

Abstract
In this study we show that postnatal development of cerebellar granule neurons (GNs) is defective in Npc1−/− mice. Compared to age-matched wild-type littermates, there is an accelerated disappearance of the external granule layer (EGL) in these mice. This is due to a premature exit from the cell cycle of GN precursors residing at the level of the EGL. As a consequence, the size of cerebellar lobules of these mice displays a 20%–25% reduction compared to that of age-matched wild-type mice. This size reduction is detectable at post-natal day 28 (PN28), when cerebellar GN development is completed while signs of neuronal atrophy are not yet apparent.
Based on the analysis of EGL thickness and the determination of proliferating GN fractions at increasing developmental times (PN8–PN14), we trace the onset of this GN developmental defect during the second postnatal week. We also show that during this developmental time Shh transcripts undergo a significant reduction in Npc1−/− mice compared to age-matched wild-type mice. In light of the mitogenic activity of Shh on GNs, this observation further supports the presence of defective GN proliferation in Npc1−/− mice. A single injection of hydroxypropyl-β-cyclodextrin at PN7 rescues this defect, restoring the normal patterns of granule neuron proliferation and cerebellar lobule size.
To our knowledge, these findings identify a novel developmental defect that was underappreciated in previous studies. This defect was probably overlooked because Npc1 loss-of-function does not affect cerebellar foliation and causes the internal granule layer and molecular layer to decrease proportionally, giving rise to a normally appearing, yet harmoniously smaller, cerebellum.
Keywords: Granule neuron proliferation, Cerebellum, Niemann Pick C1, Hydroxypropyl-β-cyclodextrin
Highlights
-
•
Cerebellar lobules of adult Npc1−/− mice display a 20–25% reduction in size compared to wild-type age-matched mice.
-
•
The proliferation of granule neuron (GN) precursors in the developing cerebellum of Npc1−/− mice is defective.
-
•
Npc1−/− GN precursors of the external granule layer (EGL) undergo a premature exit from the cell cycle.
-
•
The EGL of Npc1−/− mice is thinner and persists for a shorter time.
-
•
A single injection of hydroxypropyl-β-cyclodextrin at PN7 rescues these defects.
Introduction
Niemann–Pick C1 (NPC1) is a multipass transmembrane protein residing in late endosomes and lysosomes (Garver et al., 2000, Higgins et al., 1999, Neufeld et al., 1999) and is essential to prevent the storage of cholesterol in those compartments (Morris et al., 1982, Pentchev et al., 1986). Working with NPC2, NPC1 mediates the egress of LDL-derived cholesterol from the endolysosomal compartment (Sleat et al., 2004) by a mechanism(s) not yet fully characterized, although great progress is being made (Cheruku et al., 2006, Infante et al., 2008). The widely accepted model is that NPC2 binds free cholesterol after lysosomal hydrolysis of LDL cholesteryl esters and transfers it to NPC1, which mediates the exit of cholesterol from lysosomes (Deffieu and Pfeffer, 2011, Kwon et al., 2009). Mutations of NPC1 and NPC2 genes in our species are associated with Niemann–Pick disease type C (95% and 5% of patients, respectively) (Vanier, 2010).
A prominent feature of NPC1 disease is the massive loss of cerebellar Purkinje cells (PCs) (Higashi et al., 1993, Ong et al., 2001). A similar feature is also observed in several mouse models of NPC1 disease, in which PC degeneration and loss begin at 28–40 days of postnatal age (PN28–40) and become very pronounced at PN60 (Higashi et al., 1993). Several approaches, including chimeric mice, conditional knock-out models and various neuron/glial-specific transgenic rescue experiments (Borbon et al., 2012, Elrick et al., 2010, Yu et al., 2011, Zhang et al., 2008) have been used to investigate the contribution of specific cell types to NPC1 neuropathology. Using chimeras of Npc1−/− and normal cells, it was proposed that Npc1−/− PC death is a cell-autonomous property (Ko et al., 2005), occurring even when PCs are surrounded by wild-type Bergmann glia (BG). Moreover, wild-type PCs survive well although surrounded by Npc1−/− BG (Ko et al., 2005). Despite these observations, the role astroglial cells exert in NPC neuropathology is still controversial. Exploiting a neuron-glia co-culture system, it was observed that Npc1-deficient astrocytes do not support neurite growth as efficiently as wild-type astrocytes do (Chen et al., 2007). A transgenic line re-expressing Npc1 from a glial fibrillary acidic protein promoter in Npc1−/− mice displayed lower levels of neuronal cholesterol accumulation and lived longer (Zhang et al., 2008). On the other hand, the transgenic re-expression of Npc1 in neurons or in astrocytes of Npc1−/− mice led to the conclusion that neuronal rescue only reduced neurological symptoms (Lopez et al., 2011). Moreover, the presence of a conditional Npc1 null allele in neurons was shown to mostly recapitulate the neuropathology typical of NPC (Yu et al., 2011), but the deletion in oligodendrocytes resulted in the impairment of myelin formation and maintenance (Yu and Lieberman, 2013).
Because neuropathological signs first appear in 5-week-old Npc1−/− mice, it is currently believed that developmental processes are not affected by the loss-of-function of the Npc1 gene. However, a study by Baudry et al. (2003) has shown that 2-week-old Npc1−/− mice display a significant level of microglia activation in several brain areas, including cerebellum. Microglia regulate neuronal proliferation and differentiation (Banati and Graeber, 1994), raising the question as to whether these processes may be influenced by microglia activation. At 2 weeks of age, in fact, cerebellar cortical development is still ongoing with active granule neuron (GN) proliferation/differentiation/migration and PC dendritic expansion, making the study of postnatal cerebellar development in Npc1−/− mice worthy of further investigation.
Among cerebellar cells, GNs have been studied little in murine models of NPC1 disease, perhaps because their development takes place during the first 3 weeks of postnatal life (Altman and Bayer, 1997, Sotelo, 2004, Wang and Zoghbi, 2001), whereas signs of cerebellar pathology occur in older mice. Indeed, GNs are progressively generated from E18.5 until PN16.5 from a progenitor layer covering the cerebellar surface, which is named the external granule layer (EGL) (Solecki et al., 2001). During early postnatal development, the coexistence of two GN precursor populations splits the EGL into two sub-regions, the outer EGL (containing mitotic cells; oEGL) and the inner EGL (containing post-mitotic cells; iEGL). GN precursors forming the oEGL and iEGL can be distinguished from each other by the expression of different protein markers, such as PCNA for mitotic and p27Kip1 for post-mitotic cells (Argenti et al., 2005, Miyazawa et al., 2000). Besides these two GN populations, an additional one, named “intermediate progenitors” and residing in a region of overlap between the oEGL and iEGL, has been recently characterized (Xenaki et al., 2011).
At around the time of birth some GNs exit from the cell cycle and migrate inward to form the internal granule layer (IGL) that represents their final destination, determining the gradual disappearance of the EGL (Altman and Bayer, 1997). GN proliferation in postnatal cerebellum EGL is sustained by various factors, including Sonic Hedgehog (Shh) secreted by PCs (Dahmane et al., 1999, Vaillant and Monard, 2009, Wechsler-Reya and Scott, 1999) and several microglia-released growth factors that activate the Notch signaling pathway (Morgan et al., 2004). Upon exit from the cell cycle, GNs migrate radially along the Bergmann glia and through the PC layer to reach the internal granule layer (IGL) (Adams et al., 2002) where they contribute to the formation of cerebellar glomeruli, a complex of synapses with mossy fiber endings converging on GN dendrites (Landis et al., 1983). Mossy fiber inputs are then transmitted to PCs via synaptic contacts between GN parallel fibers and PC dendrites.
Even though a prominent effect of Npc1 loss-of-function on GN development has not been reported, membranous inclusions in the cytoplasm of these cells were observed by electron microscopy in cerebella of 5-week-old Npc1−/− mice (Tanaka et al., 1988). Moreover, in a more recent study, the IGL of a Npc1−/− cat was found to be thinner compared to that of wild-type controls, opening the possibility that GNs were generated in a lower number during postnatal cerebellar development or died because of an increased level of apoptosis (Vite et al., 2008). An additional line of evidence enlightening the possible contribution of a GN defect to NPC neuropathology derives from a recent study showing that the synaptic transmission from parallel fibers to PCs is increased and that long-term depression (LTD) is deficient in 3-week-old Npc1−/− mice (Sun et al., 2001).
In this study we show that the EGL of Npc1-deficient mice has fewer GNs and persists for a shorter time compared to age-matched wild-type mice and that a single treatment with hydroxypropyl-β-cyclodextrin (HPBCD) at PN7 re-establishes the normal EGL thickness/GN number in Npc1−/− cerebella. We also show that the level of Shh transcripts of Npc1−/− cerebella undergoes a significant reduction during the second post-natal week, likely affecting the proliferation of GN precursors. To our knowledge, these findings provide the first evidence of a developmental GN defect in Npc1−/− mice, enlarging the perspective for future studies on the molecular mechanisms that underlie NPC1 disease.
Materials and methods
Animals and treatments
Npc1−/− mice on the BALB/cJ background were generated by mating heterozygous animals. Genotypes were identified from tail DNA by PCR as described (Loftus et al., 1997). Treatment with HPBCD (Sigma Aldrich, Milan, Italy) was performed in 7-day-old mice by a subcutaneous injection of 4 g HPBCD/kg body weight. BrdU labeling was achieved by intraperitoneal injection with 5-bromodeoxyuridine (60 mg BrdU/g body weight, Sigma Aldrich Inc., Milan, Italy). Mice were maintained in our animal facility in accordance with Sapienza University guidelines for the care and use of laboratory animals. Experimental protocols and related procedures were approved by the Italian Ministry of Public Health.
Histology and immunohistochemistry
Npc1+/+ and Npc1−/− mouse brains of postnatal days 11, 14 and 28 (PN11–28), either treated or untreated with HPBCD, were quickly dissected, fixed overnight at 4 °C in 4% PFA in 0.1 M phosphate buffer (PBS), dehydrated and embedded in Paraplast Plus Tissue Embedding Medium (Società Italiana Chimici, Rome; Italy). Paraffin tissue blocks were serially sectioned (slice thickness 8 μm), obtaining parasagittal sections that were mounted on Superfrost Excell adhesion slides (Thermo Scientific, Milan, Italy), de-waxed with xylene, re-hydrated and washed in PBS. For gross morphology of cerebellar lobules, sections encompassing the middle of the cerebellar vermis were stained with Nuclear Fast Red Solution (0.1% nuclear fast red, 5% aluminum sulfate; Sigma Aldrich Inc., Milan, Italy) for 30 min. Determination of the region of interest (ROI) in histological sections for area measurements (total cerebellum, total lobule IV/V, and IGL and ML of lobule IV/V) was independently performed by two investigators on three consecutive mid-sagittal sections of at least five mice per group, using ImageJ NIH software version 1.47v (National Institutes of Health, Bethesda, MD). Images to be compared were carefully selected paying attention that they were the closest possible to the parasagittal position, as inferred by the deeper white matter tracts matching very well in each image. The region of interest (ROI) in each section was outlined using the “selection brush” or the “polygon tool”, obtaining similar results.
To determine the number of GN cells, sections from PN11 and PN14 mice were stained with 0.5 μg/mL Hoechst (Hoechst-33258, Invitrogen, Milan, Italy) for 4 min. To assess GN proliferation, brains were dissected from Npc1+/+ and Npc1−/− mice, either treated or untreated with HBCD and that had received a single BrdU injection at PN10, PN12 and PN14, and processed as described above, obtaining parasagittal sections to be stained with anti-BrdU antibodies. Epitopes were unmasked by heating sections 2 × 5 min in 0.1 M citric acid, pH 6.0, in a microwave oven. Tissue permeabilization was achieved by incubation in trypsin solution (0.05% trypsin, 0.1% CaCl2 in water) for 15 min at room temperature (RT) and then in 2 M HCl for 30 min. After washing with PBS, sections were incubated overnight at 4 °C with a monoclonal anti-BrdU antibody (Immunological Science, Rome, Italy; 1:100 dilution in PBS supplemented with 0.5% Tween 20), and then for 45 min with an anti-mouse IgG antibody (Alexa Fluor-555 InVitrogen, Milan, Italy; 1:1500 final dilution). Sections were finally counterstained with Hoechst-33258, mounted with Prolong Gold Antifade Reagent and analyzed with an epifluorescence Zeiss microscope. For quantification of BrdU-immunopositive cells, ROIs were randomly selected from bases and crowns of the anterior (between lobules II and III), middle (between lobules VI and VII) and posterior (between lobules IX and X) lobules of 4 cerebella for each developmental stage. The total number of cells in each region was calculated by determining the number of nuclei based on Hoechst staining.
For p27Kip1 immunodetection, histological sections were subjected to unmasking and permeabilization procedures similar to those described for BrdU immunostaining. Following an incubation with 5% hydrogen peroxide for 15 min to inhibit endogenous peroxidase, sections were incubated with a buffer made of 6% goat serum and 0.3% Triton-X100 in PBS for 30 min to block non-specific binding. Immunohistochemistry was performed using the avidin–biotin horseradish peroxidase complex (ABC) method. Briefly, sections were incubated with rabbit monoclonal primary antibody against p27Kip1 (Epitomics, Burlingame, CA, USA; 1:75 final dilution) overnight at 4 °C, washed 3 times with PBS, and stained using the rabbit Vectastain Elite ABC Kit (Vector Laboratories Inc., Burlingame, CA, USA) first, allowing the specific ABC complex development, and then the DAB Peroxidase Substrate Kit (Vector Laboratories Inc., Burlingame, CA, USA), according to the protocol supplied by the manufacturer. Nuclear counterstaining was performed by incubating sections with 0.5% methyl green solution (0.5% methyl green, 0.1 M sodium acetate buffer in water, pH 4.2) for 5 min at 60 °C.
In situ Terminal deoxynucleotidyl transferase(TdT)-mediated dUTP Nick-End Labeling (TUNEL)
Cell death in the developing cerebellum of Npc1+/+ and Npc1−/− mice was assessed by the in situ TUNEL method selective for apoptotic cells. Sections were covered with 20 μg/mL Proteinase K for 1 h at 37 °C to permeabilize the tissue. After 2 washes in PBS for 5 min each, sections were incubated with 100 μL Tunel Equilibration Buffer for 5 min and then with 100 μL TUNEL Reaction Buffer containing 2% TdT enzyme for 2 h at 37 °C (Immunological Science, Rome, Italy). Sections were washed 3 times for 5 min each in PBS containing 0.1% Triton X-100 and 5 mg/mL BSA. Slides were then stained with 0.5 μg/mL Hoechst (Hoechst-33258, Invitrogen, Milan, Italy) for 4 min, mounted with Prolong Gold Antifade Reagent (InVitrogen, Milan, Italy) and examined using an epifluorescence Zeiss microscope. Images were acquired with a Cool Snap K4 Photometrics camera.
RNA preparation and real-time RT-PCR
Total RNA was extracted from PN8, PN12, Npc1+/+ and Npc1−/− cerebella, either treated or untreated with HPBCD, using TRIzol (Life technologies, CA, USA) and treated with DNase I (Ambion, Life technologies, CA, USA) for 15 min at 37 °C to remove genomic DNA. cDNA was synthesized using the GoScript Reverse Transcription System (Promega, WI, USA) following the protocol supplied by the manufacturer. Real-time PCR was performed using QuantiFast Sybr Green PCR Master Mix (Qiagen Inc., CA, USA) with the following cycling conditions: 95 °C for 10 min, then 35 cycles of 95 °C for 10 s and 62 °C for combined annealing/extension for 40 s. The QuantiFast PCR primers were: Mm_Shh_1_SG Cat No. QT00122479 (Shh), Mm_Actb_2_SG Cat No. QT01136772 (β-actin), and Mm_Rps16_QT0009256 (S16 ribosomal protein). The level of Shh gene was normalized to the expression level of the housekeeping genes β-actin and rps16.
Statistics
Statistical analyses were performed using GraphPad Prism version 5.0d (GraphPad, La Jolla, CA). Data are expressed as mean values ± SEM. Statistically significant differences were analyzed using one-way ANOVA followed by Bonferroni's post hoc test. p values less than 0.05 were considered significant.
Results
Cerebellar lobule size is reduced in Npc1−/− mice and is rescued by a single HPBCD injection
During postnatal development, the increase of cerebellum volume is largely due to the proliferation of GNs at the level of the EGL (Solecki et al., 2001). In the mouse, GN proliferation is mainly regulated by Shh expression and activity that, thereby, determine final cerebellar size and shape (Ruiz et al., 2002, Vaillant and Monard, 2009, Wallace, 1999, Wechsler-Reya and Scott, 1999). Reasoning that cholesterol is of particular relevance for both Shh signal generation and transduction in receiving cells, we analyzed the size and anatomy of cerebellar lobules in PN28 Npc1−/− and age-matched Npc1+/+ mice. At this time, the cerebellar cortex has already achieved the conformation of the adult, consisting of a so-called molecular layer (ML), mainly formed by fibers with few scattered cells, and the internal granule layer (IGL), mainly containing GNs. Gross morphological analysis revealed that the overall cerebellar size of Npc1−/− mice was reduced compared to that of age-matched wild-type mice (Figs. 1A,B). To quantify such reduction in size, the total areas of parasagittal sections of wild-type, Npc1−/− and HBPCD-treated Npc1−/− cerebella were measured after nuclear fast red staining, observing that Npc1−/− mice displayed a 20% reduction and that a single subcutaneous injection of HPBCD at PN7 largely rescued this phenotype (Figs. 1B,C).
Fig. 1.

The cerebellum of adult Npc1−/− mice is reduced in size. (A) Gross morphological comparison of PN28 Npc1+/+ and Npc1−/− mouse cerebella (brackets). Cx: cortex; Cb: cerebellum; Bs: brain stem. (B) Sagittal sections of cerebellar vermis from PN28 Npc1+/+ and Npc1−/− mice, either untreated or treated with a single injection of HPBCD at PN7. Representative sections encompassing sagittal midline of vermis are shown in the figure. Superimposition of total vermis tracings indicates an overall size reduction in Npc1−/− mice compared to wild-type mice that was largely rescued by HPBCD treatment. (C) Histograms indicate total areas (mean ± SEM; 5–6 mice/group) measured on representative sections of Npc1+/+ (empty bar), Npc1−/− (full bar) and HPBCD-treated Npc1−/− mice (dashed bar). Asterisk indicates statistically significant differences (*p < 0.05). Scale bars indicate 2 mm (panel A) and 0.5 mm (panel B).
Parasagittal sections encompassing the middle part of the entire cerebellar vermis were also useful for visualizing the morphology of cerebellar lobules, showing that both fissuration and lobule/sublobule formation were normal and that the final step of lobule growth was actually impaired by Npc1 loss-of-function. This feature extended to all lobules without graded antero-posterior or postero-anterior patterns (Fig. 2A). To further investigate the issue of cerebellar size reduction, total, IGL and ML areas of a representative lobule were determined, as performed by Peeters et al. (2013). Lobule IV/V was chosen because of its conspicuous length and less elaborate shape, making area measurements more reliable. These analyses indicated that the total area of lobule IV/V displayed a 24% size reduction and that IGL and ML displayed a similar extent of size reduction, 26% and 20%, respectively (Figs. 2A,B). IGL:ML area ratios appeared not to vary significantly in each group (wild-type mice, 0.66; Npc1−/− mice, 0.62; HPBCD-treated Npc1−/− mice, 0.69; p = 0.4), leading to the conclusion that Npc1 loss-of-function determines an “in-proportion” size reduction, which is in agreement with the normally appearing, yet smaller cerebellum of Npc1−/− mice. HPBCD treatment fully rescued the reduction in size, even determining a slight size increase.
Fig. 2.

Both anterior and posterior lobules of Npc1−/− cerebellum display a reduced size. (A) Parasagittal histological sections of PN28 mouse cerebella of Fig. 1 were examined at higher magnification to evaluate the effect of genotype and/or HPBCD treatment on foliation and single lobule size. Representative sections are shown in the figure. Npc1−/− mice displayed an overall normal foliation pattern. However, the size of both anterior and posterior lobules was impaired and a single injection of HPBCD to PN7 Npc1−/− mice re-established the normal lobule size. (B) The extent of size reduction was measured in detail on lobule IV/V. Histograms indicate the total lobule and IGL and ML areas (mean ± SEM of all sections examined; 5–6 mice/group) measured on randomly selected parasagittal sections of Npc1+/+ (empty bars), Npc1−/− (full bars) and HPBCD-treated Npc1−/− mice (dashed bars). Asterisks indicate statistically significant differences (*p < 0.05; **p < 0.005). Scale bar indicates 0.5 mm.
Npc1−/− mice display a shorter-lasting EGL
Starting at around the time of birth, some GN precursors exit the cell cycle and begin to differentiate into mature GNs (Altman and Bayer, 1997). Then post-mitotic GNs migrate through the single row of PCs to reach the IGL. This determines the gradual thinning and disappearance of EGL by the third post-natal week. We investigated the issue of reduced cerebellar lobule size of Npc1−/− by determining the EGL thickness in parasagittal sections obtained from cerebella of mice at increasing post-natal days of development, namely PN8, PN11 and PN14. The EGL was visualized by staining GN nuclei with Hoechst, while the number of GNs was determined by counting nuclei in randomly selected regions of 100 μm2 each (Fig. 3). This analysis showed that EGL thickness and GN densities were similar in PN8 (not shown) and PN11 Npc1+/+ and Npc1−/− mice, whereas in PN14 Npc1−/− mice, the EGL was mostly undetectable compared to that of age-matched Npc1+/+ mice. Indeed, only a few scattered GNs, not even forming a single row of cells, were observed in both crowns and fissures of either the anterior or posterior lobules of Npc1−/− mice. In agreement with the rescuing effect HPBCD had on cerebellar lobule size, a single HPBCD injection at PN7 doubled the number of GNs in the EGL of PN14 Npc1−/− mice.
Fig. 3.

Npc1−/− mice display reduced granule neuron density in the EGL that is rescued by the HPBCD treatment. (A,C) Cerebellar sections of PN11, PN14 Npc1+/+, and Npc1−/− mice, either untreated or treated with a single injection of HPBCD at PN7, were stained with Hoechst 33258. Representative sections are shown in the figure. Higher magnification fields of EGL base or crown of lobules II and X are shown on the right of low magnification fields. No major differences were detected at PN11. At PN14 however, the EGL of both anterior and posterior lobules of Npc1−/− mice was thinner than that of age-matched Npc1+/+, while HPBCD treatment fully rescued this phenotype. (B,D) Histograms represent granule neuron densities (mean ± SEM of all sections examined; 4–5 mice/group; 3–4 sections/mouse) determined in 100 μm2 regions of the crowns of the anterior (I–V) and posterior (VI–X) lobules of Npc1+/+ (empty bars), Npc1−/− (full bars) and HPBCD-treated Npc1−/− (dashed bars) mice. Asterisks indicate statistically significant differences (*p < 0.05; **p < 0.005). Scale bars indicate 250 μm (panels) and 50 μm (inserts).
Loss of Npc1 function results in GN proliferation defect
The finding that Npc1−/− mice displayed a shorter-lasting EGL compared to age-matched Npc1+/+ mice despite the presence of normal EGL thickness/GN numbers until PN11, suggested that the clock driving the proliferation of GN precursors was somehow dysregulated in Npc1−/− mice, leading these cells to prematurely exit from the cell cycle. Alternatively, an increase in the number of cells undergoing apoptosis could also account for the reduction of GNs forming the EGL. In either case, the time window in which these events take place likely corresponded to very late stages of GN mitotic expansion i.e., between PN11 and PN14.
To address these questions, we determined the number of cells incorporating BrdU 20 h after a single BrdU injection of PN10, PN12 or PN14 Npc1+/+ and Npc1−/− mice, either untreated or treated with HPBCD. While similar in PN10 mice, the number of BrdU-positive cells in both crowns and fissures of the anterior cerebellar lobules was significantly reduced in PN12 and PN14 Npc1−/− mice compared to age-matched Npc1+/+ mice. In posterior cerebellar lobules a reduction of BrdU-positive cells was only observed in fissures of PN14 Npc1−/− mice.
HPBCD treatment led Npc1−/− mice to display a number of BrdU-positive cells similar to that of Npc1+/+ mice (Fig. 4). The possibility of apoptosis was then investigated by analyzing midsagittal sections of PN12, PN14 and PN28 mouse cerebella with TUNEL. This assay showed that few TUNEL-positive cells were observed in cerebella of Npc1+/+ and Npc1−/− mice, either treated or untreated with HPBCD (Figs. 5A,B). The number of TUNEL-positive cells decreased with age but neither varied significantly between Npc1+/+ and Npc1−/− nor was affected by the treatment with HPBCD (Fig. 5B). Noteworthy, TUNEL-positive cells were scarcely observed in the EGL of either Npc1+/+ or Npc1−/− PN12 and PN14 mice, with only 1–2 TUNEL-positive cells per lobule. These observations indicate that increased cell death is not the primary cause of the reduction in GN number, instead suggesting an earlier exit of these cells from the mitotic cycle as a more likely possibility. This possibility was further strengthened by the observation that Shh transcript levels were significantly reduced in cerebella of PN8 and PN12 Npc1−/− compared to age-matched Npc1+/+ mice and returned to normal levels after HPBCD treatment (Fig. 6).
Fig. 4.

Npc1−/− mice display reduced number of proliferating granule neurons that is rescued by the HPBCD treatment. (A) Histograms represent the number of BrdU-positive cells (mean ± SEM; 4–5 mice/group; 3–4 sections/mouse) determined in 100 μm2 regions corresponding to the bases and crowns of the anterior (I–V) and posterior (VI–X) lobules of Npc1+/+ (empty bars), Npc1−/− (full bars) and HPBCD-treated Npc1−/− (dashed bars) mice. Asterisks indicate statistically significant differences (*p < 0.05; **p < 0.005). Note that the number of BrdU-positive GNs was significantly reduced in both crowns and fissures of the anterior cerebellar lobules of PN12 and PN14 Npc1−/− mice compared to age-matched Npc1+/+ mice. (B) A representative field showing BrdU-positive cells (red) of fissure between lobules I and II of PN12 Npc1+/+ (left), Npc1−/− (middle) and HPBCD-treated Npc1−/− mice (right). Note that HPBCD treatment re-established the normal number of proliferating cells. Scale bar indicates 50 μm.
Fig. 5.

Cerebella of Npc1−/− and Npc1+/+ display comparable numbers of apoptotic GNs. (A) Cerebellar sections of PN12, PN14 and PN28 Npc1+/+ and Npc1−/− mice, either untreated or treated with a single injection of HPBCD at PN7, were stained with TUNEL. A representative field showing TUNEL-positive cells (red) of lobule II of PN14 Npc1+/+ (top), Npc1−/− (middle) and HPBCD-treated Npc1−/− mice (bottom). Arrowheads indicate TUNEL-positive cells. Scale bar indicates 50 μm. (B) Histograms represent the number of TUNEL-positive cells (mean ± SEM; 4–5 mice/group; 3–4 sections/mouse) determined in 1 mm2 regions corresponding to the crown of lobule II of Npc1+/+ (empty bars), Npc1−/− (full bars) and HPBCD-treated Npc1−/− (dashed bars) mice.
Fig. 6.
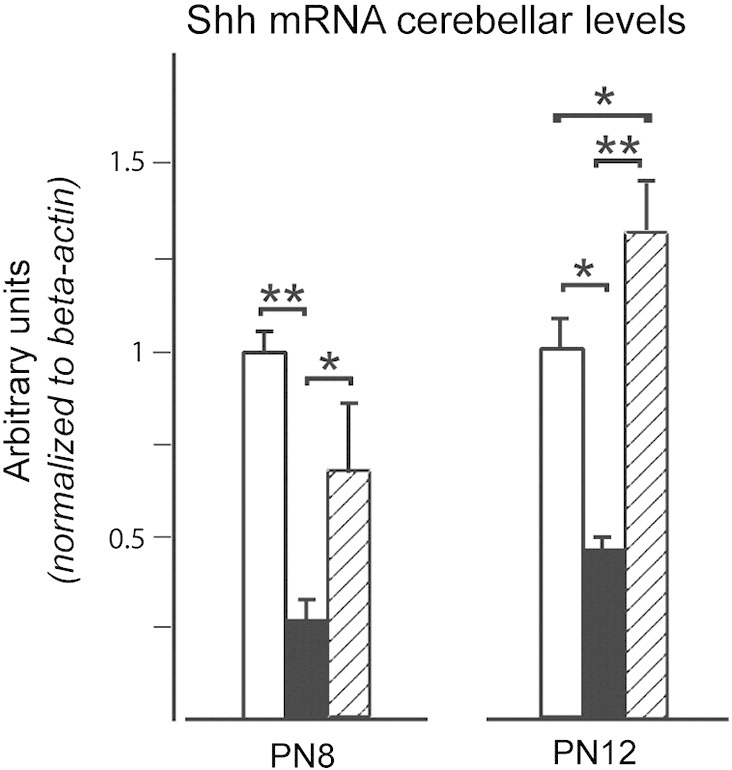
Npc1−/− mice cerebella display a significantly reduced level of Shh transcripts. Semiquantitative real-time RT-PCR was performed on cerebella of PN8 and PN12 Npc1+/+ (empty bars), Npc1−/− (full bars) and HPBCD-treated Npc1−/− (dashed bars). Histograms indicate the mean ± SEM of values obtained in three independent assays. Note that HPBCD re-established the normal level of Shh transcripts. Asterisks indicate statistically significant differences (**p < 0.005).
Loss of Npc1 function results in disorganized cellular architecture of the EGL
As stated in the Introduction section, under normal developmental conditions a sharp boundary separates the population of mitotically-active p27Kip1-negative GNs forming the outer EGL from the p27Kip1-positive post-mitotic GNs forming the inner EGL. Accordingly, when we performed p27Kip1 immunostaining coupled to a counterstaining of nuclei with methyl green on PN8 cerebella, this boundary was easily identified in both Npc1+/+ and Npc1−/− mice (Fig. 1SM). By contrast, p27Kip1 immunostaining of PN11 cerebella of Npc1−/− mice revealed that the stereotyped and ordered EGL architecture in the inner and outer sub-regions markedly differed between Npc1+/+ and Npc1−/− mice (Fig. 7). Indeed, p27Kip1-immunopositive cells were scattered along the entire EGL and apparently represented the largest population of GNs. Therefore, at PN11, the niche of mitotically active, p27Kip1-negative GNs (identifying the oEGL) of Npc1+/+ mice is strongly reduced or even lost in Npc1−/− mice. Interestingly, HPBCD treatment of Npc1−/− mice also reduced the fraction of p27Kip1-immunopositive cells but did not fully re-established the sharp boundary between the outer and inner EGLs (Fig. 7). A scheme comparing GN proliferation in Npc1+/+ and Npc1−/− EGLs, based on the findings of our study, is shown in Fig. 8.
Fig. 7.

Cerebellar granule neurons of Npc1−/− mice exit the cell cycle earlier than those of wild-type mice. Cerebellar sections of PN11 Npc1+/+ and Npc1−/− mice, either untreated or treated with a single injection of HPBCD at PN7 (3–4 mice/group; 3–4 sections/mouse) were immunostained with anti-p27Kip1 antibodies (brown). Nuclei were stained with methyl green. A representative field of fissure between lobules II and III is shown. Note the presence of scattered p27Kip1-positive GNs along the entire EGL of Npc1−/−. Low and high magnification of the same fields is shown in the right and left panels, respectively. The HPBCD treatment partially re-established the p27Kip1 expression pattern in Npc1−/− mice. Numbers (mean ± SEM) of p27Kip-positive GNs/100 μm2 were: Npc1+/+, 17.20 ± 0.81; Npc1−/−, 21.8 ± 0.79; HPBCD-treated Npc1−/−, 19.20 ± 0.95 (Npc1+/+ vs Npc1−/−, p = 0.03; HPBCD-treated Npc1−/− vs Npc1+/+, p = 0.15; HPBCD-treated Npc1−/− vs Npc1−/−, p = 0.09). oEGL: outer External Granule Layer; iEGL: inner External Granule Layer. Scale bars indicate 50 μm (left) and 25 μm (right).
Fig. 8.

A model of defective GN proliferation in the Npc1−/−.
Discussion
This study shows that Npc1 deficiency affects GN expansion during postnatal cerebellum development and that this leads first to a significant reduction of Npc1−/− IGL and finally of the entire cerebellar volume in Npc1−/− adult mice. The reduction of IGL size is also accompanied by a decrease of ML thickness, likely arising from the lower number of GN parallel fibers projecting to this area. Present observations agree with a recent report based on in vivo magnetic resonance that the volumes of several brain regions of Npc1−/− mice, including cerebellum, are smaller than those of age-matched wild-type mice since as early as 3 weeks of age (Totenhagen et al., personal communication).
In contrast to the approximately 16.5-day period of GN proliferation in the post-natal cerebellum of mice, we have found that GN precursors of Npc1−/− mice undergo a premature exit from the cell cycle, giving rise to a shortening of EGL duration in these mice. On the other hand, we have also observed that EGL thickness is normal until PN11 in Npc1−/− mice, meaning that the reduced proliferation of GN precursors does not affect the entire population of GN precursors, which apparently display timely proliferation and cell cycle exit patterns before PN11. In fact, under normal conditions, even in the presence of a consistent population of proliferating GN precursors, a fraction of these cells exit the cell cycle as early as PN4 (Altman and Bayer, 1997, Espinosa and Luo, 2008), indicating that not all GN precursors exit the cell cycle at the same time. Therefore, our observations rule out the possibility that the development of the overall GN precursor population is defective in Npc1−/−, suggesting that only longer-lasting GN precursors undergo a reduction in proliferation activity. This conclusion is supported by the finding that the EGL of PN11 Npc1−/− is consistently enriched with post-mitotic, p27Kip1-positive GNs, distributed along the entire EGL, whereas the EGL of age-matched wild-type mice still hosts a significant fraction of dividing, p27Kip1-negative GNs in the outer EGL. The lack of a boundary between the oEGL and iEGL in PN11 Npc1−/− mice indicates that the function of locally-acting signals (Xenaki et al., 2011) is severely perturbed in the EGL of these mice, likely dysregulating the balance between GN's proliferation and exit from the cell cycle and accounting for the shorter-lasting EGL and the decreased GN's proliferation rate observed in these mice.
An additional, but not alternative, explanation for the shorter lasting EGL of Npc1−/− mice, is that molecular signals sustaining the proliferation of GN precursors become a limiting factor in Npc1−/−. For instance, the constant ratio of 175 GNs for each PC (Wetts and Herrup, 1983) led researchers to postulate that the number of GNs is somehow regulated by the number and the length of time PCs secrete Shh. Such a model, in which the reduced expansion of GNs is consequence of reduced availability of Shh, is in agreement with the reduced level of Shh transcripts that we measured in PN8 and PN12 Npc1−/− mice. This would also explain why only the proliferation activity of later-lasting GN precursors is affected. In fact, while there is no loss of PCs until after P35 (Sarna et al., 2003), PCs of Npc1−/− mice display a significant cholesterol accumulation as early as at PN9 (Reid et al., 2004), making the hypothesis that the Shh signal becomes defective around this stage reasonable. However, in addition to Shh signal generation, mechanisms involved in Shh signal reception might also be involved as they are altered by cholesterol (reviewed in Eaton, 2008), cholesterol being an essential component of lipid raft microdomains containing receptors and/or adhesion molecules. For instance, the hedgehog receptor Patched has recently been implicated in cholesterol transport (Bidet et al., 2011). Key molecules controlling Shh-induced proliferation are expressed in a finely regulated spatio-temporal manner by a sub-population of GNs residing in a region of overlap between the oEGL and iEGL. Among these, TAG1 and F3, belonging to the contactin family, favor GN proliferation and cell cycle exit, respectively (Xenaki et al., 2011). In light of the complex spatial arrangement of GN subtypes at the level of the EGL, our finding that HPBCD treatment partially re-established the p27Kip1 expression pattern in Npc1−/− mice is worth of further investigation.
The very specific developmental defect identified by this study provides novel insights for a better understanding of the cerebellar neuropathology of NPC1 disease. In light of the relevance that the numerical matching between synaptic partners plays in circuit formation (Sotelo, 1990), our observations now raise the question of whether/how the reduction of GN number negatively affects the survival/differentiation of their synaptic partner, the PCs, and thus contributes to their final degeneration. However, although this possibility is attractive, at the moment it is not supported by the present observation that the reduction of GN number occurs similarly in both the anterior and posterior lobules rather than displaying the anterior-to-posterior gradient of PC degeneration typical of Npc1−/− mice (Sarna et al., 2003). The interest in the potential contribution given to PC degeneration by a GN defect(s) has also been raised by a recent study showing that Npc1−/− mice display an enhanced parallel fiber to PC synaptic transmission and deficient long-term depression (LTD) (Sun et al., 2001). As a consequence, the increased glutamatergic transmission and LTD deficiency may cause PC chronic excitotoxicity, finally resulting in neurodegeneration.
Therapies available for NPC1 disease are limited. Among these, the treatment with the cyclic oligomers of 7-glucose HPBCDs was shown to rescue cholesterol defects and extend life in mouse models of the human Niemann–Pick C1 disease (Camargo et al., 2001, Davidson et al., 2009, Ramirez et al., 2010). This correction requires the endocytosis of the drug (Rosenbaum et al., 2010) and its action is quite similar to that of NPC2 (McCauliff et al., 2011). Here we have shown that a single subcutaneous HPBCD injection given at PN7 rescues the GN expansion defect of Npc1−/−, re-establishing normal patterns of EGL thickness and also increasing the total number of GNs compared to age-matched wild-type mice. In vitro studies (Peake and Vance, 2012) have also shown the efficacy of HPBCD in correcting the cholesterol storage abnormality in GNs. In vivo, a single HPBCD injection starting at PN7 Npc1−/− is a frequently used treatment, even though, more recently, the continuous intraventricular administration of HPBCDs in adult Npc1−/− mice has also been exploited resulting in the maintenance of cerebellar structure if started at day 7 (Aqul et al., 2011). Interestingly, our results show that a single HPBCD treatment apparently counteracts the premature cell cycle exit of GN precursors, but has a limited efficacy in re-establishing the sharp boundary between the outer and inner EGL typical of the normally developing cerebellum. The ability of HPBCD to rescue the developmental defects of Npc1−/− GNs can be explained by several mechanisms. Sonic hedgehog (Shh) contains covalently-bonded cholesterol and either the generation or the transduction of this signaling could be defective (Incardona and Eaton, 2000). Cholesterol is needed for the transition of cells from G1 to S phase (Singh et al., 2013) and a relative deficiency of cholesterol at the correct sites in the dividing GNs could slow their divisions.
The findings of this study pose the question as to whether, besides the cerebellum, neuron generation is also affected in additional brain regions, either during embryonic or postnatal life, as also suggested by a study showing that Npc1 deficiency impairs the self-renewal ability of neural stem cells (Yang et al., 2006). Increasing knowledge on this specific issue may contribute to the identification of novel strategies for NPC1 disease treatment in our species.
Conflict of interest
The authors declare that they have no conflict of interest.
The following is the supplementary data related to this article.
P27Kip1 immunostaining of cerebellar sections of Npc1 +/+ and −/− mice showing that, Npc1 −/− mice display well defined inner and outer EGLs, similar to those of Npc1 +/+ mice. oEGL, outer External Granular Layer; iEGL, inner External Granular Layer. Scale bars indicate 50 μm (panels) and 25 μm (inserts).
Acknowledgment
The financial supports of Telethon Foundation — Italy (grant no. GGP13183 to M.T.F.) and the Ateneo La Sapienza (C26V127RC3) are gratefully acknowledged.
References
- Adams N.C., Comoda T., Cooper M., Dietz G., Hatten M.E. Mice that lack astrotactin have slowed neuronal migration. Development. 2002;129:965–972. doi: 10.1242/dev.129.4.965. [DOI] [PubMed] [Google Scholar]
- Altman J., Bayer S.A. CRC; Boca Raton, FL: 1997. Development of the Cerebellar System in Relation to its Evolution, Structure and Function. [Google Scholar]
- Aqul A., Liu B., Ramirez C.M., Pieper A.A., Estill S.J., Burns D.K., Liu B., Repa J.J., Turley S.D., Dietschy J.M. Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment. J. Neurosci. 2011;31:9404–9413. doi: 10.1523/JNEUROSCI.1317-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argenti B., Gallo R., Di Marcotullio L., Ferretti E., Napolitano M., Canterini S., De Smaele E., Greco A., Fiorenza M.T., Maroder M., Screpanti I., Alesse E., Gulino A. Hedgehog antagonist REN(KCTD11) regulates proliferation and apoptosis of developing granule cell progenitors. J. Neurosci. 2005;25:8338–8346. doi: 10.1523/JNEUROSCI.2438-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banati R.B., Graeber M.B. Surveillance, intervention and cytotoxicity: is there a protective role of microglia? Dev. Neurosci. 1994;16:114–127. doi: 10.1159/000112098. [DOI] [PubMed] [Google Scholar]
- Baudry M., Yao Y., Simmons D., Liu J., Bi X. Postnatal development of inflammation in a murine model of Niemann–Pick type C disease: immunohistochemical observations of microglia and astroglia. Exp. Neurol. 2003;184:887–903. doi: 10.1016/S0014-4886(03)00345-5. [DOI] [PubMed] [Google Scholar]
- Bidet M., Joubert O., Lacombe B., Ciantar M., Nehmé R., Mollat P., Brétillon L., Faure H., Bittman R., Ruat M., Mus-Veteau I. The hedgehog receptor patched is involved in cholesterol transport. PLoS One. 2011;6:e23834. doi: 10.1371/journal.pone.0023834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbon I., Totenhagen J., Fiorenza M.T., Canterini S., Ke W., Trouard T., Erickson R.P. Niemann–Pick C1 mice, a model of “juvenile Alzheimer's disease”, with normal gene expression in neurons and fibrillary astrocytes show long term survival and delayed neurodegeneration. J. Alzheimers Dis. 2012;30:875–887. doi: 10.3233/JAD-2012-120199. [DOI] [PubMed] [Google Scholar]
- Camargo F., Erickson R.P., Garver W.S., Hossain G.S., Carbone P.N., Hedienreich R.A., Blanchard J. Cyclodextrins in the treatment of a mouse model of Niemann–Pick C disease. Life Sci. 2001;70:131–142. doi: 10.1016/s0024-3205(01)01384-4. [DOI] [PubMed] [Google Scholar]
- Chen G., Li H.M., Chen Y.R., Gu X.S., Duan S. Decreased estradiol release from astrocytes contributes to the neurodegeneration in a mouse model of Niemann–Pick disease type C. Glia. 2007;55:1509–1518. doi: 10.1002/glia.20563. [DOI] [PubMed] [Google Scholar]
- Cheruku S.R., Xu Z., Dutia R., Lobel P., Storch J. Mechanism of cholesterol transfer from the Niemann–Pick type C2 protein to model membranes supports a role in lysosomal cholesterol transport. J. Biol. Chem. 2006;281:31594–31604. doi: 10.1074/jbc.M602765200. [DOI] [PubMed] [Google Scholar]
- Dahmane N., Ruiz I., Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126:3089–3100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- Davidson C.D., Ali N.F., Micsenyi M.C., Stephney G., Renault S., Dobrenis K., Ory D.S., Vanier M.T., Walkley S.U. Chronic cyclodextrin treatment of murine Niemann–Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One. 2009;4:e6951. doi: 10.1371/journal.pone.0006951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deffieu M.S., Pfeffer S.R. Niemann–Pick type C function requires luminal domain residues that mediate cholesterol-dependent NPC2 binding. Proc. Natl. Acad. Sci. U. S. A. 2011;108:18932–18936. doi: 10.1073/pnas.1110439108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton S. Multiple roles for lipids in the Hedgehog signalling pathway. Nat. Rev. Mol. Cell Biol. 2008;9:437–445. doi: 10.1038/nrm2414. [DOI] [PubMed] [Google Scholar]
- Elrick M.J., Pacheco C.D., Yu T., Dadgar N., Shakkottai V.G., Ware C., Paulson H.L., Lieberman A.P. Conditional Niemann–Pick C mice demonstrate cell autonomous Purkinje cell neurodegeneration. Hum. Mol. Gen. 2010;19:837–847. doi: 10.1093/hmg/ddp552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa J.S., Luo L. Timing neurogenesis and differentiation: insights from quantitative clonal analyses of cerebellar granule cells. J. Neurosci. 2008;28:2301–2312. doi: 10.1523/JNEUROSCI.5157-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garver W.S., Heidenreich R.A., Erickson R.P., Thomas M.A., Wilson J.M. Localization of the murine Niemann–Pick C1 protein to two distinct intracellular compartments. J. Lipid Res. 2000;41:673–687. [PubMed] [Google Scholar]
- Higashi A., Murayama S., Pentchev P.G., Suzuki K. Cerebellar degeneration in the Niemann–Pick type C mouse. Acta Neuropathol. 1993;85:175–184. doi: 10.1007/BF00227765. [DOI] [PubMed] [Google Scholar]
- Higgins M.E., Davies J.P., Chen F.W., Ioannou Y.A. Niemann–Pick C1 is a late endosome-resident protein that transiently associates with lysosomes and the trans-Golgi network. Mol. Genet. Metab. 1999;68:1–13. doi: 10.1006/mgme.1999.2882. [DOI] [PubMed] [Google Scholar]
- Incardona J.P., Eaton S. Cholesterol in signal transduction. Curr. Opin. Cell Biol. 2000;12:193–203. doi: 10.1016/s0955-0674(99)00076-9. [DOI] [PubMed] [Google Scholar]
- Infante R.E., Wang M.L., Radhakrishnan A., Kwon H.J., Brown M.S., Goldstein J.L. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl. Acad. Sci. U. S. A. 2008;105:15287–15292. doi: 10.1073/pnas.0807328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko D.C., Milenkovic L., Beier S.M., Manuel H., Buchanan J., Scott M.P. Cell-autonomous death of cerebellar Purkinje neurons with autophagy in Niemann–Pick type C disease. PLoS Genet. 2005;1:81–95. doi: 10.1371/journal.pgen.0010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H.J., Abi-Mosleh L., Wang M.L., Deisenhofer J., Goldstein J.L., Brown M.S., Infante R.E. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137:1213–1224. doi: 10.1016/j.cell.2009.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis D.M.D., Weinstein L.A., Halperin J.J. Development of synaptic junctions in cerebellar glomeruli. Brain Res. 1983;284:231–245. doi: 10.1016/0165-3806(83)90008-1. [DOI] [PubMed] [Google Scholar]
- Loftus S.K., Morris J.A., Carstea E.D., Gu J.Z., Cummings C., Brown A., Ellison J., Ohno K., Rosenfeld M.A., Tagle D.A., Pentchev P.G., Pavan W.J. Murine model of Niemann–Pick C disease: mutation in a cholesterol homeostasis gene. Science. 1997;277:232–235. doi: 10.1126/science.277.5323.232. [DOI] [PubMed] [Google Scholar]
- Lopez M.E., Klein A.D., Dimbil U.J., Scott M.P. Anatomically defined neuron-based rescue of neurodegenerative Niemann–Pick type C disorder. J. Neurosci. 2011;31:4367–4378. doi: 10.1523/JNEUROSCI.5981-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauliff L.A., Xu Z., Storch J. Sterol transfer between cyclodextrin and membranes: similar but not identical mechanism to NPC2-mediated cholesterol transfer. Biochemistry. 2011;50:7341–7349. doi: 10.1021/bi200574f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa K., Himi T., Garcia V., Yamagishi H., Sato S., Ishizaki Y. A role for p27/Kip1 in the control of cerebellar granule cell precursor proliferation. J. Neurosci. 2000;20:5756–5763. doi: 10.1523/JNEUROSCI.20-15-05756.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan S.C., Taylor D.L., Pocock J.M., Tau S.C. Microglia release activators of neuronal proliferation mediated by activation of mitogen-activated protein kinase, phosphatidylinositol-3-kinase/Akt and delta-Notch signalling cascades. J. Neurochem. 2004;90:89–101. doi: 10.1111/j.1471-4159.2004.02461.x. [DOI] [PubMed] [Google Scholar]
- Morris M.D., Bhuvaneswaran C., Shio H., Fowler S. Lysosome lipid storage in NCTR-BALBc mice. I. Description of the disease and genetics. Am. J. Pathol. 1982;108:140–149. [PMC free article] [PubMed] [Google Scholar]
- Neufeld E.B., Wastney M., Patel S., Suresh S., Cooney A.M., Dwyer N.K., Roff C.F., Ohno K., Morris J.A., Carstea E.D. The Niemann–Pick C1 protein resides in a vesicular compartment linked to retrograde transport of multiple lysosomal cargo. J. Biol. Chem. 1999;274:9627–9635. doi: 10.1074/jbc.274.14.9627. [DOI] [PubMed] [Google Scholar]
- Ong W.Y., Kumar U., Switzer R.C., Sidhu A., Suresh G., Hu C.Y., Patel S.C. Neurodegeneration in Niemann–Pick type C disease mice. Exp. Brain Res. 2001;141:218–231. doi: 10.1007/s002210100870. [DOI] [PubMed] [Google Scholar]
- Peake K.B., Vance J.E. Normalization of cholesterol homeostasis by 2-hydroxypropyl-β-cyclodextrin in neurons and glia from Niemann–Pick C1 (NPC1)-deficient mice. J. Biol. Chem. 2012;287:9290–9298. doi: 10.1074/jbc.M111.326405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters R.P., Hernandez A., Ng L., Ma M., Sharlin D.S., Pandey M., Simonds W.F., St. Germain D.L., Forrest D. Cerebellar abnormalities in mice lacking type 3 deiodinase and partial reversal of phenotype by deletion of thyroid hormone receptor alpha1. Endocrinology. 2013;154:550–561. doi: 10.1210/en.2012-1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentchev P.G., Comly M.E., Kruth H.S., Patel S., Proestel M., Weintroub H. The cholesterol storage disorder of the mutant BALB/c mouse. A primary genetic lesion closely linked to defective esterification of exogenously derived cholesterol and its relationship to human type C Niemann–Pick disease. J. Biol. Chem. 1986;261:2772–2777. [PubMed] [Google Scholar]
- Ramirez C.M., Liu B., Taylor A.M., Repa J.J., Burns D.K., Weinberg A.G., Turley S.D., Dietschy J.M. Weekly cyclodextrin administration normalizes cholesterol metabolism in nearly every organ of the Niemann–Pick type C1 mouse and markedly prolongs life. Pediatr. Res. 2010;68:309–315. doi: 10.1203/PDR.0b013e3181ee4dd2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid P.C., Sakashita N., Sugli S., Ohno-Iwashita Y., Shimada Y., Hickey W.F., Chang T.Y. A novel cholesterol stain reveals early neuronal cholesterol accumulation in the Niemann–Pick type C1 mouse brain. J. Lipid Res. 2004;45:582–591. doi: 10.1194/jlr.D300032-JLR200. [DOI] [PubMed] [Google Scholar]
- Rosenbaum A.I., Zhang Z., Warren J.D., Maxfield F.R. Endocytosis of beta-cyclodextrins is responsible for cholesterol reduction in Niemann–Pick type C mutant cells. Proc. Natl. Acad. Sci. U. S. A. 2010;107:5477–5482. doi: 10.1073/pnas.0914309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz I., Altaba A., Palma V., Dahmane N. Hedgehog–Gli signalling and the growth of the brain. Nat. Rev. Neurosci. 2002;3:24–33. doi: 10.1038/nrn704. [DOI] [PubMed] [Google Scholar]
- Sarna J.R., Larouche M., Marzban H., Sillitoe R.V., Rancourt D.E., Hawkes R. Patterned of Purkinje cell degeneration in mouse models of Niemann–Pick type C disease. J. Comp. Neurol. 2003;456:279–291. doi: 10.1002/cne.10522. [DOI] [PubMed] [Google Scholar]
- Singh P., Saxena R., Srinivas G., Pande G., Chattopadhyay A. Cholesterol biosynthesis and homeostasis in regulation of the cell cycle. PLoS One. 2013;8:e58833. doi: 10.1371/journal.pone.0058833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleat D.E., Wiseman J.A., El-Banna M., Price S.M., Verot L., Shen M.M., Tint G.S., Vanier M.T., Walkley S.U., Lobel P. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc. Natl. Acad. Sci. U. S. A. 2004;101:5886–5891. doi: 10.1073/pnas.0308456101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solecki D.J., Liu X.L., Comoda F., Fang Y., Hatten M.E. Activated Notch2 signaling inhibits differentiation of cerebellar granule neuron precursors by maintaining proliferation. Neuron. 2001;31:557–568. doi: 10.1016/s0896-6273(01)00395-6. [DOI] [PubMed] [Google Scholar]
- Sotelo C. Cerebellar synaptogenesis: what we can learn from mutant mice. J. Exp. Biol. 1990;153:225–249. doi: 10.1242/jeb.153.1.225. [DOI] [PubMed] [Google Scholar]
- Sotelo C. Cellular and genetic regulation of the development of the cerebellar system. Prog. Neurobiol. 2004;72:295–339. doi: 10.1016/j.pneurobio.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Sun X., Marks D.L., Park W.D., Wheatley C.L., Puri V., O'Brien J.F., Kraft D.L., Lundquist P.A., Patterson M.C., Pagano R.E., Snow K. Niemann–Pick C variant detection by altered sphingolipid trafficking and correlation with mutations within a specific domain of NPC1. Am. J. Hum. Genet. 2001;68:1361–1372. doi: 10.1086/320599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka J., Nakamura H., Miyawaki S. Cerebellar involvement in murine sphingomyelinosis: a new model of Niemann–Pick disease. J. Neuropathol. Exp. Neurol. 1988;47:291–300. doi: 10.1097/00005072-198805000-00008. [DOI] [PubMed] [Google Scholar]
- Vaillant C., Monard D. SHH pathway and cerebellar development. Cerebellum. 2009;8:291–301. doi: 10.1007/s12311-009-0094-8. [DOI] [PubMed] [Google Scholar]
- Vanier M.T. Niemann–Pick disease type C. Orphanet J. Rare Dis. 2010;5:4–16. doi: 10.1186/1750-1172-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vite C.H., Ding W., Bryan C., O'Donnel P., Cullen K., Aleman D., Haskins M.E., Van Winkle T. Clinical, electrophysiological, and serum biochemical measures of progressive neurological and hepatic dysfunction in feline Niemann–Pick type C disease. Pediatr. Res. 2008;64:544–549. doi: 10.1203/PDR.0b013e318184d2ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace V.A. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr. Biol. 1999;9:445–448. doi: 10.1016/s0960-9822(99)80195-x. [DOI] [PubMed] [Google Scholar]
- Wang V.Y., Zoghbi H.Y. Genetic regulation of cerebellar development. Nat. Rev. Neurosci. 2001;2:484–491. doi: 10.1038/35081558. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya R.-J., Scott M.-P. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- Wetts R., Herrup K. Direct correlation between Purkinje and granule cell number in the cerebella of lurcher chimeras and wild-type mice. Brain Res. 1983;312:41–47. doi: 10.1016/0165-3806(83)90119-0. [DOI] [PubMed] [Google Scholar]
- Xenaki D., Martin I.B., Yoshida L., Ohyama K., Gennarini G., Grumet M., Sakurai T., Furley A.J. F3/contactin and TAG1 play antagonistic roles in the regulation of sonic hedgehog-induced cerebellar granule neuron progenitor proliferation. Development. 2011;138:519–529. doi: 10.1242/dev.051912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.-R., Kim S.-Y., Byun K.-H., Hutchinson B., Lee B.-H., Mikikawa M., Lee Y.-S., Kang K.-S. NPC1 gene deficiency leads to lack of neural stem cell self-renewal and abnormal differentiation through activation of p38 mitogen-activated protein kinase signaling. Stem Cells. 2006;24:292–298. doi: 10.1634/stemcells.2005-0221. [DOI] [PubMed] [Google Scholar]
- Yu T., Lieberman A.P. Npc1 acting in neurons and glia is essential for the formation and maintenance of CNS myelin. PLoS Genet. 2013;9:e1003462. doi: 10.1371/journal.pgen.1003462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T., Shakkottai V.G., Chung C., Lieberman A.P. Temporal and cell-specific deletion establishes that neuronal Npc1 deficiency is sufficient to mediate neurodegeneration. Hum. Mol. Gen. 2011;20:4440–4451. doi: 10.1093/hmg/ddr372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M., Strnatka D., Donohue C., Hallows J.L., Vincent I., Erickson R.P. Astrocyte-only Npc1 reduces neuronal cholesterol and triples life span of Npc1−/− mice. J. Neurosci. Res. 2008;86:2848–2856. doi: 10.1002/jnr.21730. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
P27Kip1 immunostaining of cerebellar sections of Npc1 +/+ and −/− mice showing that, Npc1 −/− mice display well defined inner and outer EGLs, similar to those of Npc1 +/+ mice. oEGL, outer External Granular Layer; iEGL, inner External Granular Layer. Scale bars indicate 50 μm (panels) and 25 μm (inserts).