; Carlson E. E.; Cao J.; Bennett C. J.; McElveen E.; Taylor M.; Luedtke R. R.; Newman A. H. Novel heterocyclic trans olefin analogues of N-{4-[4-(2,3-dichlorophenyl)piperazin-1-yl]butyl}arylcarboxamides as selective probes with high affinity for the dopamine D3 receptor. J. Med. Chem. 2005, 48, 839–848. [DOI] [PubMed] [Google Scholar]
- Higley A. E.; Spiller K.; Grundt P.; Newman A. H.; Kiefer S. W.; Xi Z. X.; Gardner E. L. PG01037, a novel dopamine D3 receptor antagonist, inhibits the effects of methamphetamine in rats. J. Psychopharmacol. 2011, 25, 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheli F.; Arista L.; Bonanomi G.; Blaney F. E.; Braggio S.; Capelli A. M.; Checchia A.; Damiani F.; Di-Fabio R.; Fontana S.; Gentile G.; Griffante C.; Hamprecht D.; Marchioro C.; Mugnaini M.; Piner J.; Ratti E.; Tedesco G.; Tarsi L.; Terreni S.; Worby A.; Ashby C. R. Jr.; Heidbreder C. 1,2,4-Triazolyl azabicyclo[3.1.0]hexanes: a new series of potent and selective dopamine D(3) receptor antagonists. J. Med. Chem. 2010, 53, 374–391. [DOI] [PubMed] [Google Scholar]
- Roman V.; Gyertyan I.; Saghy K.; Kiss B.; Szombathelyi Z. Cariprazine (RGH-188), a D(3)-preferring dopamine D (3)/D (2) receptor partial agonist antipsychotic candidate demonstrates anti-abuse potential in rats. Psychopharmacology (Berlin, Ger.) 2013, 226, 285–293. [DOI] [PubMed] [Google Scholar]
- Heidbreder C. Selective antagonism at dopamine D3 receptors as a target for drug addiction pharmacotherapy: a review of preclinical evidence. CNS Neurol. Disord.: Drug Targets 2008, 7, 410–421. [DOI] [PubMed] [Google Scholar]
- Lacroix L. P.; Hows M. E.; Shah A. J.; Hagan J. J.; Heidbreder C. A. Selective antagonism at dopamine D3 receptors enhances monoaminergic and cholinergic neurotransmission in the rat anterior cingulate cortex. Neuropsychopharmacology 2003, 28, 839–849. [DOI] [PubMed] [Google Scholar]
- Gurevich E. V.; Bordelon Y.; Shapiro R. M.; Arnold S. E.; Gur R. E.; Joyce J. N. Mesolimbic dopamine D3 receptors and use of antipsychotics in patients with schizophrenia. A postmortem study. Arch. Gen. Psychiatry 1997, 54, 225–232. [DOI] [PubMed] [Google Scholar]
- Millan M. J.; Loiseau F.; Dekeyne A.; Gobert A.; Flik G.; Cremers T. I.; Rivet J. M.; Sicard D.; Billiras R.; Brocco M. S33138 (N-[4-[2-[(3aS,9bR)-8-cyano-1,3a,4,9b-tetrahydro[1]benzopyrano[3,4-c]pyrrol-2(3H)-yl)-ethyl]phenyl-acetamide), a preferential dopamine D3 versus D2 receptor antagonist and potential antipsychotic agent: III. Actions in models of therapeutic activity and induction of side effects. J. Pharmacol. Exp. Ther. 2008, 324, 1212–1226. [DOI] [PubMed] [Google Scholar]
- Millan M. J.; Buccafusco J. J.; Loiseau F.; Watson D. J.; Decamp E.; Fone K. C.; Thomasson-Perret N.; Hill M.; Mocaer E.; Schneider J. S. The dopamine D3 receptor antagonist, S33138, counters cognitive impairment in a range of rodent and primate procedures. Int. J. Neuropsychopharmacol. 2010, 13, 1035–1051. [DOI] [PubMed] [Google Scholar]
- Agai-Csongor E.; Nogradi K.; Galambos J.; Vago I.; Bielik A.; Magdo I.; Ignacz-Szendrei G.; Keseru G. M.; Greiner I.; Laszlovszky I.; Schmidt E.; Kiss B.; Saghy K.; Laszy J.; Gyertyan I.; Zajer-Balazs M.; Gemesi L.; Domany G. Novel sulfonamides having dual dopamine D2 and D3 receptor affinity show in vivo antipsychotic efficacy with beneficial cognitive and EPS profile. Bioorg. Med. Chem. Lett. 2007, 17, 5340–5344. [DOI] [PubMed] [Google Scholar]
- Agai-Csongor E.; Domany G.; Nogradi K.; Galambos J.; Vago I.; Keseru G. M.; Greiner I.; Laszlovszky I.; Gere A.; Schmidt E.; Kiss B.; Vastag M.; Tihanyi K.; Saghy K.; Laszy J.; Gyertyan I.; Zajer-Balazs M.; Gemesi L.; Kapas M.; Szombathelyi Z. Discovery of cariprazine (RGH-188): a novel antipsychotic acting on dopamine D3/D2 receptors. Bioorg. Med. Chem. Lett. 2012, 22, 3437–3440. [DOI] [PubMed] [Google Scholar]
- Gyertyan I.; Kiss B.; Saghy K.; Laszy J.; Szabo G.; Szabados T.; Gemesi L. I.; Pasztor G.; Zajer-Balazs M.; Kapas M.; Csongor E. A.; Domany G.; Tihanyi K.; Szombathelyi Z. Cariprazine (RGH-188), a potent D3/D2 dopamine receptor partial agonist, binds to dopamine D3 receptors in vivo and shows antipsychotic-like and procognitive effects in rodents. Neurochem. Int. 2011, 59, 925–935. [DOI] [PubMed] [Google Scholar]
- Newman A. H.; Grundt P.; Nader M. A. Dopamine D3 receptor partial agonists and antagonists as potential drug abuse therapeutic agents. J. Med. Chem. 2005, 48, 3663–3679. [DOI] [PubMed] [Google Scholar]
- Heidbreder C. A.; Newman A. H. Current perspectives on selective dopamine D(3) receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann. N.Y. Acad. Sci. 2010, 1187, 4–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lober S.; Hubner H.; Tschammer N.; Gmeiner P. Recent advances in the search for D3- and D4-selective drugs: probes, models and candidates. Trends Pharmacol. Sci. 2011, 32, 148–157. [DOI] [PubMed] [Google Scholar]
- Micheli F. Recent advances in the development of dopamine D3 receptor antagonists: a medicinal chemistry perspective. ChemMedChem 2011, 6, 1152–1162. [DOI] [PubMed] [Google Scholar]
- Newman A. H.; Blaylock B. L.; Nader M. A.; Bergman J.; Sibley D. R.; Skolnick P. Medication discovery for addiction: translating the dopamine D3 receptor hypothesis. Biochem. Pharmacol. 2012, 84, 882–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye N.; Neumeyer J. L.; Baldessarini R. J.; Zhen X.; Zhang A. Update 1 of: Recent progress in development of dopamine receptor subtype-selective agents: potential therapeutics for neurological and psychiatric disorders. Chem. Rev. 2013, 113, PR123–PR178. [DOI] [PubMed] [Google Scholar]
- Micheli F.; Heidbreder C. Dopamine D3 receptor antagonists: a patent review (2007–2012). Expert Opin. Ther. Pat. 2013, 23, 363–381. [DOI] [PubMed] [Google Scholar]
- Leopoldo M.; Berardi F.; Colabufo N. A.; De Giorgio P.; Lacivita E.; Perrone R.; Tortorella V. Structure–affinity relationship study on N-[4-(4-arylpiperazin-1-yl)butyl]arylcarboxamides as potent and selective dopamine D(3) receptor ligands. J. Med. Chem. 2002, 45, 5727–5735. [DOI] [PubMed] [Google Scholar]
- Newman A. H.; Cao J.; Bennett C. J.; Robarge M. J.; Freeman R. A.; Luedtke R. R. N-(4-[4-(2,3-Dichlorophenyl)piperazin-1-yl]butyl, butenyl and butynyl)arylcarboxamides as novel dopamine D(3) receptor antagonists. Bioorg. Med. Chem. Lett. 2003, 13, 2179–2183. [DOI] [PubMed] [Google Scholar]
- Banala A. K.; Levy B. A.; Khatri S. S.; Furman C. A.; Roof R. A.; Mishra Y.; Griffin S. A.; Sibley D. R.; Luedtke R. R.; Newman A. H. N-(3-Fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl)piperazine-1-yl)butyl)arylcarboxamides as selective dopamine D3 receptor ligands: critical role of the carboxamide linker for D3 receptor selectivity. J. Med. Chem. 2011, 54, 3581–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman A. H.; Beuming T.; Banala A. K.; Donthamsetti P.; Pongetti K.; Labounty A.; Levy B.; Cao J.; Michino M.; Luedtke R. R.; Javitch J. A.; Shi L. Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J. Med. Chem. 2012, 55, 6689–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien E. Y.; Liu W.; Zhao Q.; Katritch V.; Han G. W.; Hanson M. A.; Shi L.; Newman A. H.; Javitch J. A.; Cherezov V.; Stevens R. C. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science (New York, N.Y.) 2010, 330, 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobrath J. V.; Wang S. Computational elucidation of the structural basis of ligand binding to the dopamine 3 receptor through docking and homology modeling. J. Med. Chem. 2006, 49, 4470–4476. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Mach R. H.; Luedtke R. R.; Reichert D. E. Subtype selectivity of dopamine receptor ligands: insights from structure and ligand-based methods. J. Chem. Inf. Model. 2010, 50, 1970–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campiani G.; Butini S.; Trotta F.; Fattorusso C.; Catalanotti B.; Aiello F.; Gemma S.; Nacci V.; Novellino E.; Stark J. A.; Cagnotto A.; Fumagalli E.; Carnovali F.; Cervo L.; Mennini T. Synthesis and pharmacological evaluation of potent and highly selective D3 receptor ligands: inhibition of cocaine-seeking behavior and the role of dopamine D3/D2 receptors. J. Med. Chem. 2003, 46, 3822–3839. [DOI] [PubMed] [Google Scholar]
- Chu W.; Tu Z.; McElveen E.; Xu J.; Taylor M.; Luedtke R. R.; Mach R. H. Synthesis and in vitro binding of N-phenyl piperazine analogs as potential dopamine D3 receptor ligands. Bioorg. Med. Chem. 2005, 13, 77–87. [DOI] [PubMed] [Google Scholar]
- Newman A. H.; Grundt P.; Cyriac G.; Deschamps J. R.; Taylor M.; Kumar R.; Ho D.; Luedtke R. R. N-(4-(4-(2,3-Dichloro- or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with functionalized linking chains as high affinity and enantioselective D3 receptor antagonists. J. Med. Chem. 2009, 52, 2559–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettinetti L.; Schlotter K.; Hubner H.; Gmeiner P. Interactive SAR studies: rational discovery of super-potent and highly selective dopamine D3 receptor antagonists and partial agonists. J. Med. Chem. 2002, 45, 4594–4597. [DOI] [PubMed] [Google Scholar]
- Hocke C.; Prante O.; Lober S.; Hubner H.; Gmeiner P.; Kuwert T. Synthesis and radioiodination of selective ligands for the dopamine D3 receptor subtype. Bioorg. Med. Chem. Lett. 2004, 14, 3963–3966. [DOI] [PubMed] [Google Scholar]
- Leopoldo M.; Lacivita E.; De Giorgio P.; Colabufo N. A.; Niso M.; Berardi F.; Perrone R. Design, synthesis, and binding affinities of potential positron emission tomography (PET) ligands for visualization of brain dopamine D3 receptors. J. Med. Chem. 2006, 49, 358–365. [DOI] [PubMed] [Google Scholar]
- Taylor M.; Grundt P.; Griffin S. A.; Newman A. H.; Luedtke R. R. Dopamine D3 receptor selective ligands with varying intrinsic efficacies at adenylyl cyclase inhibition and mitogenic signaling pathways. Synapse (New York, N.Y.) 2010, 64, 251–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundt P.; Prevatt K. M.; Cao J.; Taylor M.; Floresca C. Z.; Choi J. K.; Jenkins B. G.; Luedtke R. R.; Newman A. H. Heterocyclic analogues of N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)arylcarboxamides with functionalized linking chains as novel dopamine D3 receptor ligands: potential substance abuse therapeutic agents. J. Med. Chem. 2007, 50, 4135–4146. [DOI] [PubMed] [Google Scholar]
- Geneste H.; Backfisch G.; Braje W.; Delzer J.; Haupt A.; Hutchins C. W.; King L. L.; Kling A.; Teschendorf H. J.; Unger L.; Wernet W. Synthesis and SAR of highly potent and selective dopamine D(3)-receptor antagonists: 1H-pyrimidin-2-one derivatives. Bioorg. Med. Chem. Lett. 2006, 16, 490–494. [DOI] [PubMed] [Google Scholar]
- Geneste H.; Amberg W.; Backfisch G.; Beyerbach A.; Braje W. M.; Delzer J.; Haupt A.; Hutchins C. W.; King L. L.; Sauer D. R.; Unger L.; Wernet W. Synthesis and SAR of highly potent and selective dopamine D3-receptor antagonists: variations on the 1H-pyrimidin-2-one theme. Bioorg. Med. Chem. Lett. 2006, 16, 1934–1937. [DOI] [PubMed] [Google Scholar]
- Geneste H.; Backfisch G.; Braje W.; Delzer J.; Haupt A.; Hutchins C. W.; King L. L.; Lubisch W.; Steiner G.; Teschendorf H. J.; Unger L.; Wernet W. Synthesis and SAR of highly potent and selective dopamine D(3)-receptor antagonists: quinolin(di)one and benzazepin(di)one derivatives. Bioorg. Med. Chem. Lett. 2006, 16, 658–662. [DOI] [PubMed] [Google Scholar]
- Michino M.; Donthamsetti P.; Beuming T.; Banala A.; Duan L.; Roux T.; Han Y.; Trinquet E.; Newman A. H.; Javitch J. A.; Shi L. A single glycine in extracellular loop 1 is the critical determinant for pharmacological specificity of dopamine D2 and D3 receptors. Mol. Pharmacol. 2013, 84, 854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilon C.; Levesque D.; Dimitriadou V.; Griffon N.; Martres M. P.; Schwartz J. C.; Sokoloff P. Functional coupling of the human dopamine D3 receptor in a transfected NG 108-15 neuroblastoma-glioma hybrid cell line. Eur. J. Pharmacol. 1994, 268, 129–139. [DOI] [PubMed] [Google Scholar]
- Bassoni D. L.; Raab W. J.; Achacoso P. L.; Loh C. Y.; Wehrman T. S. Measurements of beta-arrestin recruitment to activated seven transmembrane receptors using enzyme complementation. Methods Mol. Biol. 2012, 897, 181–203. [DOI] [PubMed] [Google Scholar]
- Johnson M.; Antonio T.; Reith M. E.; Dutta A. K. Structure–activity relationship study of N(6)-(2-(4-(1H-indol-5-yl)piperazin-1-yl)ethyl)-N(6)-propyl-4,5,6,7-tetrahydrobenzo[d]thiazole-2,6-diamine analogues: development of highly selective D3 dopamine receptor agonists along with a highly potent D2/D3 agonist and their pharmacological characterization. J. Med. Chem. 2012, 55, 5826–5840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S.; Zhang S.; Fernandez F.; Ghosh B.; Zhen J.; Kuzhikandathil E.; Reith M. E.; Dutta A. K. Further structure–activity relationships study of hybrid 7-{[2-(4-phenylpiperazin-1-yl)ethyl]propylamino}-5,6,7,8-tetrahydronaphthalen-2-ol analogues: identification of a high-affinity D3-preferring agonist with potent in vivo activity with long duration of action. J. Med. Chem. 2008, 51, 101–117. [DOI] [PubMed] [Google Scholar]
- Gogoi S.; Antonio T.; Rajagopalan S.; Reith M.; Andersen J.; Dutta A. K. Dopamine D(2)/D(3) agonists with potent iron chelation, antioxidant and neuroprotective properties: potential implication in symptomatic and neuroprotective treatment of Parkinson’s disease. ChemMedChem 2011, 6, 991–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sally E. J.; Xu H.; Dersch C. M.; Hsin L. W.; Chang L. T.; Prisinzano T. E.; Simpson D. S.; Giuvelis D.; Rice K. C.; Jacobson A. E.; Cheng K.; Bilsky E. J.; Rothman R. B. Identification of a novel “almost neutral” mu-opioid receptor antagonist in CHO cells expressing the cloned human mu-opioid receptor. Synapse (New York, N.Y.) 2010, 64, 280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban J. D.; Clarke W. P.; von Zastrow M.; Nichols D. E.; Kobilka B.; Weinstein H.; Javitch J. A.; Roth B. L.; Christopoulos A.; Sexton P. M.; Miller K. J.; Spedding M.; Mailman R. B. Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 2007, 320, 1–13. [DOI] [PubMed] [Google Scholar]
- Kobilka B. K.; Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol. Sci. 2007, 28, 397–406. [DOI] [PubMed] [Google Scholar]
- Shukla A. K.; Xiao K.; Lefkowitz R. J. Emerging paradigms of beta-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem. Sci. 2011, 36, 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschammer N.; Elsner J.; Goetz A.; Ehrlich K.; Schuster S.; Ruberg M.; Kuhhorn J.; Thompson D.; Whistler J.; Hubner H.; Gmeiner P. Highly potent 5-aminotetrahydropyrazolopyridines: enantioselective dopamine D3 receptor binding, functional selectivity, and analysis of receptor–ligand interactions. J. Med. Chem. 2011, 54, 2477–2491. [DOI] [PubMed] [Google Scholar]
- Allen J. A.; Yost J. M.; Setola V.; Chen X.; Sassano M. F.; Chen M.; Peterson S.; Yadav P. N.; Huang X. P.; Feng B.; Jensen N. H.; Che X.; Bai X.; Frye S. V.; Wetsel W. C.; Caron M. G.; Javitch J. A.; Roth B. L.; Jin J. Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 18488–18493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Sassano M. F.; Zheng L.; Setola V.; Chen M.; Bai X.; Frye S. V.; Wetsel W. C.; Roth B. L.; Jin J. Structure–functional selectivity relationship studies of beta-arrestin-biased dopamine D(2) receptor agonists. J. Med. Chem. 2012, 55, 7141–7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galaj E.; Ananthan S.; Saliba M.; Ranaldi R. The effects of the novel DA D3 receptor antagonist SR 21502 on cocaine reward, cocaine seeking and cocaine-induced locomotor activity in rats. Psychopharmacology (Berlin, Ger.) 2014, 231, 501–510. [DOI] [PubMed] [Google Scholar]
- Hachimine P.; Seepersad N.; Ananthan S.; Ranaldi R. The novel dopamine D3 receptor antagonist, SR 21502, reduces cocaine conditioned place preference in rats. Neurosci. Lett. 2014, 569, 137–141. [DOI] [PubMed] [Google Scholar]
- Luedtke R. R.; Mishra Y.; Wang Q.; Griffin S. A.; Bell-Horner C.; Taylor M.; Vangveravong S.; Dillon G. H.; Huang R. Q.; Reichert D. E.; Mach R. H. Comparison of the binding and functional properties of two structurally different D2 dopamine receptor subtype selective compounds. ACS Chem. Neurosci. 2012, 3, 1050–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvano E.; Millan M. J.; Mannoury la Cour C.; Han Y.; Duan L.; Griffin S. A.; Luedtke R. R.; Aloisi G.; Rossi M.; Zazzeroni F.; Javitch J. A.; Maggio R. The tetrahydroisoquinoline derivative SB269,652 is an allosteric antagonist at dopamine D3 and D2 receptors. Mol. Pharmacol. 2010, 78, 925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H.; Daly J. W.; Creveling C. R. A radioisotopic method for measuring the formation of adenosine 3′,5′-cyclic monophosphate in incubated slices of brain. J. Neurochem. 1969, 16, 1609–1619. [DOI] [PubMed] [Google Scholar]
- Sherman W.; Day T.; Jacobson M. P.; Friesner R. A.; Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.