

Highlights
-
•
The aromatic acid transporter PcaK was recombinantly expressed and purified.
-
•
PcaK is a stable homotrimer in n-dodecyl-β-d-maltoside.
-
•
A reconstituted assay shows asymmetric transport.
-
•
The electrical component of the proton gradient drives transport.
-
•
Unexpectedly, PcaK is active in transporting 2-hydroxybenzoates.
Keywords: Enzyme purification, Recombinant protein expression, Membrane proteins, Membrane transport, Reconstitution of membrane transporters
Abstract
The aromatic acid:H+ symporter family of integral membrane proteins play an important role in the microbial metabolism of aromatic compounds. Here, we show that the 4-hydroxybenzoate transporter from Acinetobacter sp. ADP1, PcaK, can be successfully overexpressed in Escherichia coli and purified by affinity chromatography. Affinity-purified PcaK is a stable, monodisperse homotrimer in the detergent n-dodecyl-β-d-maltopyranoside supplemented with cholesteryl hemisuccinate. The purified protein has α-helical secondary structure and can be reconstituted to a functional state in synthetic proteoliposomes. Asymmetric substrate transport was observed when proteoliposomes were energized by applying an electrochemical proton gradient or a membrane potential (ΔΨ) but not by ΔpH alone. PcaK was selective in transporting 4-hydroxybenzoate and 3,4-dihydroxybenzoate over closely related compounds, confirming previous reports on substrate specificity. However, PcaK also showed an unexpected preference for transporting 2-hydroxybenzoates. These results provide the basis for further detailed studies of the structure and function of this family of transporters.
Introduction
The microbial metabolism of aromatic hydrocarbons is of broad interest. This stems from the discovery of novel enzymes capable of organic transformations, with potential in biotechnology and bioprocessing [1], the drive to develop efficient bioremediation strategies in response to a variety of aromatic pollutants [2], the desire to better appreciate microbial adaptation and diversity [3] and the need to understand the breakdown and utilization of the complex aromatic polymer lignin, which constitutes approximately 25% of the land-based biomass on Earth and is a key component of the carbon cycle [4], [5]. The benzene ring is one of the most abundant chemical structures in the biosphere and aromatic compounds are common microbial growth substrates [3].
The major facilitator superfamily of membrane transporters includes a protein family that have been identified as specific Aromatic Acid:H+ Symporters (AAHS)1 [6]. The AAHS transporters are predicted to be composed of ∼450 amino acids arranged as a bundle of 12 transmembrane α-helices, although very little experimental evidence of protein structure is available. Members of the AAHS family include transporters specific for 4-hydroxybenzoate (4-HB) and 3,4-dihydroxybenzoate/protocatechuate (protein name PcaK), 3-hydroxybenzoate (MhbT), 2,5-dihydroxybenzoate/gentisate (GenK), benzoate (BenK) and 4-hydroxy-3-methoxybenzoate/vanillate (VanK). A BLAST search identifies AAHS homologs across bacterial phyla including the Proteobacteria, Actinobacteria, and Firmicutes suggesting that the transport of aromatic acids is relatively common.
Current evidence for the transport activity and specificity of AAHS transporters comes largely from studies of gain-of-function in recombinant cell lines and loss-of-function in knockout cells [7], [8], [9], [10], [11], [12], [13], [14]. Among the best-studied of the AAHS family is PcaK. A recombinant Escherichia coli cell line expressing PcaK from Pseudomonas putida selectively transported protocatechuate and 4-HB with Vmax for 4-HB of 25 nmol/min/mg protein and KM of 6 μM. Uptake was apparently driven by the transmembrane electrochemical proton gradient independent of ATP [8], [15]. A pcaK− strain of P. putida was deficient in 4-HB transport, and transport by PcaK in rescued cell lines was abolished upon site-directed mutagenesis of amino acid residues that are conserved within AAHS and the wider superfamily [16], [17].
Here, we establish methods for the recombinant expression and purification of PcaK that enable initial studies of structure and function in vitro. We have chosen to study a PcaK homolog from Acinetobacter, a genus of gammaproteobacteria that are ubiquitous in the environment and which are relevant to human disease, biotechnology and ecology. Our results provide some of the first direct evidence of the structure and function of this relatively uncharacterized membrane protein family.
Materials and methods
Materials
Isopropyl-β-d-thiogalactopyranoside (IPTG), Tris(2-carboxyethyl)phosphine (TCEP), n-dodecyl-β-d-maltopyranoside (DDM) and precast SDS–PAGE gels were from Generon. All aromatic acids, l-arabinose, cholesteryl hemisuccinate Tris salt (CHS), 0.4–0.6 mm acid-washed glass beads, valinomycin and Proteinase K-agarose were from Sigma–Aldrich. HisTrap columns, PD-10 and PD SpinTrap G-25 gel filtration columns, Superdex 200 10/300 GL column, size exclusion column standards, and nitrocellulose membrane were from GE Healthcare. Chemiluminescence reagents (LumiGLO) were from Cell Signaling Technologies. E. coli strain BL21-AI, LDS loading buffer, V5-HRP, pyranine and gels and reagents for blue native PAGE were from Life Technologies. Centrifugal concentrators were from Millipore. The detergent compatible Lowry assay was purchased in kit format from ThermoFisher. E. coli polar lipid extract and egg phosphatidylcholine were from Avanti Polar Lipids.
Plasmid construct
cDNA encoding Acinetobacter sp. ADP1 PcaK (NCBI gene ID: 2879440) with a c-terminal V5 epitope (GKPIPNPLLGLDST) was made as a synthetic gene (Eurofins MWG) flanked by restriction sites for NcoI and XhoI. This was cloned by cohesive end ligation into the multiple cloning site of pET28a, in frame with a plasmid-borne His10 tag. The construct was verified by sequencing.
Protein expression
Recombinant PcaK was expressed in E. coli BL21-AI. For small-scale expression cultures, a single colony from an agar plate was used to inoculate 10 ml of LB media supplemented with 25 μg/ml kanamycin. After overnight growth at 37 °C with agitation at 220 rpm, 1 ml of this primary culture was used to inoculate 100 ml of the same media in a baffled conical flask and cultures grown to OD600 of 0.7. Protein expression was induced by the addition of 1 mM IPTG and 0.1% (w/v) l-arabinose and growth continued for 2 h at 37 °C, 220 rpm. Cells were harvested at 3300g, resuspended in 0.5 ml of 50 mM sodium phosphate pH 7.4, 5 mM EDTA, and transferred to a 1.5 ml eppendorf tube with an equivalent volume of 0.4–0.6 mm glass beads. Cell lysates were prepared by 5 cycles of 30 s vortex followed by a 30 s ice rest. A final centrifugation at 13,000g for 1 min was used to pellet beads and unlysed cells and the cell lysate was removed by pipetting. Total lysate protein concentration was determined with the detergent-compatible Lowry assay. For protein purification, the expression protocol was scaled so that the primary culture was 100 ml and induction cultures were a total volume of 6 l. Cells were harvested at 3300g and resuspended in 50–100 ml of 1× phosphate-buffered saline with 5 mM TCEP.
Protein purification
Harvested cells were lysed by one passage through a cell disrupter (Constant Systems) at 25 KPSI. Unlysed cells and large debris were removed by centrifugation at 10,000g for 15 min. Cell membranes were obtained from this lysate by ultracentrifugation at 150,000g for 1 h at 4 °C. The supernatant, corresponding to the cytoplasmic fraction, was removed and pellets were resuspended in 40 ml of 50 mM sodium phosphate pH 7.4, 150 mM NaCl, 5% glycerol, 5 mM TCEP, 2% w/v DDM, 0.05% CHS when included. Solubilization was carried out for 1 h at 4 °C with constant mixing. Insoluble membranes were pelleted at 150,000g, 1 h, 4 °C. The supernatant, corresponding to DDM-soluble membranes, was supplemented with 20 mM imidazole and applied to an equilibrated 1 ml HisTrap column at a flow rate of 1 ml/min via a peristaltic pump. Unbound material flowing through the resin was collected for analysis. The column was washed with 40 volumes of Column Buffer (50 mM sodium phosphate pH 7.4, 150 mM NaCl, 5% glycerol, 5 mM TCEP, 0.05% w/v DDM, 0.01% CHS when included) with 75 mM imidazole before elution in 2.5 column volumes of Column Buffer containing 0.5 M imidazole at a flow rate of 0.15 ml/min. The sample was immediately desalted into Column Buffer using a PD-10 gel filtration column and concentrated in a centrifugal concentrator with 100 kDa molecular weight cutoff. Protein concentration was determined with the detergent-compatible Lowry assay and, for purified protein, with absorbance at 280 nm using a calculated extinction coefficient of 76,890 M−1 cm−1 and a theoretical mass of 52.2 kDa (web.expasy.org/protparam/).
Protein analysis
For SDS–PAGE, samples were diluted into LDS loading buffer, heated to 95 °C for 10 min, and applied to a 12% Tris-Glycine gel. For subsequent Western blotting, transfer to a nitrocellulose membrane was performed in 12 mM Tris, 96 mM Glycine, 20% Methanol, 0.01% SDS, pH 8.3. The nitrocellulose was blocked for 1 h in 1× PBS, 0.05% Tween-20 and 5% low-fat powdered milk. The V5 antibody, directly conjugated to horseradish peroxidase, was introduced at 1:10,000 dilution in the same buffer. Membranes were washed five times in 1× PBS, 0.05% Tween-20 before incubation with commercial enhanced chemiluminescence reagents and imaging on photographic film. For direct immunoblots (“dot blots”) 2 µl of sample was added directly to nitrocellulose membranes by pipetting and allowed to dry for 30 min before blocking and developing as above.
Size exclusion chromatography was carried out on a Superdex S200 10/300 GL column equilibrated in 1.5 column volumes of Column Buffer and loaded with 2 ml of purified protein. The flow rate was 0.5 ml/min at 4 °C and 1 ml fractions were collected.
Circular Dichroism was carried out on an Aviv instrument in a 2 mm path length cell at protein concentration of 1 mg/ml. The approximate α-helical content was determined using Eq. (1) [18], [19]:
| (1) |
Blue native PAGE was performed using a 4–16% gradient gel according to the manufacturers instructions with 0.006% Coomassie G-250 added to the sample before loading.
Multi-angle static light scattering coupled to size-exclusion chromatography (SEC-MALS) was carried out essentially as described [20]. The column was Superdex 200 10/300 GL loaded with 95 µl of PcaK at 3 mg/ml and run at 0.8 ml/min. The buffer was 50 mM sodium phosphate pH 7.4, 150 mM NaCl, 5% glycerol, 5 mM TCEP, 0.03% DDM, 0.003% CHS. Detectors were Wyatt Dawn Heleos II and Optilab rEX. The three-detector method in combination with the software package Astra 6.0.5 (Wyatt Technologies) was used to calculate molecular masses [20]. A BSA standard was used for calibration.
Reconstitution
To form liposomes, E. coli polar lipid mix and egg phosphatidylcholine were dissolved at 3:1 weight ratio in 1:1 chloroform:methanol and allowed to dry. Mixed lipids were resuspended at a final lipid concentration of 40 mg/ml in 5 mM potassium phosphate pH 7.4, 0.25 M K2SO4. The lipid suspension was sonicated in a bath-type sonicator for 15 min before undergoing 5 freeze/thaw cycles. The sonication and freeze/thaw process was repeated. Liposomes were prepared by extrusion to 0.4 µm diameter in a benchtop extruder (Avanti Polar Lipids) at room temperature.
For reconstitution, 500 µl extruded liposomes at 40 mg/ml were mixed with 45 µl of 20% (w/v) sodium cholate and 55 µl of purified and size-exclusion polished PcaK at 1.6–2.0 mg/ml. After incubation at room temperature for 30 min with occasional gentle agitation, cholate was removed with a PD SpinTrap G-25 column. The recovered proteoliposomes were centrifuged at 200,000g for 1 h and the supernatant removed. The liposome pellet was resuspended in 500 µl of 5 mM potassium phosphate pH 7.4, 0.25 M K2SO4, 0.2 mM valinomycin, 1 mM pyranine and frozen. The same protocol was followed for preparing lipid-only controls but DDM/CHS buffer was used in place of protein.
For transport assays, proteoliposome or liposome samples were thawed and diluted 4-fold into either 5 mM potassium phosphate pH 7.4, 0.25 M K2SO4 (influx) or 5 mM potassium phosphate pH 6.0, 0.25 M Na2SO4 and substrate (efflux). Samples were frozen, thawed, and sonicated twice for 5 s with 15 s rest. The freeze/thaw and sonication steps were repeated and external pyranine was removed by gel filtration.
Sucrose flotation assays were carried out on a discontinuous sucrose gradient. Samples in 60 % sucrose were layered with 4.5 volumes of 40% sucrose and 1 volume of buffer. Gradients were centrifuged at 173,000g for 1 h and aliquots removed by pipetting for dot blot analysis as above.
For protease digest assays, 5 mU of Proteinase K-agarose was incubated with 100 μl undiluted proteoliposomes or equivalent volume of protein at 100 μg/ml. Proteolysis was stopped at specified timepoints by pelleting Proteinase K-agarose for 10 s at 13,000g and removing an aliquot of digested protein into SDS–PAGE loading buffer. Samples were boiled before loading on SDS–PAGE.
Influx assay
Substrate influx was initiated by diluting 30 µl of pyranine-loaded proteoliposomes (approximately 1 μg protein) with an internal buffer of 5 mM potassium phosphate pH 7.4 and 0.25 M K2SO4 into 3 ml of 5 mM potassium phosphate pH 6.0, 0.25 M Na2SO4, 0.1 mM 4-hydroxybenzoate. Substrate was prepared either as a 10 mM stock in acetonitrile or directly in the aqueous buffer at low concentrations and allowed to dissolve completely at room temperature for >1 h. The pH-dependent pyranine fluorescence was measured with excitation at 460 nm and emission at 508 nm. Fluorescence was recorded over 5 min with 1 s sampling intervals at 25 °C. Nigericin was added at 1 μM to the assay buffer as required.
Efflux assay
Substrate efflux was established by diluting 30 µl of pyranine-loaded proteoliposomes with an internal buffer of 5 mM potassium phosphate pH 6.0, 0.25 M Na2SO4, 0.1 mM substrate into 3 ml of 5 mM potassium phosphate pH 7.4, 0.25 M K2SO4. Pyranine fluorescence was followed as above.
Results
Protein overexpression
The cDNA for Acinetobacter sp. ADP1 PcaK with a C-terminal V5 epitope and His10 tag (hereafter, PcaK) was cloned into the vector pET28a for recombinant expression in E. coli. Fig. 1 shows the results of a small-scale expression test. Induced cell lines carrying PcaK showed a band upon Western blotting at ∼32 kDa. This is substantially lower than the theoretical weight of the PcaK construct of 52.2 kDa. It was previously shown that [35S]methionine-labeled PcaK runs at a lower apparent molecular weight close to 30 kDa on SDS–PAGE gels [8]. Gel shifting has been observed previously for integral membrane proteins and likely arises from atypical SDS binding and conformational effects [21].
Fig. 1.
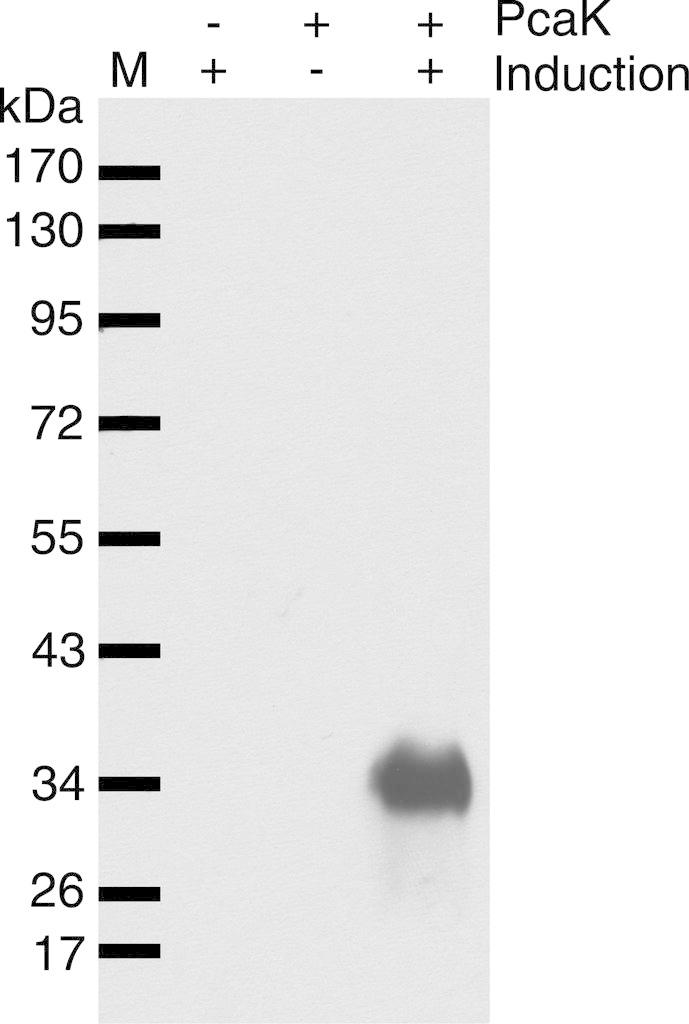
Small-scale expression analysis of recombinant PcaK in Escherichia coli. SDS–PAGE gels were Western blotted and probed with a V5 antibody directed toward an artificial C-terminal V5 epitope. M, molecular weight marker in kDa as shown. 10 μg total protein was loaded in each lane.
Recombinant protein purification
PcaK was purified by immobilized metal affinity chromatography (IMAC) after solubilization in n-dodecyl-β-d-maltopyranoside (DDM) with typical yields approximately 1 mg protein per litre bacterial culture. Cell fractions were analysed by SDS–PAGE. Gel staining with Coomassie brilliant blue (Fig. 2a) confirmed the purification of a single major protein species at ∼32 kDa, with a minor band at 70 kDa. The major band at 32 kDa was excised from the gel and confirmed as PcaK by peptide sequencing. Western blotting against the V5 epitope was used to specifically track PcaK through the purification process and confirmed that the band at 70 kDa was immunopositive and could be assigned to a minor PcaK dimer (Fig. 2b). Collectively, Fig. 2a and b shows that PcaK was targeted to the E. coli plasma membrane upon expression, was successfully solubilized in DDM and was efficiently bound to, and eluted from, the IMAC resin.
Fig. 2.
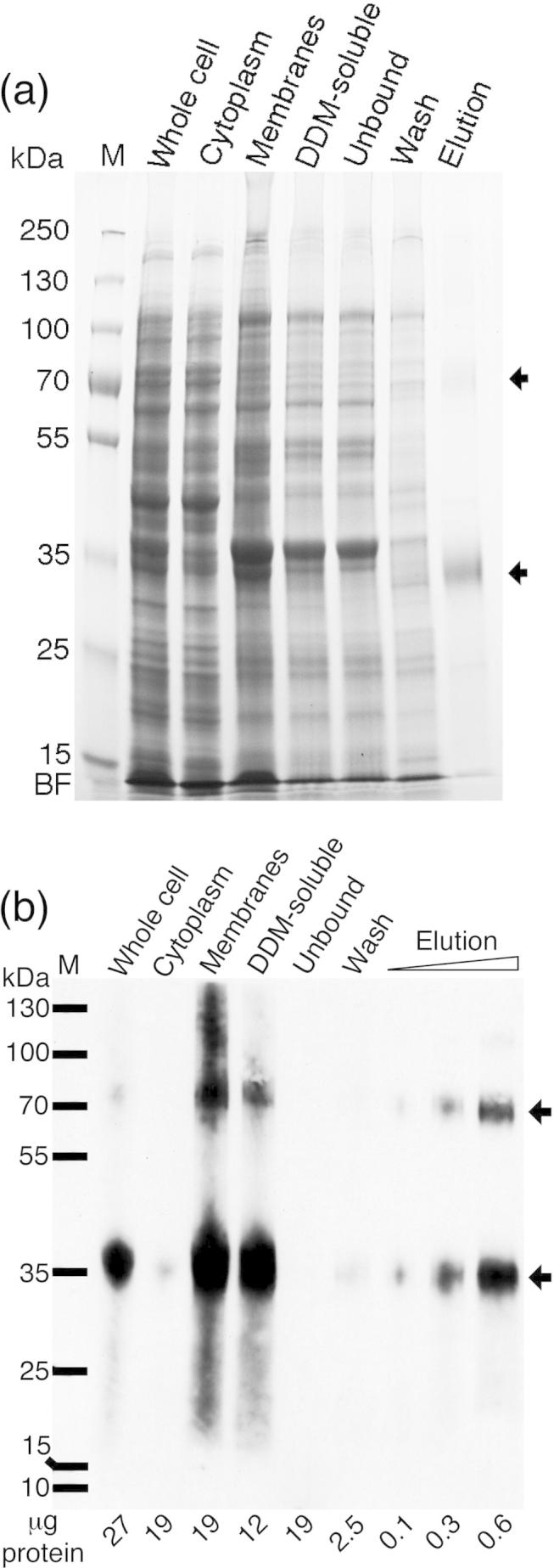
Purification of PcaK. (a) Cell fractions and column purification fractions were loaded onto SDS–PAGE gels and stained with Coomassie. (b) Equivalent gel subject to Western blotting with anti-V5. Purified PcaK migrated as two bands corresponding to the apparent molecular weights of PcaK monomer (32 kDa) and dimer (70 kDa). Fractions are: Whole cell, total cell lysate; Cytoplasm, supernatant after ultracentrifugation; Membranes, pellet after ultracentrifugation; DDM-soluble, membranes solubilized in detergent DDM, supernatant after ultracentrifugation; Unbound, IMAC column flow-through; Wash, column wash fraction; Elution, column eluent. M, molecular weight marker in kDa as shown. μg protein, μg total protein loaded per lane. BF, buffer front.
Protein analysis
Size-exclusion chromatography is a final polishing step in protein purification that additionally provides information on protein purity, monodispersity, and molecular size. When PcaK was purified in DDM a substantial amount of the protein eluted in the column void and the chromatogram was highly heterogenous, probably indicating non-specific protein aggregation (Fig. 3b). The inclusion of cholesteryl hemisuccinate (CHS) in purification buffers substantially improved the performance of PcaK in column chromatography (Fig. 3a). No protein was observed in the void volume in DDM/CHS samples, and a single well-resolved peak was observed at an apparent molecular weight of 180 kDa. After a single pass through the column, fractions underneath the peak were pooled and reapplied to confirm the peak position. Direct immunoblotting (‘dot blot’) of column fractions confirmed that the peak corresponded to V5-tagged PcaK. Size exclusion chromatography thus confirms that IMAC-purified PcaK in DDM/CHS is pure, monodisperse and stable with an apparent molecular weight of 180 kDa.
Fig. 3.
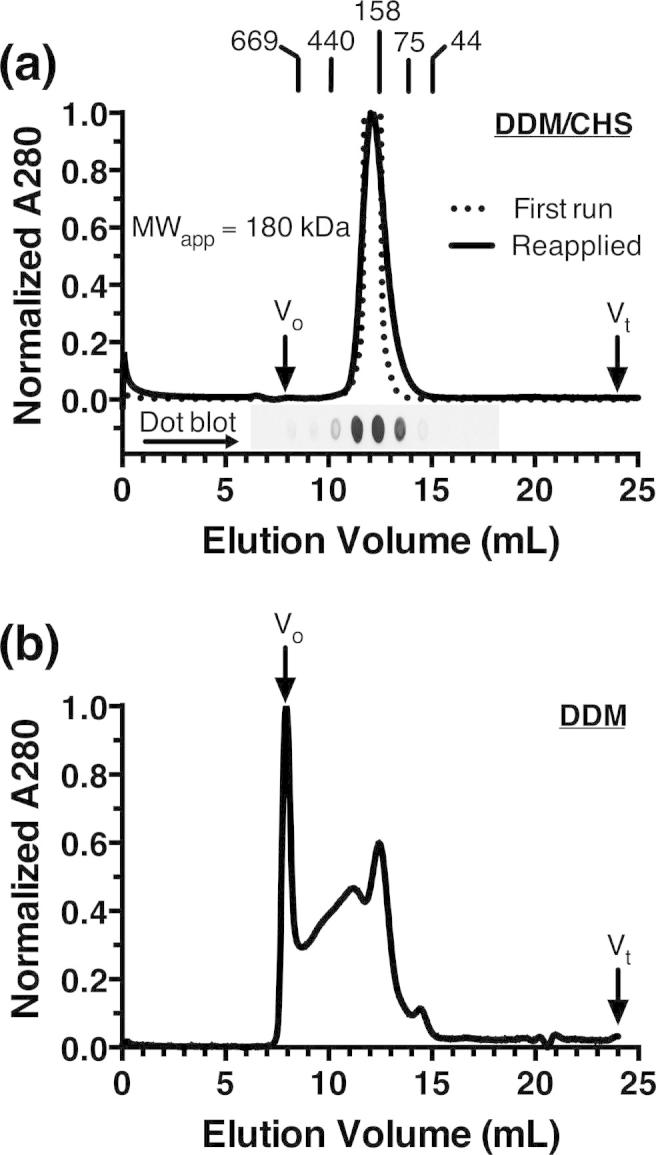
Size-exclusion chromatography of purified PcaK. (a) In DDM/CHS mixed micelles a single well-resolved peak is observed at an apparent molecular weight of 180 kDa relative to known protein standards as shown. Immunoblot of column fractions (‘dot blot’) confirms that the peak corresponds to PcaK. (b) In contrast, chromatograms collected under conditions of DDM alone, without CHS added to purification buffers or the size exclusion buffer, show substantial heterogeneity.
A non-denaturing blue native PAGE gel of PcaK recovered after size exclusion chromatography is shown in Fig. 4. When <7 μg protein was loaded per lane a diffuse smear was observed. However, loading ⩾14 μg per lane resolved a single major band at approximately 180 kDa, in agreement with the apparent molecular weight of the peak observed in size exclusion chromatography. Minor bands are observed at higher molecular weights, most obviously at approximately 250 kDa, which presumably result from protein aggregation in the native PAGE running buffer or differential Coomassie binding.
Fig. 4.
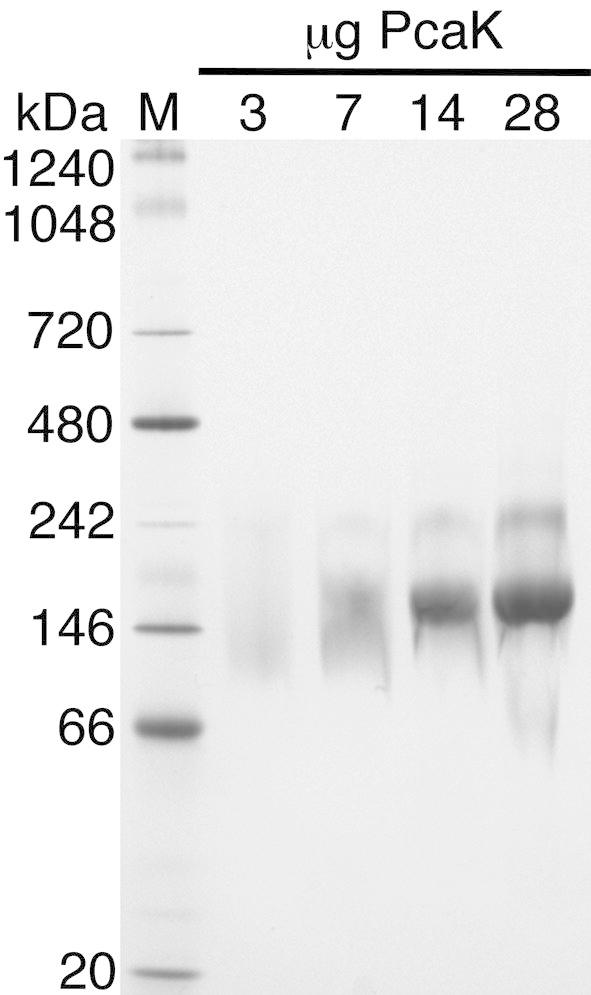
Blue native PAGE gel of PcaK after size exclusion chromatography. Total weight of loaded protein in each lane is shown. When sufficient protein is loaded a single major band is observed at approximately 180 kDa, in agreement with size-exclusion chromatograms. M, molecular weight marker in kDa.
The absolute molecular weight of the protein-detergent complex was determined by multi-angle static light scattering coupled to size-exclusion chromatography (SEC-MALS). The combined results from simultaneous measurements of light scattering, refractive index and absorbance at 280 nm (UV280) are shown in Fig. 5. The protein-detergent complex was confirmed to be monodisperse with Mw/Mn of 1.00 ± 0.01. The protein:detergent mass ratio was 2:1 and the molecular mass of the total complex was found to be 226 ± 2 kDa. Using the three-detector method [20] the molecular mass for the protein component was found to be 152 ± 2 kDa and the micelle mass was 74 ± 2 kDa, in agreement with literature values for DDM [22], [23]. Since the PcaK monomer has a theoretical mass of 52.2 kDa, these results are consistent with the protein being a homotrimer in the protein-detergent complex. These results illustrate the well-known difficulties in estimating the oligomeric state of solubilized membrane proteins using hydrodynamic methods such as native PAGE and SEC [20].
Fig. 5.
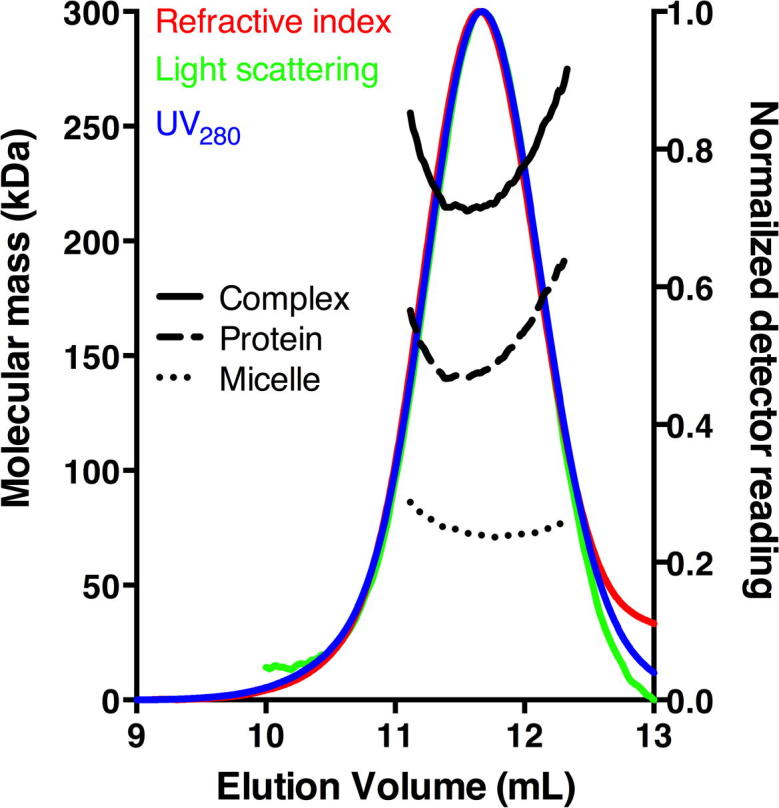
Molecular mass determination from size-exclusion chromatography with in-line multi-angle static light scattering (SEC-MALS). Normalized outputs from refractive index, light scattering and absorbance detectors are overlaid. Black lines show the molecular mass calculations across the peak for the protein-detergent complex (solid, 226 ± 2 kDa), protein component (dashed, 152 ± 2 kDa) and micelle component (dotted, 74 ± 2 kDa).
Sequence-based predictions of the secondary structure of PcaK suggest that it will comprise 12 transmembrane α-helices similar to many other members of the major facilitator superfamily [6]. Circular dichroism (CD) spectroscopy was used to assess the secondary structure content of purified PcaK (Fig. 6). A characteristic α-helical spectrum was observed, with negative deflections in ellipticity at 222 nm and 210 nm. As expected, the CD data show no evidence for any extensive β-structure but allow for the presence of unstructured loops outside the membrane. The magnitude of the 222 nm peak suggests that 45% of amino acids in PcaK will be within α-helical segments (Eq. (1)). This is in reasonable agreement with sequence-based predictions that approximately 252 of 483 amino acids (52%) will be within α-helices.
Fig. 6.
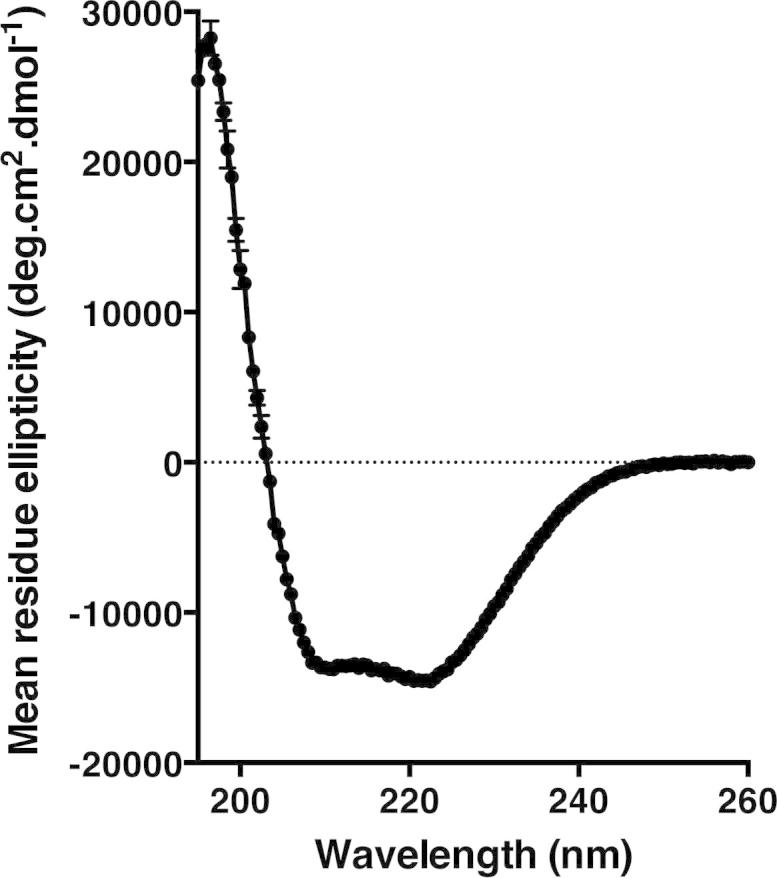
Circular dichroism spectrum of PcaK displaying a classical alpha-helical signature.
Reconstitution into proteoliposomes
PcaK was reconstituted into proteoliposomes by a cholate dilution method as described in Materials and Methods [24], [25], [26]. The migration (‘flotation’) of proteoliposomes on a discontinuous sucrose gradient was used to determine the efficiency of reconstitution. Fig. 7 shows that upon reconstitution, almost all of the protein becomes associated with proteoliposomes and floats to the top of the gradient under centrifugation (Fig. 7, PL, fraction 1). A protein only control (P) confirms the retention of unreconstituted protein at the bottom of the gradient.
Fig. 7.
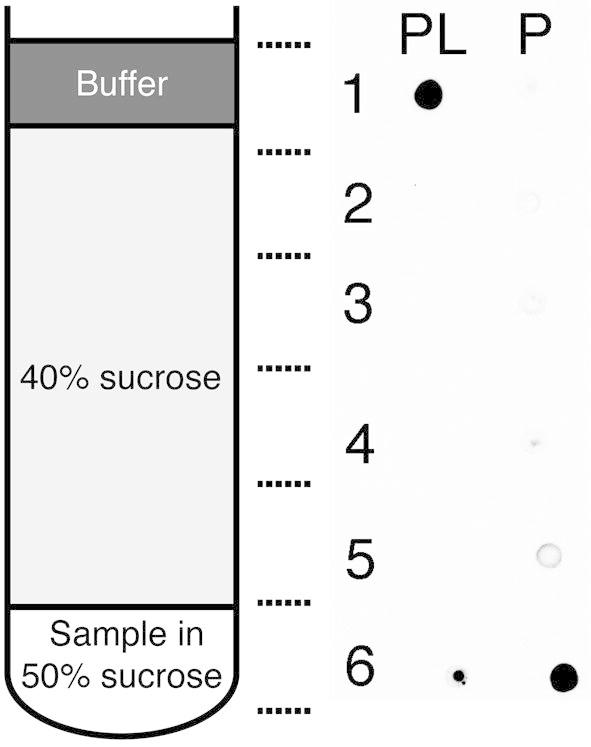
Sucrose flotation assay demonstrating reconstitution of PcaK. Fractions were removed from a discontinuous sucrose gradient as indicated and the presence of PcaK determined by dot blot with anti-V5. Proteoliposomes (PL) migrate to the buffer interface under ultracentrifugation while unreconstituted protein remains at the bottom of the gradient, confirmed by a protein-only control (P).
A proteolytic digest assay was used to determine the orientation of reconstituted PcaK. Samples were incubated with Proteinase K and Western blotted to assess whether the C-terminal V5 epitope had been degraded by the protease. PcaK in DDM/CHS micelles was rapidly digested by Proteinase K, with significant proteolysis within the 10 s deadtime of the experiment (Fig. 8). The band for full-length PcaK was substantially diminished and a partial cleavage product was observed at approximately 20 kDa. Reconstituted PcaK in proteoliposomes was equally susceptible to proteolysis, suggesting that the C-terminal V5 tag is exposed at the liposome exterior rather than being sequestered in the lumen. Amino acid sequence analysis (http://topcons.cbr.su.se) [27] predicts that PcaK will adopt Nin/Cin topology in vivo. PcaK is thus apparently reconstituted in an ‘inside-out’ orientation, whereby the side of the protein that faces the cell interior in vivo faces the liposome exterior in vitro.
Fig. 8.
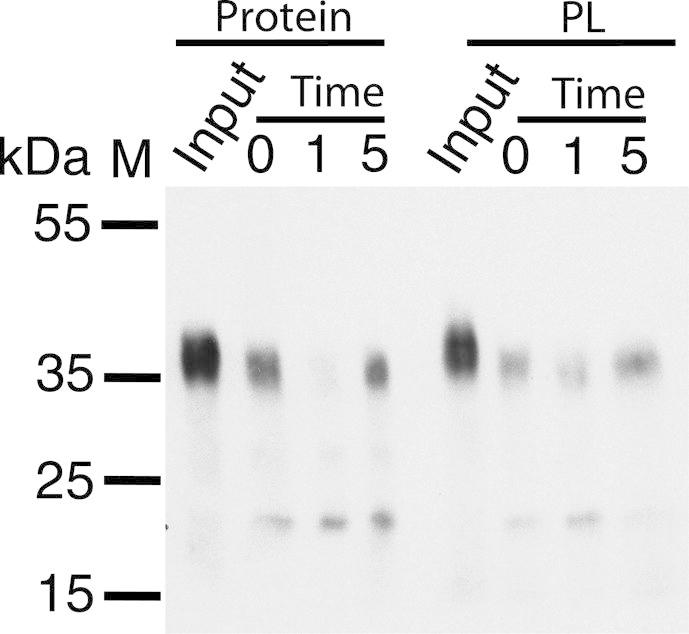
Susceptibility of the C-terminal V5 tag to proteolysis as a measure of protein orientation. Western blotting shows that Proteinase K immediately digests the C-terminal V5 epitope of PcaK in detergent micelles (Protein) and after reconstitution into proteoliposomes (PL), suggesting that the C-terminal of the reconstituted protein is exposed at the proteoliposome exterior. Time in minutes.
Substrate translocation assays
Substrate translocation was measured indirectly by fluorescence of the pH-responsive fluorescent dye pyranine [28], [29], [30]. The dye was efficiently incorporated into proteoliposomes by freeze-thaw and sonication, evidenced by the visible fluorescence from pyranine-containing proteoliposomes. Translocation of 4-HB was initiated as described in Materials and Methods by diluting pyranine-loaded proteoliposomes and liposome controls into buffers that establish either an inward-directed or outward-directed proton gradient . Throughout, the assays were complicated by high background signals arising from the spontaneous diffusion of protons and aromatic acids across the membrane. All experiments described here were carried out at <250 μM substrate to minimize substrate diffusion. For unknown reasons, liposome samples consistently exhibited ∼1.3× higher fluorescence than proteoliposomes.
Fig. 9 shows the results of influx assays under inward-directed (interior alkaline and negative) in the presence of 4-HB at concentrations between 0 and 250 μM. Influx is expected to cause acidification of the lumen, resulting in a decrease in pyranine fluorescence. Experiments in the absence of 4-HB showed a substantial decrease in pyranine fluorescence as a result of proton diffusion across the membrane and subsequent acidification of the lumen (Fig. 9a and b). The relative rate and magnitude of this acidification was not altered by including 4-HB in the external buffer (Fig. 9a). The KM of PcaK for 4-HB was previously determined to be 6 μM [8]; thus the lowest concentration range used here is twice KM. No differences were observed between proteoliposome samples and liposome controls, and representative data from these experiments at 100 μM 4-HB are shown in Fig. 9b. The introduction of the proton ionophore nigericin immediately collapses the proton gradient and abolishes the diffusion signal (Fig. 9b).
Fig. 9.

Influx assays under applied (inside negative and alkaline). (a) Influx assays in proteoliposomes at different concentrations of 4-HB as shown. (b) Comparison of data from proteoliposomes (PL) to control experiments in the absence of protein (L). Traces are the mean of at least two replicates with error bars omitted for clarity.
The data presented in Fig. 9 suggest that reconstituted PcaK is not active as an influx transporter. We thus investigated whether we could establish substrate efflux in our reconstituted system. For efflux assays, proteoliposomes were preloaded with 4-HB and an outward-directed (interior acidic and positive) was applied. Efflux is expected to cause alkalization of the lumen and a corresponding increase in pyranine fluorescence.
Proteoliposomes showed a modest but reproducible increase in the rate of alkalization over liposome controls (Fig. 10a), demonstrating PcaK-catalysed efflux of substrate and H+ above the rate of diffusion. Adjusting buffer conditions as described in Materials and Methods allowed us to dissect the relative contribution of ΔpH and ΔΨ to transport. Fig. 10b and c suggest that ΔΨ alone, but not ΔpH alone, is capable of driving substrate transport in agreement with a previous report [8]. No evidence of substrate efflux was observed in the absence of energetic gradients (Fig. 10d).
Fig. 10.

Substrate efflux (inside positive and acidic). Proteoliposomes, grey smoothed line on black error bars; liposome controls, black smoothed line on grey error bars. Experiments were performed in the presence of (a) , (b) ΔpH only, (c) ΔΨ only or (d) in the absence of any energetic gradient. All traces are mean ± range of at least two replicates after subtracting relevant control experiments absent substrate.
Substrate selectivity
Nichols and Harwood [8] used direct measurements of transport activity, competition experiments and knock-out growth studies to show that PcaK selectively transports 4-HB and protocatechuate. A competition screen of a range of aromatic and non-aromatic compounds suggested that 4-HB transport was partially inhibited by competition with 3-hydroxybenzoate and certain halogenated benzoates but was not substantially inhibited by competition with, among others, 2-hydroxybenzoate/salicylate and vanillate. Benzoate binds competitively to PcaK but is not transported. These results are of interest since benzoate and vanillate are proposed to have their own transporters (BenK and VanK, respectively) and so might not have overlapping specificity with PcaK. We sought to test the substrate specificity of PcaK in our reconstituted assay system, using the most closely related benzoate compounds. Assays were carried out in the presence of ΔΨ only to minimize any background signal from H+ diffusion. The results are shown in Fig. 11.
Fig. 11.

Substrate specificity of reconstituted PcaK under outward-directed ΔΨ. Data are mean ± range of at least two independent experiments with different protein and proteoliposome preparations, relative to a control absent substrate.
Under the relatively permissive conditions of the assay (100 μM each substrate) we observed no significant difference between the transport of 4-HB and protocatechuate. Relative to 4-HB, there was a trend of reduced transport in the series protocatechute > 3-HB > vanillate > gentisate >> benzoic acid. Unexpectedly, there was relatively higher transport of 2,4-dihydroxybenzoate (2,4-DHB) and salicylate was found to be transported most effectively.
Discussion
The genus Acinetobacter are ubiquitous gammaproteobacteria that express at least the four AAHS transporters PcaK, BenK, VanK and MucK enabling the mineralization of aromatic compounds via the β-ketoadipate pathway [31], [32]. The emergence of multidrug-resistant strains of Acinetobacter has become a public health concern [33], [34], [35]. Acinetobacter are also target organisms for bioremediation [2] and industrial biotechnology [36] because of their remarkable metabolic versatility. Understanding the structure and function of archetypal AAHS transporter homologs from Acinetobacter will help to illuminate a previously unstudied transporter family and provide specific insight into the biochemistry of a microorganism that is relevant to human disease, biotechnology and ecology.
The data presented here show that Acinetobacter baylyi PcaK can be recombinantly expressed, purified and reconstituted in vitro. We found that the addition of the cholesterol derivative CHS to purification buffers was essential for stabilizing PcaK. This might arise from specific sterol-protein interactions or from changes to the shape, size or physicochemistry of the micelle. Light scattering and small-angle X-ray scattering data [37] suggest that mixtures of DDM and CHS form bicelle-like discoids that are larger than the oblate ellipsoid micelles formed by DDM alone. This change in micelle structure was proposed to arise largely from the interaction of CHS with the detergent acyl chain region, allowing closer packing and resulting in a thicker hydrophobic core and lower curvature than a DDM micelle. These bicelle-like structures are likely to be closer membrane mimics that can better accommodate the hydrophobic surface of the protein and promote sterol binding to specific protein sites, leading to increased stability and reduced aggregation.
Transport assays presented here suggest that PcaK is functionally asymmetric and prefers to transport 4-HB in a single direction. In reconstituted proteoliposomes, only efflux is observed. Because proteolysis assays suggest that PcaK reconstitutes in an ‘inside-out’ orientation, this corresponds to substrate import being the dominant pathway in vivo. Previous competition assays in the presence of 4-HB [8] suggested that PcaK transports protocatechuate and, with lower efficiency, 3-HB, and that PcaK can bind to, but not transport, benzoate. Of the numerous compounds tested in the prior study, neither salicylate nor vanillate were found to inhibit 4-HB uptake and so were not considered to be substrates for PcaK. In contrast the data presented here suggest that under the relatively high substrate concentrations required for our in vitro assay, PcaK can transport a variety of aromatic acids with hydroxyl substitutions at the 2-, 3- and 4-positions, with the 2- and 4-positions being favoured. The reduced transport of vanillate versus protocatechuate implies that hydroxyl substitutions are generally favoured over methoxyl groups. Surprisingly, hydroxyl substitutions at the 2-position appeared to be an important determinant for transport.
Considering these results, it is of note that a relatively narrow substrate specificity appears to be a feature of the AAHS transporters. P. putida MhbT was found to be selective for 3-HB over 2-HB and 4-HB and is inhibited by, but does not transport, gentisate, with almost no activity toward benzoate and protocatechuate [7]. The relative growth rates of Acinetobacter strains in which benK was disrupted suggested that BenK is specific in transporting benzoate and benzaldehyde but not 4-HB, 2-aminobenzoate or benzyl alcohol [38]. Where it has been determined, KM for AAHS transporters is in the range 5–10 μM [7], [8], [9], [13] consistent with relatively selective substrate binding. We were prevented from studying transport at concentrations closer to values of KM because of decreased signal-to-noise at lower substrate concentrations. To resolve the apparent discrepancy between our results in vitro and cell-based competition assays it will be informative in the future to determine the substrate binding affinity of different aromatic acids in vitro, for example by isothermal titration calorimetry, and to make direct measurements of radiolabelled substrate transport at low concentrations.
We thus present here an experimental platform for studying the structure and function of PcaK in vitro. We anticipate that further work based upon these results will characterize in full the molecular basis for substrate transport and selectivity. The methods described and validated here can potentially be extended for biophysical studies of other transporters within the AAHS family.
Acknowledgments
We thank Heather E. Findlay and Paula J. Booth for the kind gift of expression vector pET28a-His10.
Footnotes
This work was supported by European Research Council Starting Grant 282101 under the European Union’s Seventh Framework Programme (FP7/2007-2013) to P.C. L.S. is supported by Engineering and Physical Sciences Research Council Doctoral Training Centre Grant EP/G036780/1.
Abbreviations used: AAHS, aromatic acid: H+ transporter family; CD, circular dichroism spectroscopy; CHS, cholesteryl hemisuccinate Tris salt; DDM, n-dodecyl-β-d-maltopyranoside; 2,4-DHB, 2,4-dihydroxybenzoate; 4-HB, 4-hydroxybenzoate; 3-HB, 3-hydroxybenzoate; IMAC, immobilized metal affinity chromatography; PAGE, polyacrylamide gel electrophoresis; SEC-MALS, size exclusion chromatography coupled to multi-angle laser light scattering.
References
- 1.Gibson D.T., Parales R.E. Aromatic hydrocarbon dioxygenases in environmental biotechnology. Curr. Opin. Biotechnol. 2000;11:236–243. doi: 10.1016/s0958-1669(00)00090-2. [DOI] [PubMed] [Google Scholar]
- 2.Cao B., Nagarajan K., Loh K.-C. Biodegradation of aromatic compounds: current status and opportunities for biomolecular approaches. Appl. Microbiol. Biotechnol. 2009;85:207–228. doi: 10.1007/s00253-009-2192-4. [DOI] [PubMed] [Google Scholar]
- 3.Fuchs G., Boll M., Heider J. Microbial degradation of aromatic compounds – from one strategy to four. Nat. Rev. Microbiol. 2011;9:803–816. doi: 10.1038/nrmicro2652. [DOI] [PubMed] [Google Scholar]
- 4.Kirk T.K., Farrell R.L. Enzymatic “combustion”: the microbial degradation of lignin. Annu. Rev. Microbiol. 1987;41:465–505. doi: 10.1146/annurev.mi.41.100187.002341. [DOI] [PubMed] [Google Scholar]
- 5.Bugg T.D.H., Ahmad M., Hardiman E.M., Rahmanpour R. Pathways for degradation of lignin in bacteria and fungi. Nat. Prod. Rep. 2011;28:1883–1896. doi: 10.1039/c1np00042j. [DOI] [PubMed] [Google Scholar]
- 6.Pao S., Paulsen I.T., Saier M.H. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 1998;62:1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu Y., Gao X., Wang S.-H., Liu H., Williams P.A., Zhou N.-Y. MhbT is a specific transporter for 3-hydroxybenzoate uptake by gram-negative bacteria. Appl. Environ. Microbiol. 2012;78:6113–6120. doi: 10.1128/AEM.01511-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nichols N.N., Harwood C.S. PcaK, a high-affinity permease for the aromatic compounds 4-hydroxybenzoate and protocatechuate from Pseudomonas putida. J. Bacteriol. 1997;179:5056–5061. doi: 10.1128/jb.179.16.5056-5061.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leveau J.H.J., Zehnder A.J.B., van der Meer J.R. The tfdk geneproduct faciliates uptake of 2,4-dichlorophenoxyacetate by Ralstonia eutropha JMP134(pJP4) J. Bacteriol. 1998;180:2237–2243. doi: 10.1128/jb.180.8.2237-2243.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaudry M.T., Huang Y., Shen X.-H., Poetsch A., Jiang C.-Y., Liu S.-J. Genome-wide investigation of aromatic acid transporters in Corynebacterium glutamicum. Microbiology. 2007;153:857–865. doi: 10.1099/mic.0.2006/002501-0. [DOI] [PubMed] [Google Scholar]
- 11.Merkens H., Beckers G., Wirtz A., Burkovski A. Vanillate metabolism in Corynebacterium glutamicum. Curr. Microbiol. 2005;51:59–65. doi: 10.1007/s00284-005-4531-8. [DOI] [PubMed] [Google Scholar]
- 12.Xu Y., Chen B., Chao H., Zhou N.-Y. MhpT enodes an active transporter involved in 3-(3-hydroxyphenyl)propionate catabolism by Escherichia coli K-12. Appl. Environ. Microbiol. 2013;79:6362–6368. doi: 10.1128/AEM.02110-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu Y., Wang S.H., Chao H.J., Liu S.J., Zhou N.-Y. Biochemical and molecular characterization of the gentisate transporter GenK in Corynebacterium glutamicum. PLoS One. 2012;7:e38701. doi: 10.1371/journal.pone.0038701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams P.A., Shaw L.E. MucK, a gene in Acinetobacter calcoaceticus ADP1 (BD413), encodes the ability to grow on exogenous cis, cis-muconate as the sole carbon source. J. Bacteriol. 1997;179:5935. doi: 10.1128/jb.179.18.5935-5942.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harwood C.S., Nichols N.N., Kim M.K., Ditty J.L., Parales R.E. Identification of the pcaRKF gene cluster from Pseudomonas putida: involvement in chemotaxis, biodegradation, and transport of 4-hydroxybenzoate. J. Bacteriol. 1994;176:6479–6488. doi: 10.1128/jb.176.21.6479-6488.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ditty J.L., Harwood C.S. Conserved cytoplasmic loops are important for both the transport and chemotaxis functions of PcaK, a protein from Pseudomonas putida with 12 membrane-spanning regions. J. Bacteriol. 1999;181:5068–5074. doi: 10.1128/jb.181.16.5068-5074.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ditty J.L., Harwood C.S. Charged amino acids conserved in the aromatic acid/H+ symporter family of permeases are required for 4-hydroxybenzoate transport by PcaK from Pseudomonas putida. J. Bacteriol. 2002;184:1444–1448. doi: 10.1128/JB.184.5.1444-1448.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morrow J.A., Segall M.L., Lund-Katz S., Phillips M.C., Knapp M., Rupp B., Weisgraber K.H. Differences in stability among the human apolipoprotein E isoforms determined by the amino-terminal domain. Biochemistry. 2000;39:11657–11666. doi: 10.1021/bi000099m. [DOI] [PubMed] [Google Scholar]
- 19.MacRaild C.A., Hatters D.M., Howlett G.J., Gooley P.R. NMR structure of human apolipoprotein C-II in the presence of sodium dodecyl sulfate. Biochemistry. 2001;40:5414–5421. doi: 10.1021/bi002821m. [DOI] [PubMed] [Google Scholar]
- 20.Slotboom D.J., Duurkens R.H., Olieman K., Erkens G.B. Static light scattering to characterize membrane proteins in detergent solution. Methods. 2008;46:73–82. doi: 10.1016/j.ymeth.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 21.Rath A., Glibowicka M., Nadeau V.G., Chen G., Deber C.M. Detergent binding explains anomalous SDS–PAGE migration of membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 2009;106:1760–1765. doi: 10.1073/pnas.0813167106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strop P., Brunger A.T. Refractive index-based determination of detergent concentration and its application to the study of membrane proteins. Protein Sci. 2005;14:2207–2211. doi: 10.1110/ps.051543805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oliver R.C., Lipfert J., Fox D.A., Lo R.H., Doniach S., Columbus L. Dependence of micelle size and shape on detergent alkyl chain length and head group. PLOS One. 2013;8:e62488. doi: 10.1371/journal.pone.0062488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rigaud J.L., Levy D. Reconstitution of membrane proteins into liposomes. Methods Enzymol. 2003;372:65–86. doi: 10.1016/S0076-6879(03)72004-7. [DOI] [PubMed] [Google Scholar]
- 25.Lee C., Kang H.J., von Ballmoos C., Newstead S., Uzdavinys P., Dotson D.L., Iwata S., Beckstein O., Cameron A.D., Drew D. A two-domain elevator mechanism for sodium/proton antiport. Nature. 2013;501:573–577. doi: 10.1038/nature12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Page M.G.P., Rosenbusch J.P., Yamato I. The effects of pH on proton sugar symport activity of the lactose permease purified from Escherichia coli. J. Biol. Chem. 1988;263:15897–15905. [PubMed] [Google Scholar]
- 27.Bernsel A., Viklund H., Hennerdal A., Elofsson A. TOPCONS: consensus prediction of membrane protein topology. Nucleic Acids Res. 2009;37:W465–W468. doi: 10.1093/nar/gkp363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seigneuret M., Rigaud J.-L. Use of the fluorescent pH probe pyranine to detect heterogenous directions of proton movement in bacteriorhodopsin reconstituted large liposomes. FEBS Lett. 1985;188:101–106. [Google Scholar]
- 29.Kano K., Fendler J.H. Pyranine as a sensitive pH probe for liposomes interiors and surfaces. pH gradients across phospholipid vesicles. Biochim. Biophys. Acta. 1978;509:289–299. doi: 10.1016/0005-2736(78)90048-2. [DOI] [PubMed] [Google Scholar]
- 30.Clement N.R., Gould J.M. Pyranine (8-hydroxyl-1,3,6-pyrenetrisulfonate) as a probe of internal aqueous hydrogen ion concentration in phospholipid vesicles. Biochemistry. 1981;20:1534–1538. doi: 10.1021/bi00509a019. [DOI] [PubMed] [Google Scholar]
- 31.Barbe V., Vallenet D., Fonknechten N., Kreimeyer A., Oztas S., Labarre L., Cruveiller S., Robert C., Duprat S., Wincker P., Ornston L.N., Weissenbach J., Marlière P., Cohen G.N., Médigue C. Unique features revealed by the genome sequence of Acinetobacter sp. ADP1, a versatile and naturally transformation competent bacterium. Nucleic Acids Res. 2004;32:5766–5779. doi: 10.1093/nar/gkh910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harwood C.S., Parales R.E. The β-ketoadipate pathway and the biology of self-identity. Annu. Rev. Microbiol. 1996;50:553–590. doi: 10.1146/annurev.micro.50.1.553. [DOI] [PubMed] [Google Scholar]
- 33.Peleg A.Y., Paterson D.L. Multidrug-resistant Acinetobacter: a threat to the antibiotic era. Int. Med. J. 2006;36:479–482. doi: 10.1111/j.1445-5994.2006.01130.x. [DOI] [PubMed] [Google Scholar]
- 34.García-Quintanilla M., Pulido M.R., López-Rojas R., Pachón J., McConnell M.J. Emerging therapies for multidrug resistant Acinetobacter baumannii. Trends Microbiol. 2012;21:157–163. doi: 10.1016/j.tim.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 35.McConnell M.J., Actis L., Pachón J. Acinetobacter baumannii: human infections, factors contributing to pathogenesis and animal models. FEMS Microbiol. Rev. 2013;37:130–155. doi: 10.1111/j.1574-6976.2012.00344.x. [DOI] [PubMed] [Google Scholar]
- 36.Gutnick D.L., Bach H. In: Acinetobacter Molecular Biology. Gerischer U., editor. Caister Scientific Press; Norfolk, UK: 2008. Potential Application of Acinetobacter in Biotechnology. [Google Scholar]
- 37.Thompson A., Liu J., Chun E., Wacker D., Wu H., Cherezov V., Stevens R. GPCR stabilization using the bicelle-like architecture of mixed sterol-detergent micelles. Methods. 2011;55:310–317. doi: 10.1016/j.ymeth.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Collier L.S., Nichols N.N., Neidle E.L. BenK encodes a hydrophobic permease-like protein involved in benzoate degradation by Acinetobacter sp. strain ADP1. J. Bacteriol. 1997;179:5943–5946. doi: 10.1128/jb.179.18.5943-5946.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]