

Abstract
Introduction
Diabetes mellitus (DM) is a prothrombotic and proinflammatory state. Hyperglycemia (HG) is encountered even in patients without DM. We have shown that combined HG and hyperinsulinemia (HI) in healthy non-diabetic subjects increased circulating tissue factor (TF) and thrombin generation. To understand the changes in platelet and monocyte pathways induced by combined HG and HI in healthy non-diabetic state, we performed whole genome expression profiling of leukocyte-depleted platelets and monocytes before and after 24 hours of combined HG (glucose ∼200 mg/dL) and HI by glucose infusion clamp in a healthy non-diabetic subject.
Results
We defined time-dependent differential mRNA expression (24 versus 0 hour fold change (FC) ≥2) common to platelets and monocytes. Ingenuity Pathways Analysis revealed alterations in canonical insulin receptor signaling and coagulation pathways. A preliminary group of 9 differentially expressed genes was selected for qRT-PCR confirmation. Platelet 24 hour sample was compared to the 0 hour sample plus 4 controls. Five transcripts in platelets and 6 in monocytes were confirmed. Platelet GSK3B and PTPN1 were upregulated, and STXBP4 was downregulated in insulin signaling, and F3 and TFPI were upregulated in coagulation pathways. Monocyte, PIK3C3, PTPN11 and TFPI were downregulated. Platelet GSKβ3 and PTPN11 protein and TF antigen in platelets and monocytes was increased.
Conclusions
Even in non-diabetic state, HG+HI for 24 hours induces changes in platelets and monocytes. They suggest downregulation of insulin signaling and upregulation of TF. Further studies are needed to elucidate cellular alterations leading to the prothrombotic and proinflammatory state in DM.
Keywords: Diabetes mellitus, Hyperglycemia-hyperinsulinemia, Platelets, Monocytes, Insulin signaling, Tissue factor
Introduction
Patients with diabetes mellitus (DM) have increased incidence of acute vascular events [1]. This is due in part to a prothrombotic and proinflammatory state in these patients who have elevated plasma coagulation factors and enhanced platelet responsiveness [2-5]. The hallmarks of type 2 DM (T2DM) - hyperglycemia (HG) and hyperinsulinemia (HI) – are both independent risk factors for mortality [6-8]. Our studies in healthy non-diabetic subjects using infusion clamps showed that selective HI and HG, and more so the combination of HI+HG, increased circulating membrane-bound tissue factor-procoagulant activity (TF-PCA), plasma coagulation factor (F) VIII and markers of thrombin generation [9]. In addition, HI+HG induced platelet and monocyte activation, and upregulated monocyte TF [10]. Our studies in type 1 DM and type 2 DM patients [11, 12] showed that TF-PCA in whole blood and FVII in plasma were elevated even under basal conditions.
To obtain insights into the wide-ranging changes that may be induced by HG+HI in platelets and monocytes, and contributing to the prothrombotic and proinflammatory state, we performed expression profiling of leukocyte-depleted platelets and monocytes before and after 24 hour of HG+HI clamping in a healthy non-diabetic subject. This is an unbiased approach likely to yield unanticipated findings. Moreover, alterations induced by acutely elevating glucose may be different from those seen in DM with chronic elevations in glucose and insulin. Acute elevations in glucose are encountered in hospitalized patients, including those without known DM [6-8]. To our knowledge studies on the effects of HG-HI using expression profiling of platelets and monocytes in healthy non-diabetic subjects have not been reported. Our present studies provide evidence that 24 hours of HG+HI, even in the non-diabetic healthy state, induces demonstrable and substantial changes in platelets and monocytes, including alterations in insulin-signaling and coagulation related pathways. They lay the foundation for future detailed studies to explore changes in various cellular pathways in DM.
Methods
Glucose Infusion Study
This study was performed in the General Clinical Research Center in a 42 year old black male after obtaining informed consent and approval by the Institutional Review Board. Plasma glucose was maintained ∼200 mg/dl by glucose infusion as described [9]. This elevates endogenous insulin levels to induce HI, thus creating combined HG and HI, with levels close to those encountered in T2DM. The infusion clamps studies began at ∼8AM after an overnight fast. A 20% glucose solution was infused intravenously and adjusted to maintain plasma glucose at ∼200 mg/dl (∼11 mmol/L). The insulin levels rise to ∼1,000 pmol/L in response to the HG [9]. Small blood samples (0.25 ml) were collected every 30-60 min initially and every 1-2 hours later for measurement of glucose concentrations. Plasma electrolytes were monitored every 6 hours, body weight every 12 hours and fluid balances every 6 hours. Potassium (20 mg) and magnesium (400 mg) were given PO every 12 hours. Blood samples were obtained at baseline and at 24 hours.
Chemicals and Reagents
All chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA). Human CD45 antibody conjugated magnetic microbeads was purchased from Miltenyi Biotec Inc. (Auburn, CA, USA). TRIZOL reagent was from Invitrogen (Carlsbad, CA, USA) and, APC conjugated CD45 and fluorescein isothiocyanate (FITC) conjugated CD41 antibodies were from eBioscience (San Diego, CA, USA).
Platelet and Monocyte Purification
Platelets were purified according to Gnatenko et al [13] with modifications. Whole blood (45 ml) from human subjects drawn into 5 ml of EDTA (100 mM) was incubated with carbacyclin (50 nM) at room temperature and centrifuged at 180g for 20 min to generate platelet rich plasma (PRP). Upper two-thirds of the PRP was passed through Sepharose 2B gel filtration columns equilibrated with HEPES-buffered modified Tyrode's (HBMT) buffer (10 mM HEPES, 150 mM NaCl, 2.5 mM KCl, 0.3 mM NaH2PO4, 12 mM NaHCO3, 0.2% bovine serum albumin, 0.1% glucose, 2mM EDTA). Gel-filtered platelets from the column were centrifuged at 2000g for 8 min. Platelet pellet was gently resuspended in 8 ml of HBMT buffer and labeled with 40 μl of human CD45 antibody conjugated to magnetic microbeads on a rotating platform at 4°C. After 20 min platelet suspension was passed through magnetic separation column on MACS II separator to capture CD45+ cells (leukocytes). The platelet suspension was centrifuged and the pellet was resuspended in HBMT buffer (8 ml). The magnetic labeling and immunodepletion were repeated as described above to get leukocyte-free platelets. The platelets were subjected to centrifugation at 2000g, and the pellet was resuspended in TRIZOL (1ml) reagent for RNA extraction. The final product contained up to 3 leukocytes per 1× 105 platelets as determined by flow cytometric analysis.
Mononuclear cells were isolated from whole blood by the Ficol-Paque TM Plus gradient method according to the manufacturer's instructions (Amersham, Piscataway, NJ). Monocytes were positively selected using CD14 microbeads per 107 mononuclear cell according the manufacturer's protocol (Miltenyi Biotech, Auburn, CA) [9].
Expression Profiling Methods and Data Analysis
We performed expression profiling of platelets and monocytes before and after 24 hour of HG+HI clamping in a healthy non-diabetic subject. The platelets and monocytes were collected and resuspended in TRIzol (Life Technologies, Grand Island, NY). Total RNA was isolated using the RNA Clean-Up and Concentration Micro-Kit (Norgen Biotek, Thorold, ON, Canada).
Expression profiling of each cell type was performed using U133 Plus 2.0 GeneChips (Affymetrix, Santa Clara, CA). Profiling data was analyzed in Genomics Suite™ (Partek Inc., St. Louis, Missouri) to detect differentially expressed mRNAs between conditions. We generated lists of time-dependent differential mRNA expression (24 hour vs 0 hour fold change). The Applied Biosystems 7900HT Fast Real-Time PCR System (Life Technologies, Grand Island, NY) was used to validate the differential expression of select genes. Primers used to amplify F3 pre-mRNA were GGCTGGATCAGGTCATCTCTAAAG (forward) and TGTGCCTGGGATCCTCAATAGA (reverse). Primers used to amplify all other genes, including F3 mRNA, were obtained from Applied Biosystems (Life Technologies) (Inventoried sequences – proprietary) (Supplement Table 1). Identification of biological pathways in which gene products participate was determined in Ingenuity Pathways Analysis software (Ingenuity Systems, Inc., Redwood City, CA).
Other Assays
Immunoblotting of protein lysates for protein tyrosine phosphatase (SHP2), GSK3β and actin was performed using antibodies purchased from Cell Signaling (Danvers, MA) (SHP2, GSK3β) and Santa Cruz (B-actin). TF-PCA was measured in whole blood lysates using a two-stage clotting assay [9, 14]. TF and TFPI antigen were measured using an ELISA (Sekisui Diagnostics, Stamford, CT). Plasma coagulation factors VIII (FVIII), VII (FVIIC) and FVIIa were measured as previously described [9].
Results
Analysis of Expression Profiling Data and Significant Canonical Pathways
Hierarchical clustering of platelet (Supplemental Fig 1A) and monocyte (Supplemental Fig 1B) mRNA expression at 0 and 24 hours was performed. mRNA transcripts with a 24 hour fold change ≥ 1.5 or greater in both cells were clustered based on the expression patterns. 17,020 transcripts were >=1.5 fold up or down regulated in platelets and monocytes in the 24 hour samples compared to all normals. The findings indicate that 24 hours of HG+HI is associated with substantial changes in gene expression in both platelets and monocytes.
Biological pathways in which the altered genes may be critical players were determined using Ingenuity Pathways Analysis software. Significant canonical pathways identified in platelets include genes related to the coagulation system (intrinsic and extrinsic prothrombin activation), insulin receptor signaling, and glutamate receptor signaling. In monocytes, genes related to the Wnt-B catenin signaling, endothelin-1 signaling, and intrinsic prothrombin activation were identified (Table 1). In the present paper we focus on insulin-signaling and coagulation pathways.
Table 1. Significant Canonical Pathways.
| PLATELETS 0hr vs 24hr FC 1.5 | ||
|---|---|---|
|
| ||
| Canonical Pathways | −log10 p-value | Genes |
| Intrinsic Prothrombin Activation | 0.25 | F11,F10,COL1A1,F12,SERPINC1,KLK3,F5,COL10A1,FGA,COL18A1, THBD,F13B,F2,COL3A1 |
| Extrinsic Prothrombin Activation | 1.00 | F10,F12,SERPINC1,F5,F7,TFPI,THBD,FGA,F3,F13B,F2 |
| Glutamate Receptor Signaling | 2.01 | SLC17A8,GRM2,GRIN2A,SLC1A4,SLC17A6,GRM3,GRIA1,GRIN2D, HOMER3,GRIA4,GRIK5,GRIN2C,HOMER1,GRM6,DLG4,SLC17A1, GRIK2,GRIK1,GRIN1,GRIN2B,SLC1A6,CALML5,GRM8,GRM1, GRIA2,GRIP1,SLC1A1,GRM4,GRIN3A,GRM5,GRM7,CALM1 (includes others), GRIK4,SLC17A7,SLC1A2,GLUL, GRIA3 |
| Insulin Receptor Signaling | 0.32 | PRKACB,RAPGEF1,SOCS3,ASIC2,MAPK1,BAD,SOS2,PDPK1, LIPE,KRAS,PPP1R3A,PIK3R4,PPP1R14B,SCNN1A,MAP2K2,PPP1R7, INS,TSC2,GSK3B,ATM,RPS6KB1,AKT2,EIF2B3,PPP1R11, VAMP2,GYS2,SYNJ2,CBL,RHOQ,GAB1,PTPN11,IRS1, PRKACG,PRKACA,EIF2B5,INSR,PIK3CA,PIK3R5,PPP1CB,STXBP4, JAK2,OCRL,PTPRF,PDE3B,INPP5B,PIK3C3,SOS1,PTPN1, FOXO3,RPTOR,AKT3,PIK3R2,NRAS,PIK3C2A,MAPK8,ACLY, INPP5D,GRB10,PPP1R3D,RRAS2,PRKCI,SYNJ1,PRKA G2,PRKAR1A |
|
| ||
| MONOCYTES 0hr vs 24hr FC 1.5 | ||
|
| ||
| Canonical Pathways | −log10 p-value | Genes |
|
| ||
| Wnt-B catenin Signaling | 4.36 | CDKN2A,WNT3,TGFBR3,SOX10,SOX12,CSNK1A1,WNT16,WNT6, SOX13,RARG,CCND1,TCF7,PPM1J,WNT7B,RARA,PPM1L, WNT4,WNT5B,TP53,AXIN2,APPL2,WNT9A,TCF3,CDH2, CDH1,CDH12,CDH5,GNAO1,PPP2R2B,TGFB3,DKK4,FZD5, LEF1,SOX8,SFRP1,DVL2,FZD10,MMP7,FRZB,SOX1, PPP2R2A,LRP6,ACVR2B,HNF1A,WNT8B,H2 BFM,SOX17, JUN,NLK,DKK3,RARB,TGFB2,AKT3,SFRP5,SOX14, PPP2R2C,CTNNB1,ACVR1C,SOX5,SRC,SOX7,MDM2,SOX11, FZD8,FZD4,SOX6,WNT10A,TLE4,CD44,NR5A2,BTRC, SOX9,DVL3,DKK1,LRP1,TCF7L2,WNT5A,FZD7 |
| Endothelin-1 Signaling | 1.88 | MAPK1,PLA2R1,SHC3,KRAS,MAPK13,PIK3R4,NOS3,LCAT, MRAS,PLCB1,GNA13,PLCL1,GUCY1B3,PRKD1,PLA2G12A,PRKCQ, GUCY1A3,ITPR2,MAPK4,MAPK12,PLD1,PIK3R3,PLA2G2D,GAB1, ITPR3,GNAO1,GUCY1A2,GNAT2,PRKCH,ADCY10,GNAL,CASP5, NOS1,GUCY2C,CASP4,GNA14,PRKCZ,SHC1,JUN,PIK3C3, SOS1,PRKCE,ECE1,PNPLA3,NOS2,PLCD4,PRKCA,SRC, ADCY2,GNAS,EDNRB,GNA12,PLA2G4C,ADCY6,PLCG1,PLA2G3, PRKCG,GNAI2,PLCZ1,FOS,PLCB4,RRAS2,PLA2G4D,ADCY1,EDNRA,PTGS2,PRKCB |
| Intrinsic Prothrombin Activation | 1.40 | F11,KNG1,F10,COL1A1,F9,COL5A3,KLK3,F5,PROC,COL2A1,FGB,F GA,F13B,COL3A1 |
Insulin signaling pathways
Both platelets and monocytes possess the insulin receptors and signaling [15-17]. Fig 1 shows the genes relevant to the insulin signaling pathways that are altered in platelets and monocytes. The genes upregulated and downregulated over the 24 hour study period are shown in red and green, respectively; also shown are the fold changes. In platelets, upregulated genes include VAMP2 (vesicle associated membrane protein 2) (14 fold), PTPN11 (13 fold), which encodes for a protein tyrosine phosphatase (SHP2), and GSKB3 (glycogen synthase kinase 3β, 5.5 fold). These genes play an important role in insulin signaling. Downregulated genes included STXBP4 (syntaxin 4 binding protein, Synip, 7.7 fold), INSR (insulin receptor, 4.3 fold) and IRS1 (insulin receptor substrate 1, 7.1 fold). In monocytes, the downregulated genes included PI3K (6.3), and SHP2 (1.7 fold).
Fig 1.
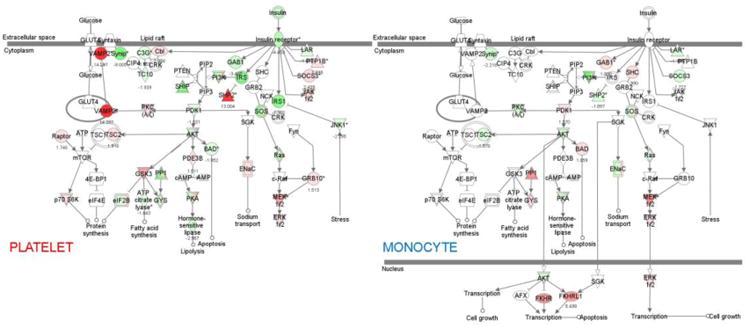
Insulin receptor signaling pathways and transcript expression in platelets (left panel) and monocytes (right panel). Genes with increased expression are shown in red and those with decreased expression in green.
A preliminary group of up or down-regulated genes was selected for qRT-PCR confirmation. Table 2 shows the fold change in platelets on expression profiling, 24 hours vs 0 hours and on qRT-PCR. For additional validity, the 24 hour platelet sample was compared to samples from 5 healthy non-diabetic subjects – the 0 hour sample from the present subject plus 4 additional healthy subjects. Notably, 5 out of the 9 selected platelet transcripts were confirmed by qRTPCR: GSKB3, PTPN11, STXBP4, F3, and TFPI (Table 2). Taking together the findings in platelets and monocytes, 8 of the 9 transcripts selected for qRT-PCR were confirmed (Table 1). Among those confirmed, the direction of changes were the same in platelets and monocytes for STXBP4 (decreased); it was opposite for PTPN11 and TFPI (Table 2), indicating that the effects of HG+HI may be distinct in these cells. PTPN11 and TFPI were upregulated in platelets and downregulated in monocytes.
Table 2. Showing the Expression Profiling and qRT-PCR Fold Changes in selected Insulin Receptor Signaling Transcripts and Coagulation.
| Gene Symbol | Gene Title | Platelets | Monocytes | ||
|---|---|---|---|---|---|
|
| |||||
| Fold Change Expression (24 vs 0 hrs) | Fold Change qRT-PCR (24 vs 0 hrs and 4 NLS) | Fold Change Expression (24 vs 0 hrs) | Fold Change qRT-PCR (24 vs 0 hrs and 4 NLS) | ||
|
|
|||||
| IRS1 | Insulin receptor substrate 1 | 0.14 | 2.45 (2.27, 2.64) | 0.73 | 0.88 (0.72, 1.08) |
| INSR | Insulin receptor | 0.23 | 1.00 (0.83, 1.21) | 0.89 | 0.88* (0.86, 0.91) |
| GSK3B | Glycogen synthase kinase 3 beta | 5.51 | 1.62* (1.52, 1.74) | 1.05 | 0.87 (0.83, 0.91) |
| PTPN11 | Protein tyrosine phosphatase, non-receptor type II | 13.00 | 1.97* (1.56, 2.49) | 0.60 | 0.65* (0.65, 0.65) |
| PIK3C3 | Phosphoinositide-3-kinase class 3 | 0.31 | 1.37 (1.31, 1.44) | 0.16 | 0.81* (0.75, 0.87) |
| VAMP2 | Vesicle-associated membrane protein 2 | 14.29 | 0.82 (0.75, 0.89) | 0.86 | 0.85* (0.83, 0.86) |
| STXBP4 | Syntaxin binding protein 4 | 0.13 | 0.87* (0.87, 0.88) | 0.97 | 0.91* (0.83, 0.99) |
| F3 | Tissue factor | 3.74 | 7.08* (1.08, 46.32) | 0.40 | 1.12 (1.01, 1.23) |
| TFPI | Tissue factor pathway inhibitor | 4.48 | 1.30* (1.26, 1.35) | 0.79 | 0.66* (0.56, 0.79) |
Results of qRT-PCR are shown as mean; the 95% confidence units are shown in parenthesis.
Genes validated by qRT-PCR are shown with an asterisk and in bold font.
In platelets, GSK3B was increased 5.51 fold on profiling and 1.62 fold by qRT-PCR. PTPN11 was increased 13 fold on profiling and 1.97 fold on qPCR. STXBP4 (Synip) was decreased (fold change, FC 0.13), with a smaller fold change on qPCR (0.87). Although on profiling studies IRIS1 and INSR showed a decreases (FC 0.14 and 0.23, respectively), these were not validated by qRT-PCR. Likewise, there was a decrease in PIK3C3 (FC 0.31) and an increase in VAMP2 (FC 14.29) over 24hrs; these were not validated.
In monocytes, 6 of the 9 transcripts selected were confirmed by qRT-PCR (Table 2). The fold change for PTPN11 was 0.60, a decrease of 40% that was validated by a similar change by qRT-PCR. This is in contrast to the findings in platelets, where there was a marked increase in PTPN11. PIK3C3 was decreased ∼6 fold (FC 0.16), and this was confirmed on qPCR. Much smaller decreases were noted on the INSR, VAMP2, and STXBP4 and the qPCR findings followed the trend (Table 2).
Coagulation System
In platelets, F3 (tissue factor) was upregulated by 3.74 fold and was validated (FC 7.08) on qRT-PCR. TFPI was increased 4.48 fold and validated (FC 1.3) by qRT-PCR. Platelets have the splicesome and cell activation induces splicing of pre-mRNA in resting platelets to mature, translatable mRNAs and subsequent protein synthesis [18, 19]. We, therefore, studied by qRTPCR F3 mRNA and pre-mRNA in platelets and monocytes (Fig 2). To assess pre-mRNA we amplified by qRT-PCR mRNA using primers specific for intron 4 [19]. TF mRNA in platelets was increased over 24 hours of HG+HI and platelet F3-pre-mRNA was decreased at 24 hours (FC ∼ 0.61 decrease). These studies are consistent with HG+HI-induced splicing of F3 pre-mRNA in platelets.
Fig 2.
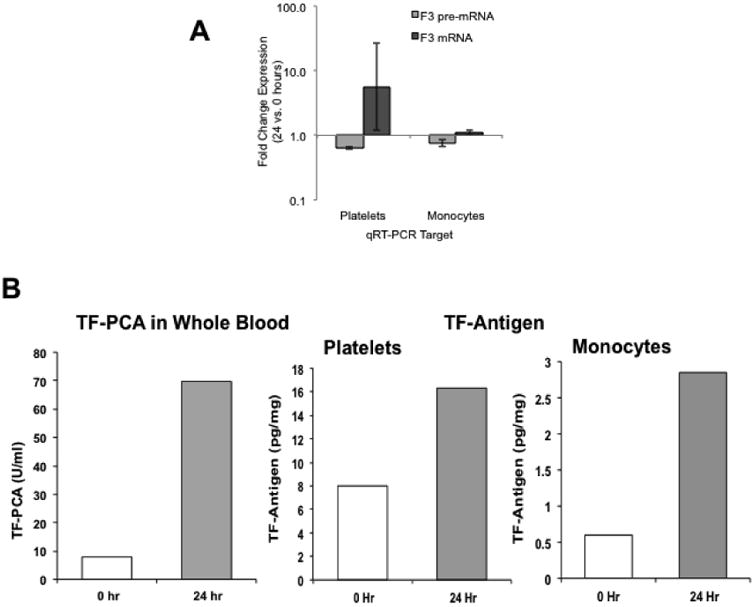
Top Panel (A): Change (24 versus 0 hours) in tissue factor (F3) pre-mRNA and F3 mRNA expression in platelets and monocytes. Shown are fold change from 0 hours.
Bottom Panel (B): Whole blood tissue factor procoagulant activity (TF-PCA), and tissue factor antigen in platelets and monocytes at 0 and 24 hours of hyperglycemia-hyperinsulinemia.
In monocytes, expression profiling showed a decrease (FC 0.40) in F3 (TF) at 24 hours. However, the qRT-PCR showed a small increase, which was consistent with the increase in TF protein measurement (Fig 3) and our previous studies showing an increase in both monocyte TF mRNA [9] and TF surface expression (FACS) [10]. Monocyte F3 pre-mRNA showed a small decrease (Fig 2). Interestingly, TFPI was downregulated (FC 0.79 by profiling and 0.66 by qPCR).
Fig 3.
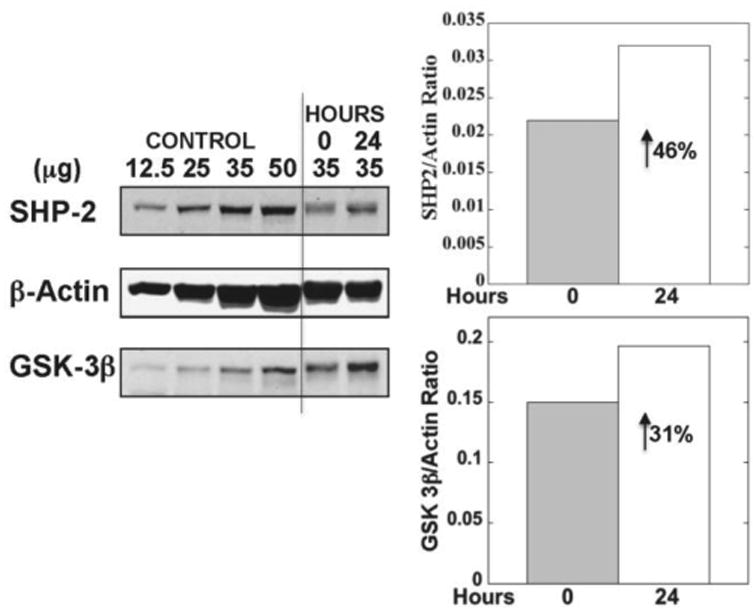
Changes in platelet protein tyrosine phosphatase (SHP2, PTPN11) and GSK-3B (GSK3B) at 0 and 24 hours. Shown in immunoblots are four dilutions of a control platelet sample and the study subject samples from 0 and 24 hours. The numbers above the lanes show the amount of protein loaded on the gel. Also shown is actin as loading control. The bars on the right show the protein levels at 0 and 24 hours normalized against actin.
Confirmation at Protein Level
Because of limited sample availability, platelet protein was assessed for only GSK3B and PTPN11 and these studies showed increases in both at 24 hour relative to baseline (Fig 3). To ensure that the findings in the present study were in line with our previous studies [9] TF procoagulant activity was measured in whole blood membranes by a two-stage clotting assay [9]; this showed a marked increase (∼7 fold) at 24 hours (Fig 3), and is in line with our previous studies. TF protein in platelets and monocytes assayed using an ELISA showed increased levels at 24 hours compared to baseline (Fig 3B). Lastly, we assessed plasma coagulation factors VIII, FVII, and VIIa – FVIII rose from 1.50 U/ml to 1.76 U/ml at 24 hours; FVIIc and FVIIa declined from 1.1 U/ml to 0.8 U/ml and from 38.8 to 23.1 U/ml, respectively. Plasma TFPI declined from 48.2 to 43.3 ng/ml. These plasma measurements are in line with our previous studies using HG+HI clamps [9].
Discussion
Our studies using expression profiling provide evidence that 24 hours of HG+HI induces substantial changes in platelet and monocyte insulin signaling and coagulation pathways even in the non-diabetic state. Platelet GSK3B and PTPN11 were upregulated and STXBP4 was downregulated (Table 2) - all 3 are important players in insulin signaling (Fig 1) and the findings suggest downregulation of insulin signaling. Platelet expression of F3 (TF) and TFPI, the principal TF pathway inhibitor) [20] was upregulated in the coagulation pathways. These are important findings in the context of the prothrombotic state in DM. On the monocyte side, the findings suggest downregulation of INSR, PTPN11, PIK3C3, VAMP2, STXBP4 – all key players in insulin signaling [20]. These are consistent with the concept that 24 hours of HG+HI induces a compensatory response to down-regulate insulin-driven pathways. In the coagulation system the combination of current findings of increased TF protein (Fig 3) and previous studies [9, 10] indicate that prothrombotic mechanisms are upregulated.
Our findings constitute proof of principle that expression profiling is a powerful tool and likely to unravel changes in other currently unrecognized mechanisms and pathways in blood cells – both in healthy subjects and DM patients. A limitation of our study is that a single subject was studied. However, these 24 hour glucose infusion studies in healthy subjects are difficult and resource-intensive studies. Moreover, the 24 hour platelet sample was compared in qRT-PCR studies against baseline samples from 5 subjects, which makes the observed changes in the 24 hours sample more robust in the context of platelets than monocytes.
Platelet GSK3B mRNA and protein (Table 2, Fig 3) were elevated following 24 hours of HG+HI. Glycogen synthase kinase 3 (GSK3) is a constitutively acting, multi-functional serine threonine kinase involved in insulin signaling and glucose metabolism [17, 20, 21]. GSK3 phosphorylates several substrates and inactivates them, including glycogen synthase and β catenin. Insulin binding to its receptor leads to its autophosphorylation, followed by binding of IRS that is phosphorylated, and subsequent activation of PI3 kinase and Akt/PKB. The latter phosphorylates GSK3 [20, 21], which attenuates glycogen synthase inhibition and eIF2B (eukaryotic initiation factor 2B) phosphorylation, leading to active forms of glycogen synthase and eIF2B and to increased glycogen and protein synthesis, respectively. Thus, elevated GSK3B would be consistent with inhibition of insulin signaling in the face of high glucose and insulin. GSK3B has been implicated in insulin resistance and in T2DM patients [22, 23] and GSK3 inhibitors improve insulin signaling and glucose levels [24].
GSK3B, the predominant platelet form, is phosphorylated by Akt and negatively regulates platelet function; reduced levels lead to enhanced platelet responsiveness [25, 26]. Thus, the elevated platelet GSKB3 levels may represent a regulatory mechanism to counter enhanced platelet/megakaryocyte activation induced by HG. Moreover, GSK3 negatively regulates TF expression in monocytes interacting with platelets [27]. Both these observation assume importance because platelet responses are enhanced in DM [4, 5] and HG+HI elevates TF even in normal subjects [9].
PTPN11 encodes for a ubiquitously expressed cytoplasmic protein tyrosine phosphatase (SHP2) that contains two SH2 domains (8). SHP2 binds IRS proteins [17, 20]. Tyrosinephosphorylated motifs on the IRS proteins bind to the SH2 domains in proteins that mediate down-stream signals, including SHP2, phosphatidylinositol 3 kinase, and GRB-2. SHP2 attenuates the phosphorylation and downstream signal transmission of IRS-1, and the interaction of IRS-1 and SHP2 is thus an important regulatory event that attenuates insulin metabolic responses and signaling [28]. Megakaryocytic-specific deletion of SHP2 reveals that it is a major regulator of platelet production and function [29]; SHP2 deletion induced hyper-responsiveness of platelets. The marked upregulation of platelet SHP2 (PTPN11) may, thus, serve to downregulate platelet hyper-responsiveness. Interestingly, monocyte PTPN11 transcript was decreased at 24 hour indicating that platelets and monocyte respond differently.
Our studies suggest that platelet and monocyte expression of STXBP4 (synip) is decreased. STXBP4 encodes for a protein involved in GLUT4 vesicle trafficking and insulin-driven facilitative glucose uptake [30, 31]. GLUT4-containing synaptic-like vesicles are enriched for the V-SNARE protein VAMP2. Insulin induces association of these vesicles with the plasma membrane through interaction of t-SNARE complex consisting of syntaxin 4 and SNAP23. STXBP4 (synip) binds to syntaxin 4 and is a positive regulator of insulin-stimulated GLUT4 translocation [32]. Thus, the observed downregulation of STXBP4 may be a response to limit cellular glucose uptake. Platelets possess vesicle trafficking machinery and future studies would provide insights into these mechanisms.
On the procoagulant side, 24 hours of HG+HI showed an increase in platelet TF mRNA (Table 2) and protein (Fig 3) associated with a decrease in F3 pre-mRNA in platelets (Fig 2), and consistent with splicing of pre-mRNA to mRNA [19]. Despite being anucleate platelets possess the ability to process pre-mRNA and stimulate protein synthesis on activation [18, 19], and this occurs in sepsis [33]. Our previous studies have shown that 24 hours of HG+HI in healthy subjects induces activation of platelets and monocytes with an increase in circulating platelet-monocyte aggregates, platelet CD40 ligand and monocyte TF [9, 10]. The upregulation of platelet TF in the present study is relevant to the prothrombotic state associated with HG+HI and DM. Monocyte TF was also increased (Fig 3) and consistent with our previous studies [9, 10]. TFPI, the principal inhibitor of the TF-VIIa, is present in platelets, synthesized by megakaryocytes and monocytes, and expressed on surface on activation [34, 35]. Cell surface TFPI is an important regulator of procoagulant activity [35]. In the face of upregulation of platelet TF (F3) the increase in TFPI expression may be a counter-regulatory mechanism to inhibit procoagulant mechanisms [34, 35]. In contrast to platelets, monocyte TFPI mRNA was downregulated at 24 hour (Table 2). Interestingly, our previous studies showed decreased plasma TFPI by 24 hours of HG+HI [9].
As is well recognized, findings in transcript profiling need to be validated by independent quantitative PCR or other methods and that not all of the alterations noted in transcript profiling are generally confirmed. This applies to some of the genes shown in Table 2. Our studies, therefore, are of no exception. A larger number of studies will be needed to define the changes in these genes. However, in the case of platelet F3, GSKB3, and PTPN11 we provide confirmation not only using qRT-PCR but also at the protein level, which allows us to make conclusions that will lay the foundation to pursue in future studies.
In summary, our studies provide evidence that 24 hour of HG+HI, even in the non-diabetic healthy state, induces changes in platelets and monocytes, including alterations in insulin-signaling and procoagulant pathways. Overall, the upregulation of platelet GSK3B and PTPN11, and downregulation of STXBP4 along with possible downregulation of IRS1 and INSR (Table 2) are consistent with downregulation of platelet insulin-signaling by HG+HI at multiple steps from initial insulin action to the late steps of glucose uptake. Similar trends were noted in monocytes as well. These studies lay the foundation for future studies to explore in an unbiased manner the effects of HG and HI in various currently unrecognized cellular pathways in blood cells that play a role thrombosis, inflammation, atherosclerosis and angiogenesis.
Supplementary Material
Highlights.
Diabetes mellitus is a prothrombotic and proinflammatory state.
Hyperglycemia (HG)-hyperinsulinemia (HI) in healthy subjects activates coagulation.
We studied effect of HG-HI on platelets and monocytes in non-diabetic state.
We used cell expression profiling before and after 24 hours of HG and HI.
Even in healthy state HG+HI induces major changes in platelet and monocytes.
Acknowledgments
This work was supported by the National Institutes of Health grant R01-HL-073367. The excellent assistance of the members of the General Clinical Research Center during the infusion studies and of Ms. Denise Tierney in the preparation of the manuscript is gratefully acknowledged.
Footnotes
Contribution: AKR designed the research and wrote the manuscript. GB designed the research. RF performed the expression profiling studies and wrote the manuscript. GJ, AS, GM, AW and PC performed various research studies.
Conflict of Interest Statement: The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Grundy SM, Howard B, Smith S, Jr, Eckel R, Redberg R, Bonow RO. Prevention Conference VI: Diabetes and Cardiovascular Disease: executive summary: conference proceeding for healthcare professionals from a special writing group of the American Heart Association. Circulation. 2002;105(18):2231–9. doi: 10.1161/01.cir.0000013952.86046.dd. [DOI] [PubMed] [Google Scholar]
- 2.Carr ME. Diabetes mellitus: a hypercoagulable state. J Diabetes Complications. 2001;15(1):44–54. doi: 10.1016/s1056-8727(00)00132-x. [DOI] [PubMed] [Google Scholar]
- 3.Alzahrani SH, Ajjan RA. Coagulation and fibrinolysis in diabetes. Diab Vasc Dis Res. 2010;7(4):260–73. doi: 10.1177/1479164110383723. [DOI] [PubMed] [Google Scholar]
- 4.El Haouari M, Rosado JA. Platelet signalling abnormalities in patients with type 2 diabetes mellitus: a review. Blood Cells Mol Dis. 2008;41(1):119–23. doi: 10.1016/j.bcmd.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 5.Kakouros N, Rade JJ, Kourliouros A, Resar JR. Platelet function in patients with diabetes mellitus: from a theoretical to a practical perspective. Int J Endocrinol. 2011 doi: 10.1155/2011/742719. 2011742719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Capes SE, Hunt D, Malmberg K, Gerstein HC. Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet. 2000;355(9206):773–8. doi: 10.1016/S0140-6736(99)08415-9. [DOI] [PubMed] [Google Scholar]
- 7.Umpierrez GE, Isaacs SD, Bazargan N, You X, Thaler LM, Kitabchi AE. Hyperglycemia: an independent marker of in-hospital mortality in patients with undiagnosed diabetes. J Clin Endocrinol Metab. 2002;87(3):978–82. doi: 10.1210/jcem.87.3.8341. [DOI] [PubMed] [Google Scholar]
- 8.Falciglia M, Freyberg RW, Almenoff PL, D'Alessio DA, Render ML. Hyperglycemia-related mortality in critically ill patients varies with admission diagnosis. Crit Care Med. 2009;37(12):3001–9. doi: 10.1097/CCM.0b013e3181b083f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vaidyula VR, Rao AK, Mozzoli M, Homko C, Cheung P, Boden G. Effects of hyperglycemia and hyperinsulinemia on circulating tissue factor procoagulant activity and platelet CD40 ligand. Diabetes. 2006;55(1):202–8. [PubMed] [Google Scholar]
- 10.Vaidyula VR, Boden G, Rao AK. Platelet and monocyte activation by hyperglycemia and hyperinsulinemia in healthy subjects. Platelets. 2006;17(8):577–85. doi: 10.1080/09537100600760814. [DOI] [PubMed] [Google Scholar]
- 11.Boden G, Vaidyula VR, Homko C, Cheung P, Rao AK. Circulating tissue factor procoagulant activity and thrombin generation in patients with type 2 diabetes: effects of insulin and glucose. J Clin Endocrinol Metab. 2007;92(11):4352–8. doi: 10.1210/jc.2007-0933. [DOI] [PubMed] [Google Scholar]
- 12.Singh A, Boden G, Homko C, Gunawardana J, Rao AK. Whole-Blood Tissue Factor Procoagulant Activity Is Elevated in Type 1 Diabetes: Effects of hyperglycemia and hyperinsulinemia. Diabetes Care. 2012;35(6):1322–7. doi: 10.2337/dc11-2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gnatenko DV, Dunn JJ, McCorkle SR, Weissmann D, Perrotta PL, Bahou WF. Transcript profiling of human platelets using microarray and serial analysis of gene expression. Blood. 2003;101(6):2285–93. doi: 10.1182/blood-2002-09-2797. [DOI] [PubMed] [Google Scholar]
- 14.Key NS, Slungaard A, Dandelet L, Nelson SC, Moertel C, Styles LA, et al. Whole blood tissue factor procoagulant activity is elevated in patients with sickle cell disease. Blood. 1998;91(11):4216–23. [PubMed] [Google Scholar]
- 15.Perveen R, Funk K, Thuma J, Ridge SW, Cao Y, Akkerman JW, et al. A novel small molecule 1,2,3,4,6-penta-O-galloyl-α-D-glucopyranose mimics the antiplatelet actions of insulin. PLoS One. 2011;6(11):e26238. doi: 10.1371/journal.pone.0026238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferreira IA, Eybrechts KL, Mocking AI, Kroner C, Akkerman JW. IRS-1 mediates inhibition of Ca2+ mobilization by insulin via the inhibitory G-protein Gi. J Biol Chem. 2004;279(5):3254–64. doi: 10.1074/jbc.M305474200. [DOI] [PubMed] [Google Scholar]
- 17.Frojdo S, Vidal H, Pirola L. Alterations of insulin signaling in type 2 diabetes: a review of the current evidence from humans. Biochim Biophys Acta. 2009;1792(2):83–92. doi: 10.1016/j.bbadis.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 18.Zimmerman GA, Weyrich AS. Signal-dependent protein synthesis by activated platelets: new pathways to altered phenotype and function. Arterioscler Thromb Vasc Biol. 2008;28(3):s17–24. doi: 10.1161/ATVBAHA.107.160218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwertz H, Tolley ND, Foulks JM, Denis MM, Risenmay BW, Buerke M, et al. Signal-dependent splicing of tissue factor pre-mRNA modulates the thrombogenicity of human platelets. J Exp Med. 2006;203(11):2433–40. doi: 10.1084/jem.20061302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7(2):85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 21.Rayasam GV, Tulasi VK, Sodhi R, Davis JA, Ray A. Glycogen synthase kinase 3: more than a namesake. Br J Pharmacol. 2009;156(6):885–98. doi: 10.1111/j.1476-5381.2008.00085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nikoulina SE, Ciaraldi TP, Mudaliar S, Carter L, Johnson K, Henry RR. Inhibition of glycogen synthase kinase 3 improves insulin action and glucose metabolism in human skeletal muscle. Diabetes. 2002;51(7):2190–8. doi: 10.2337/diabetes.51.7.2190. [DOI] [PubMed] [Google Scholar]
- 23.Nikoulina SE, Ciaraldi TP, Mudaliar S, Mohideen P, Carter L, Henry RR. Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes. 2000;49(2):263–71. doi: 10.2337/diabetes.49.2.263. [DOI] [PubMed] [Google Scholar]
- 24.Meijer L, Flajolet M, Greengard P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol Sci. 2004;25(9):471–80. doi: 10.1016/j.tips.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 25.Li D, August S, Woulfe DS. GSK3beta is a negative regulator of platelet function and thrombosis. Blood. 2008;111(7):3522–30. doi: 10.1182/blood-2007-09-111518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore SF, van den Bosch MT, Hunter RW, Sakamoto K, Poole AW, Hers I. Dual regulation of glycogen synthase kinase 3 (GSK3)alpha/beta by protein kinase C (PKC)alpha and Akt promotes thrombin-mediated integrin alphaIIbbeta3 activation and granule secretion in platelets. J Biol Chem. 2013;288(6):3918–28. doi: 10.1074/jbc.M112.429936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Santo A, Amore C, Dell'Elba G, Manarini S, Evangelista V. Glycogen synthase kinase-3 negatively regulates tissue factor expression in monocytes interacting with activated platelets. J Thromb Haemost. 2011;9(5):1029–39. doi: 10.1111/j.1538-7836.2011.04236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Myers MG, Jr, Mendez R, Shi P, Pierce JH, Rhoads R, White MF. The COOH-terminal tyrosine phosphorylation sites on IRS-1 bind SHP-2 and negatively regulate insulin signaling. J Biol Chem. 1998;273(41):26908–14. doi: 10.1074/jbc.273.41.26908. [DOI] [PubMed] [Google Scholar]
- 29.Mazharian A, Mori J, Wang YJ, Heising S, Neel BG, Watson SP, et al. Megakaryocyte-specific deletion of the protein-tyrosine phosphatases Shp1 and Shp2 causes abnormal megakaryocyte development, platelet production, and function. Blood. 2013;121(20):4205–20. doi: 10.1182/blood-2012-08-449272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pessin JE, Thurmond DC, Elmendorf JS, Coker KJ, Okada S. Molecular basis of insulin-stimulated GLUT4 vesicle trafficking. Location! Location! Location! J Biol Chem. 1999;274(5):2593–6. doi: 10.1074/jbc.274.5.2593. [DOI] [PubMed] [Google Scholar]
- 31.Watson RT, Pessin JE. Bridging the GAP between insulin signaling and GLUT4 translocation. Trends Biochem Sci. 2006;31(4):215–22. doi: 10.1016/j.tibs.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 32.Min J, Okada S, Kanzaki M, Elmendorf JS, Coker KJ, Ceresa BP, et al. Synip: a novel insulin-regulated syntaxin 4-binding protein mediating GLUT4 translocation in adipocytes. Mol Cell. 1999;3(6):751–60. doi: 10.1016/s1097-2765(01)80007-1. [DOI] [PubMed] [Google Scholar]
- 33.Rondina MT, Schwertz H, Harris ES, Kraemer BF, Campbell RA, Mackman N, et al. The septic milieu triggers expression of spliced tissue factor mRNA in human platelets. J Thromb Haemost. 2011;9(4):748–58. doi: 10.1111/j.1538-7836.2011.04208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maroney SA, Haberichter SL, Friese P, Collins ML, Ferrel JP, Dale GL, et al. Active tissue factor pathway inhibitor is expressed on the surface of coated platelets. Blood. 2007;109(5):1931–7. doi: 10.1182/blood-2006-07-037283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Basavaraj MG, Gruber FX, Sovershaev M, Appelbom HI, Osterud B, Petersen LC, et al. The role of TFPI in regulation of TF-induced thrombogenicity on the surface of human monocytes. Thromb Res. 2010;126(5):418–25. doi: 10.1016/j.thromres.2010.07.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.