

Mutations in the nuclear RNA-binding protein FUS can cause the neurodegenerative disease amyotrophic lateral sclerosis (ALS). Study of ALS patient fibroblasts reveals FUS protein aggregated in the nucleus and its regulation of RNA polymerase II disrupted. Thus mutant FUS need not be aggregated in the cytoplasm to have deleterious consequences.
Abstract
Mutations in the RNA-binding protein FUS have been shown to cause the neurodegenerative disease amyotrophic lateral sclerosis (ALS). We investigate whether mutant FUS protein in ALS patient–derived fibroblasts affects normal FUS functions in the nucleus. We investigated fibroblasts from two ALS patients possessing different FUS mutations and a normal control. Fibroblasts from these patients have their nuclear FUS protein trapped in SDS-resistant aggregates. Genome-wide analysis reveals an inappropriate accumulation of Ser-2 phosphorylation on RNA polymerase II (RNA Pol II) near the transcription start sites of 625 genes for ALS patient cells and after small interfering RNA (siRNA) knockdown of FUS in normal fibroblasts. Furthermore, both the presence of mutant FUS protein and siRNA knockdown of wild-type FUS correlate with altered distribution of RNA Pol II within fibroblast nuclei. A loss of FUS function in orchestrating Ser-2 phosphorylation of the CTD of RNA Pol II is detectable in ALS patient–derived fibroblasts expressing mutant FUS protein, even when the FUS protein remains largely nuclear. A likely explanation for this loss of function is the aggregation of FUS protein in nuclei. Thus our results suggest a specific mechanism by which mutant FUS can have biological consequences other than by the formation of cytoplasmic aggregates.
INTRODUCTION
Fused in Sarcoma (FUS) is an abundant nuclear RNA-binding protein and is also known as Translocated in Liposarcoma (TLS). The FUS protein contains an RNA recognition motif and a zinc finger, both of which are capable of binding RNA (Iko et al., 2004; Schwartz et al., 2013). At its N-terminus, FUS has a domain of low amino acid sequence complexity that can assemble into higher-order structures (Kato et al., 2012; Kwon et al., 2013).
FUS affects multiple levels of RNA biogenesis. FUS binds and recruits RNA polymerase II (RNA Pol II) directly to DNA, as well as inhibiting or redirecting the activity of multiple transcriptional activators (Zinszner et al., 1994; Immanuel et al., 1995; Hallier et al., 1998; Powers et al., 1998; Yang et al., 2000; Das et al., 2007; Wang et al., 2008; Tan and Manley, 2010; Tan et al., 2012; Kwon et al., 2013; Schwartz et al., 2012, 2013). FUS interacts with several heterogeneous nuclear ribonucleoproteins, as well as with the U1 small nuclear ribonucleoprotein complex, and is reported to affect mRNA splicing (Zinszner et al., 1994; Calvio et al., 1995; Hackl and Luhrmann, 1996; Hallier et al., 1998; Lerga et al., 2001; Hoell et al., 2011; Ishigaki, Masuda, et al., 2012; Lagier-Tourenne, Polymenidou, Hutt, et al., 2012). FUS is also reported to affect mRNA transport (Zinszner et al., 1997; Fujii, Okabe, et al., 2005; Fujii and Takumi, 2005).
One nuclear function of FUS is to bind the C-terminal domain (CTD) of RNA Pol II (Schwartz et al., 2012, 2013; Kwon et al., 2013). The CTD is important for transcription because its sequential phosphorylation on Ser-5 and Ser-2 of the repeat CTD sequence YSPTSPS regulates initiation and elongation of the polymerase and the binding of factors involved in mRNA processing. FUS regulates the phosphorylation status of the CTD at Ser-2 (Schwartz et al., 2012). Because phosphorylation of Ser2 serves coordinately to regulate RNA splicing, polyadenylation, and trafficking, FUS regulation of this RNA Pol II modification provides one simple model for the broad effects of the FUS protein on multiple levels of RNA biogenesis (Glover-Cutter, Kim, et al., 2008; Munoz et al., 2009; Schwartz et al., 2012; Gu et al., 2013).
Mutations in FUS have been shown to be a cause of the neurodegenerative disease amyotrophic lateral sclerosis (ALS). FUS mutations account for 5% of familial and 1% of sporadic ALS disease; mutations in FUS therefore have a similar frequency to those in TDP-43 but are less prominent than mutations in C9ORF72 and SOD1 as known genetic causes of the disease (Kwiatkowski et al., 2009; Vance, Rogelj, Hortobagyi, De Vos, et al., 2009). Known ALS-causing FUS mutations are mostly limited to the nuclear localization signal (NLS; Lattante et al., 2013). Histological staining after autopsy reveals cytoplasmic aggregates staining positive for FUS in motor neurons of ALS patients and cortical neurons of a subset of frontotemporal dementia patients (Mackenzie et al., 2011; Lattante et al., 2013). These observations led to a model in which disruption of cytoplasmic shuttling of this protein may play a role in disease pathology. Both loss-of-function and toxic gain-of-function models have been proposed to explain FUS-dependent pathology. For example, loss of FUS from the nucleus could disrupt the normal nuclear functions of the protein. Alternatively or in addition, the presence of FUS or its aggregates in the cytoplasm could result in a toxic gain of function. A gain of function in disease pathology does not preclude additional loss of function or its role in pathology.
Convincing counterarguments can be made for either a loss- or gain-of-function model of FUS pathology. One problem with a loss-of-function model is that several ALS-causing mutations do not cause a dramatic reduction in nuclear FUS (Bosco et al., 2010; Kwiatkowski et al., 2009; Vance et al., 2013). In certain model organisms, cytoplasmic mislocalization seems sufficient to produce toxicity without reduction of endogenous and nuclear FUS (Halliday et al., 2012). On the other hand, molecular observations upon expression of mutant FUS protein, including the loss of nuclear Gemini of coiled bodies (Gems), are consistent with a loss of FUS function (Yamazaki et al., 2012; Tsuiji et al., 2013). A zebrafish model shows that FUS- possessing, ALS-causing mutations fail to rescue a FUS knockdown, indicating a loss of function (Kabashi et al., 2011). Finally, overexpression models of FUS have produced mixed results, including cytoplasmic aggregates without toxicity or toxicity without aggregates (Halliday et al., 2012).
To provide more insight regarding this unresolved question, we investigated the physical state and function of FUS in normal and ALS patient fibroblasts. Patient-derived fibroblasts provide a powerful tool because they can be cultured and grown in the large quantities needed for molecular biology experiments, possess the native genetic background of the patient, and have not been genetically manipulated and therefore would not have genetic artifacts due to artificial levels of expression of the FUS protein. We studied fibroblasts from two ALS patients with FUS mutations, wild-type fibroblast controls, and wild-type controls after small interfering RNA (siRNA)–mediated knockdown of FUS protein levels. We measured relative FUS protein levels in the nucleus and cytoplasm, phosphorylation status of RNA Pol II near transcription start sites (TSS), and localization of active RNA Pol II. We found evidence that 1) FUS is sequestered in nuclear aggregates, 2) RNA Pol II is inappropriately phosphorylated at Ser2 for a subset of genes, and 3) RNA Pol II is mislocalized in the nucleus in these ALS patient fibroblasts.
RESULTS
FUS is present in nuclear aggregates in ALS patient–derived fibroblasts
We cultured human fibroblasts with three genetic backgrounds. The first fibroblast culture (mFUS; Figure 1A), derived from a patient with familial ALS, has a homozygous and recessive point mutation in the NLS of FUS, H517Q (Kwiatkowski et al., 2009). The second fibroblast culture (ΔNLS; Figure 1A), derived from a patient with sporadic ALS, possesses a heterozygous insertion and frameshift mutation resulting in the loss of the entire NLS, M511Nfs*6. The third (wild type; Figure 1A) was from a sex-matched, unrelated, neurologically normal control.
FIGURE 1:

ALS patient fibroblasts expressing mutant FUS show FUS predominantly in nuclei. (A) Three cell lines were tested. mFUS is homozygous for a point mutation, H517Q. ΔNLS is heterozygous for a frameshift mutation deleting the entire nuclear localization signal, M511Nfs. Wild type is a sex-matched normal control. (B) Immunofluorescence microscopy reveals that FUS is almost entirely nuclear, except for the ΔNLS cell line, which shows some cytoplasmic localization. Scale bar (lower left), 10 μm.
All experiments were performed on dividing cells that had not entered senescence. The cell culture protocol (see Materials and Methods) was important, because if the cells did not maintain contact with other cells, they quickly entered senescence. Doubling times were measured to be between 5 and 7 d for both wild-type and ALS patient-derived fibroblasts. Cells were split once every 1–1.5 wk and fed three times per week (see Materials and Methods). With careful passaging, cells could be grown for >30 passages.
By immunofluorescence (IF; Figure 1B), FUS was nuclear in wild-type fibroblasts and remained nuclear in the mFUS fibroblasts. Partial mislocalization of FUS to the cytoplasm was apparent only in the ΔNLS patient, consistent with previous literature (Kwiatkowski et al., 2009; Bosco et al., 2010). The specificity of the antibody used for IF is indicated by the absence of FUS staining after siRNA knockdown (see final section of Results).
When we lysed whole cells in 4% lithium dodecyl sulfate (LDS) loading buffer, the total cellular FUS protein levels were the same in the three genetic backgrounds (Figure 2A, left). (Even though the predicted molecular weight of FUS is 55 kDa, the band for FUS protein appeared slightly above the 62-kDa molecular weight marker, which is consistent with previous work; Crozat et al., 1993.) When we fractionated cells by hypotonic lysis and performed Western analysis after SDS–PAGE, we noted that FUS protein levels were reduced in the nucleus of mFUS and almost undetectable in ΔNLS cells (Figure 2A, right). In our fractionated samples, no FUS was observed in the cytoplasmic fraction (Figure 2B). Therefore we reasoned that FUS could exist in SDS-resistant aggregates, which may not migrate into the SDS–PAGE gel. To test whether FUS was trapped in SDS-resistant aggregates, we performed Western analysis on cytoplasmic and nuclear fractions from the wild-type and ΔNLS fibroblasts using a dot blot rather than electrophoresis and found at least 90% (wild type) or 70% (ΔNLS) of FUS protein to be present in the nucleus, consistent with our immunofluorescence observations (Figure 2C). The nuclear aggregates in ΔNLS and mFUS cells were much more insoluble after hypotonic lysis and were resistant to 1–2% SDS, 1% Triton X-100, RNase A, DNase I, and benzonase, as well as boiling in loading buffer containing 50 mM dithiothreitol (DTT) or 5 mM β-mercaptoethanol (unpublished data). However, treating nuclear lysates with formic acid followed by sonication in loading buffer did allow some FUS protein to resolve in the well during SDS–PAGE, consistent with the hypothesis that the protein existed in a higher-order protein complex (Figure 2D).
FIGURE 2:
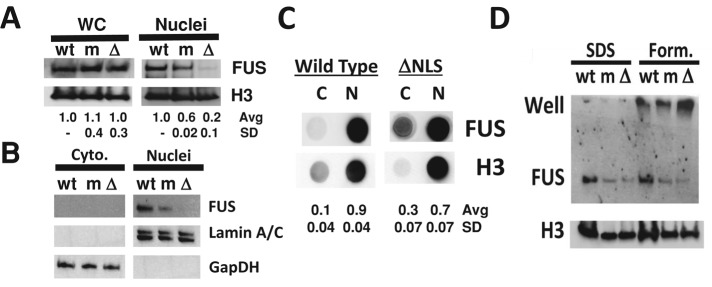
Mutant FUS is trapped in aggregates within nuclei of ALS patient fibroblasts. (A) Western analysis of whole-cell FUS protein reveals that levels between the three cell lines are the same (WC, left). For ΔNLS samples, FUS protein from isolated nuclei after hypotonic lysis fails to resolve by SDS–PAGE (Nuclei, right). (B) Cytoplasmic FUS (Cyto., left) is also not detectable by SDS–PAGE. (C) Dot blot analysis of FUS protein isolated from cytoplasmic (C) or nuclear (N) fractions reveals that FUS is predominantly nuclear, in agreement with immunofluorescence data. (D) Treatment of nuclear protein with formic acid (Form., right) partially disrupts FUS aggregates, allowing protein to accumulate in the well during SDS–PAGE.
ALS patient–derived fibroblasts have altered Ser-2 phosphorylation of RNA Pol II
The knockdown of FUS protein leads to inappropriate Ser-2 phosphorylation (Ser2P) on the CTD of RNA Pol II in HEK293T/17 cells (Schwartz et al., 2012). That is, instead of Ser2P levels being low near the TSS and increasing toward the gene terminus (a “canonical” distribution; Kim, Erickson, et al., 2010; Mayer, Lidschreiber, Siebert, et al., 2010), the loss of FUS leads to high Ser2P levels near the TSS for thousands of genes. This change was accompanied by higher RNA Pol II levels near the TSS, which could reflect delayed promoter clearance, more pausing, or premature termination (Schwartz et al., 2012). This observation is therefore taken to be a loss-of-function phenotype.
We hypothesized that the aggregation of FUS in the nuclei of the ALS fibroblasts would reduce the levels of active FUS, which would then perturb Ser-2 phosphorylation of the CTD in a manner similar to a FUS knockdown. To test this idea, we performed chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) of RNA Pol II using an antibody that selectively binds CTD possessing the Ser2P modification. For simplicity, we defined expressed genes as the 15,000 genes with the highest total Ser2P density along the gene. However, our initial experiments gave an unexpected result: the majority of genes expressed in wild-type fibroblasts showed a distribution of Ser2P signal that was higher near the TSS than near the gene terminus (Figure 3A, dashed lines). This led us to conclude that Ser2P regulation in fibroblast cells differs from its regulation in HEK293T/17 cells (Schwartz et al., 2012). Indeed, we analyzed several publicly available Ser2P ChIP-seq data sets and found that many cell lines from a variety of tissues also show noncanonical Ser2P distributions for a majority of genes (unpublished data).
FIGURE 3:

A subset of genes shows accumulation of Ser-2 phosphorylation (Ser2P) on RNA Pol II in ALS patient cells and after siRNA knockdown. (A) Average Ser2P signal for either all expressed genes (Total, dashed line) or the 625 genes at the intersect of B (Intersect, solid line) for wild-type cells, siFUS, mFUS, or ΔNLS cells. The p value is measured between normalized sum of Ser2P near the TSS for the intersect of treated compared with wild type and using the two-tailed Student's t test assuming unequal variances. (B) Using the threshold of twofold increase in Ser2P within 300 nucleotides of the transcription start site (TSS) with respect to wild-type cells, 625 genes show increased Ser2P in both ALS patient cells and after siRNA knockdown of FUS in wild-type cells (siFUS). See inset (right) for relative mRNA levels of FUS in siFUS-treated cells measured by real-time PCR. (C) Median Ser2P signal for either all expressed genes (Total) or the 625 genes at the intersect of B (Intersect). Error bars represent 25th and 75th percentiles. RPM, reads per million.
Nevertheless, for mFUS and ΔNLS fibroblasts a subset of genes did show a greater averaged Ser2P near the TSS compared with wild-type fibroblasts. We hypothesized that genes directly affected by FUS should also show increased Ser2P near their TSS after siRNA treatment (siFUS) in wild-type fibroblasts. Our siFUS treatment dramatically reduced FUS mRNA levels seen by real-time PCR (Figure 3B, inset) and protein levels seen by immunofluorescence (see preceding section). A total of 625 genes showed significantly higher Ser2P near their TSS for siFUS, mFUS, and ΔNLS fibroblasts than with untreated wild-type fibroblasts (Figure 3B). A Student's t test comparing the Ser2P signals near the TSS of each sample to wild-type fibroblasts found highly significant differences for these genes (see p value in Figure 3A). Ontological analysis of these 625 genes revealed no significant enrichment for genes of particular cellular pathways or processes. In wild-type fibroblasts, these 625 genes predominantly displayed canonical Ser2P (Figure 3A, solid line). The median of total Ser2P levels near the TSS for the 625 intersecting genes in wild-type cells was lower than that for all genes because these genes mostly have canonically distributed Ser2P, and the levels for the 625 genes were higher in siFUS, mFUS, and ΔNLS samples (Figure 3C).
FUS has a granular distribution and partially colocalizes with RNA Pol II
FUS forms higher-order assemblies that bind and recruit RNA Pol II to gene promoters through interactions with the CTD (Schwartz et al., 2012, 2013; Kwon et al., 2013). We therefore hypothesized that FUS organization in the nucleus may contribute to RNA Pol II organization in the nucleus. We observed in FUS immunofluorescence an uneven staining level throughout the nucleus and no staining in the nucleolus. This observation agrees with reports that FUS distribution is granular, with certain subnuclear regions possessing a higher local concentration of FUS signal (Alliegro and Alliegro, 1996; Kino et al., 2010).
We stained wild-type fibroblasts with 4′,6-diamidino-2-phenylindole (DAPI) to visualize chromatin, an RNA Pol II antibody specific for the phosphorylated Ser-5 (Ser5P) mark to detect transcriptionally active RNA Pol II, and an antibody specific for FUS. We noted that RNA Pol II Ser5P staining also appeared granular, with dense immunofluorescence staining within areas stained weakly for DAPI (Figure 4, A and B). This is consistent with active transcription occurring in regions of loose chromatin compaction, hence weaker staining by DAPI (Mo and Dynan, 2002; Zhu et al., 2004; Cisse et al., 2013). FUS staining also appeared granular, with dense FUS staining localized to regions of less densely compacted chromatin (Figure 4A). FUS granules somewhat colocalized with RNA Pol II granules (Figure 4, A and C), similar to published reports that FUS and its Drosophila homologue Cabeza localize to actively transcribed regions of chromosomal DNA (Immanuel et al., 1995; Kuroda et al., 2000). However, some RNA Pol II granules were largely devoid of FUS staining (Figure 4A). This is consistent with the observation that by ChIP-seq, approximately one-third of actively transcribed genes are not bound by or regulated by FUS protein (Schwartz et al., 2012).
FIGURE 4:

FUS and RNA Pol II immunofluorescence reveal their substantial colocalization and concentration in areas stained weakly by DAPI. (A) Nuclei stained with DAPI (stains DNA) and immunofluorescence for Ser5P (active RNA Pol II) and FUS. Both FUS and RNA Pol II show granular staining patterns within the nucleus of wild-type fibroblast cells. Yellow lines indicate line profiles quantified in B and C. (B) Quantification of relative fluorescence units (RFU) for Ser5P (red) and DAPI (blue) along the line in A, lower left. (C) Quantification of RFU for Ser5P (red) and FUS (green) along the line in A, lower right.
Loss of FUS or expression of mutant FUS disrupts RNA Pol II localization
We analyzed FUS and RNA Pol II Ser5P immunofluorescence in wild-type fibroblasts and ALS patient–derived fibroblasts. Owing to the complex granular staining of FUS and RNA Pol II in these fibroblasts, we were not able to determine with confidence whether any change in colocalization of FUS and RNA Pol II granules existed between wild-type and ALS patient–derived fibroblasts.
The most striking difference between these samples was in the distribution of active Ser5P RNA Pol II. In wild-type cells, Ser5P concentrated into several large granules clearly demarked by the reduced DAPI staining (Figure 5A). However, in mFUS and ΔNLS cells, these granules appeared increasingly fragmented, smaller, and more numerous (Figure 5, B–D). The number of Ser5P granules per cell was measured for two technical replicates (n > 30 cells per sample), revealing a significant difference between wild-type and ALS patient–derived cells (Figure 5E). In addition, the diameters of granules were measured (n > 200 granules per sample) to reveal that the distribution of granule diameters was substantially smaller for mFUS and ΔNLS samples (Figure 5F).
FIGURE 5:

RNA Pol II granules are more abundant and smaller in cells expressing mutant FUS or lacking FUS due to siRNA knockdown. (A–D) Four representative nuclei for wild-type, mutant FUS, or FUS knockdown. Scale bar (lower left), 5 μm. (E) The mean number of RNA Pol II-stained granules per nucleus is higher in mFUS, ΔNLS, and siFUS cells than in wild-type cells (n > 30 cells). Error bars, SD. **p < 0.0001, *p < 0.05, Student's t test. (F) The diameter of RNA Pol II granules at their widest cross section tends to be smaller in mFUS, ΔNLS, and siFUS cells than in wild-type cells (n > 200 granules). As with E, p < 0.0001 for mFUS and ΔNLS vs. wild-type and p < 0.05 for siFUS vs. wild-type.
We hypothesized that if changes in RNA Pol II distributions were due to a loss of FUS function, siFUS-treated wild-type fibroblasts should also show a similar trend. Ser5P distributions showed smaller but still discernible differences between siFUS and wild-type fibroblasts (Figure 5D). These differences were also statistically significant both in the number of granules per cell and their size (Figure 5, E and F). This again supports the hypothesis that the redistribution of RNA Pol II is due to a loss of FUS function.
DISCUSSION
We report here that FUS is sequestered in nuclear aggregates in fibroblasts derived from two ALS patients with FUS mutations We also make two new observations in the ALS patient fibroblasts that argue for a loss of FUS function: inappropriate Ser2P near the TSS of some genes and a disruption in the distribution of active RNA Pol II in the nucleus. These observations, along with reported disruptions in Gem formation and splicing defects, support the argument for a loss of normal FUS function associated with mutations in the NLS (Kabashi et al., 2011; Lagier-Tourenne, Polymenidou, Hutt, et al., 2012; Yamazaki et al., 2012; Tsuiji et al., 2013). We therefore present a model in which loss of FUS function observed here and in other published reports is due to FUS protein being trapped in nuclear aggregates. Of course, the existence of a loss of FUS function does not preclude an additional gain of function for the mutant protein.
The increase in Ser-2 phosphorylation for a subset of genes is consistent with a loss of FUS function. In our previous studies, we noted that the majority of genes in HEK293T/17 cells had much less Ser-2 phosphorylation near the TSS compared with the gene terminus. Therefore genome-wide changes in Ser2P upon loss of FUS were large and affected thousands of genes. Ser2P distributions were significantly different in fibroblast cells compared with HEK293T/17 cells, a result that we did not anticipate. Despite this difference, a subset of 625 genes was identified that showed changes in Ser2P upon mutation or siRNA knockdown of FUS. The observation that Ser2P is regulated differently in these fibroblasts provides evidence of a deficit in a nonneuronal cell type and raises further questions about how this modification may be regulated in motor neurons, the cells whose dysfunction is central to ALS disease. Ser2P is noted to affect RNA splicing, and human and mouse neuronal splicing is reported to be more affected by FUS mutations than for other cell types (Lagier-Tourenne, Polymenidou, Hutt, et al., 2012; Schwartz et al., 2012; Tsuiji et al., 2013).
FUS protein has a granular localization within the nucleus of fibroblast cells. The fact that these FUS granules overlap with sites enriched for active RNA Pol II is consistent with our previous finding in HEK293T/17 cells that FUS binds RNA Pol II near the TSS of many genes (Schwartz et al., 2012; Tan et al., 2012). That the colocalization is imperfect agrees with the fact that a significant subset of actively transcribed genes did not show enrichment for FUS in HEK293T/17 cells (Schwartz et al., 2012; Tan et al., 2012). The determinants of this specificity remain unknown. Although RNA binding is important for FUS to bind the CTD, FUS binds RNA promiscuously in vitro. The in vivo observation that regions of active RNA Pol II are spatially segregated in the nucleus from regions enriched for FUS may suggest a role for nuclear structures and organization in determining whether a specific gene is regulated by FUS (Meissner et al., 2003).
Finally, we report that the granular localization of active Ser5P RNA Pol II is altered in cells possessing FUS mutations. Granular staining of Ser5P RNA Pol II is more dramatic in fibroblasts than in HeLa or HEK293T/17 cells (unpublished data), which we hypothesize may be due to the slow cell cycle of fibroblasts compared with transformed cell lines. It is possible that these differences are even larger in nondividing neurons. However, it is not known to what extent disruptions in the granular localization of Ser5P leads to any further disruption in global transcription. We cannot exclude the possibility that these differences in Ser5P localization provide a clear marker of a loss in FUS function but have no further significance to normal cell biology.
Whereas nuclear aggregates can explain why FUS function is compromised, why NLS mutations cause nuclear aggregation of FUS is unclear. The simplest expectation for a loss of FUS function would be that NLS mutations would impede nuclear import and thereby lead to inappropriate cytoplasmic localization of FUS, which indeed is supported by some reports using overexpressed protein (Bosco et al., 2010; Kino et al., 2010; Vance et al., 2013). Alternatively, the NLS may have some additional function within the nucleus. Our in vitro studies with purified mutant FUS proteins revealed no changes in RNA binding, CTD binding, or formation of higher-order protein assemblies that might shed light on the nuclear aggregation (our unpublished data; Sun, Diaz, et al., 2011). A number of additional candidates for changes that might promote mutant FUS aggregation remain untested. For example, phosphorylation of the N-terminal low- complexity (LC) domain of the FUS protein modulates the ability to form molecular assemblies (Kato et al., 2012). Furthermore, methylation of arginines in the RGG motifs of FUS is modulated by nuclear–cytoplasmic shuttling of the FUS protein (Tradewell, Yu, et al., 2012; Yamaguchi and Kitajo, 2012; Scaramuzzino, Monaghan, et al., 2013). Whether arginine methylation affects aggregation itself is not known. Finally, the homologue of FUS, EWSR1, has been noted to be modified in its LC domain by O-GlcNAc, which regulates its ability to affect transcription (Bachmaier et al., 2009). If FUS is found to be O-GlcNAc modified like EWSR1, then modifications that have the potential to affect FUS assembly may not be limited to phosphorylation or methylation.
In summary, our observations strengthen the conclusion that FUS function is compromised by ALS disease–causing mutations. Furthermore, we present a clear mechanism for this loss of function—the entrapment of FUS protein in nuclear aggregates. Whether such nuclear aggregation also occurs in motor neurons is a key unanswered question. The mechanism by which FUS is entrapped in these nuclear aggregates also remains to be established. Nevertheless, even though the ΔNLS fibroblasts clearly show cytoplasmic FUS protein, our dot blot analysis suggests that the bulk of aggregated FUS protein in these cells is, in fact, in the nucleus. The identification and characterization of this aggregation represents a significant step forward in our understanding of normal FUS biology and the molecular consequences of ALS-causing FUS mutations.
MATERIALS AND METHODS
Cell culture
Fibroblast cells were grown in DMEM (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS), Gluta-max (Life Technologies, Carlsbad, CA), and penicillin/ampicillin antibiotics. Cells were fed with fresh media three times per week. Cells were maintained at a high confluency (no less than 70% confluency or 1.2 million cells/15-cm dish) and split 1:2 every 1–1.5 wk. At harvest, ∼2–2.5 million cells were recovered from each 15-cm dish. Cells were transfected with siRNA targeting FUS (Schwartz et al., 2012) at a 25 nM final concentration using RNAiMAX (Life Technologies; 19.2 μl/15-cm dish) using the reverse-transfection protocol. RNAiMAX and siRNA were incubated for 20 min in Optimem (Life Technologies), and then 1.2 million cells were added and the volume brought up to 20 ml with normal growth medium.
Western analysis
Cells were grown to high confluency (2.4 million cells/dish) and harvested with 0.25% trypsin (Life Technologies). Cell pellets were washed with phosphate-buffered saline (PBS) and cells counted to control for loading. Total cell lysates were prepared by adding 4× NuPage LDS sample buffer (Life Technologies) directly to cell pellets, boiling at 95ºC for 5 min, and loading equal cell numbers onto NuPage 4–12% Bis-Tris gels (Life Technologies). Nuclear and cytoplasmic fractions were separated by incubating cells with hypotonic lysis buffer (10 mM Tris-HCl, pH 7.5, 10 mM NaCl, 3 mM MgCl2, and 0.5% [vol/vol] NP-40) for 4 min on ice. Lysis was monitored by microscopy to ensure that nuclei were intact. Nuclei were pelleted by centrifugation (500 × g for 4 min) and the supernatant saved as the cytoplasmic fraction. Nuclei were lysed in lysis buffer (50 mM Tris HCl, 120 mM NaCl, 1% SDS, 1 mM EDTA, 50 mM DTT, and protease inhibitor cocktail [Roche, Indianapolis, IN]), sonicated, and boiled at 95ºC for 5 min. Each was diluted into 1× NuPage LDS sample buffer, loaded onto a NuPage 4x12% Bis-Tris gel, and run in NuPage MES SDS running buffer (Life Technologies).
Proteins were transferred to Amersham Hybond ECL membrane (GE Healthcare, Piscataway, NJ) by electroblotting using 0.5 A for 2 h. For dot blotting, lysates were added to 150 μl of transfer buffer and spotted onto the Hybond ECL membrane using a dot blot apparatus. Blots were probed with anti-FUS (4H11; sc-47711; Santa Cruz Biotechnology, Dallas, TX), anti-H3 (ab1791; Abcam, Cambridge, UK), or anti–glyceraldehyde-3-phosphate dehydrogenase (sc-137179; Santa Cruz Biotechnology).
ChIP-seq experiments
Twenty 15-cm dishes were transfected or seeded without transfection for each fibroblast cell culture. Cells were allowed to grow for 5 d after transfection before harvesting. ChIP experiments were performed essentially as described (Schwartz et al., 2012). Cells were cross-linked in 1% formaldehyde solution in PBS. Cross-linking was stopped by addition of 125 mM glycine dissolved in PBS. Cells were harvested by scraping and combined. Cells were lysed in lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1, 1× protease inhibitors [Roche]) and sonicated for 15 min with a Bioruptor (Diagenode, Denville, NJ) per the manufacturer's recommendations. Lysates were diluted 10-fold in IP buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl, 1× protease inhibitors [Roche]) and 10 μg of antibodies against Ser2P RNA Pol II as described (Schwartz et al., 2012).
Immunofluorescence
Forty thousand cells were transfected with siRNA or seeded without transfection onto poly-l-lysine–coated cover slips (Neuvitro, El Monte, CA) in 12-well dishes and grown for 5 d. Cells were cross-linked with 4% formaldehyde (Sigma-Aldrich, St. Louis, MO) in PBS for 15 min at room temperature with shaking. Cells were washed three times with PBS and then incubated for 1 h at room temperature in permeabilization/blocking buffer (0.5% Triton X-100, 3% bovine serum albumin in PBS). Antibodies were then added at 1:350 dilutions and incubated with shaking for 1–2 h at 30ºC. Slides were washed three times with PBS at room temperature. Slides were mounted with Vectashield (Vector Laboratories, Burlingame, CA) mounting medium with DAPI, sealed with nail polish, and allowed to dry overnight.
Antibodies used were anti-FUS (4H11; sc-47711; Santa Cruz Biotechnology) or anti-Ser5P (ab5131; Abcam). Secondary antibodies used were Alexa Fluor 546 donkey anti-mouse or Alexa Fluor 633 goat anti-rabbit (Life Technologies). Images were collected on a DeltaVision Elite System (GE Healthcare) using a 60× objective. Images shown are a representative slice from a z-stack (3.5- to 7.5-μm stack, 0.5-μm slices).
Image analysis
Images were analyzed using ImageJ software (National Institutes of Health, Bethesda, MD). Quantification began with randomization of images in order to blind the researcher performing the analysis. Each slice was inspected to ensure continuity of granules through the z-slices. Numbers of maxima were counted (n = 30–35 cells, two biological replicates), and the widest diameter among the slices was measured (n = 200–250 granules, two biological replicates).
Acknowledgments
We thank M. Cudkowicz and the Neurological Clinical Research Institute at Massachusetts General Hospital. We also thank R. Brown (University of Massachusetts Medical School, Worcester, MA) for the coordination of patient recruitment. J.C.S. was supported by National Institute of General Medical Sciences National Research Service Award Fellowship 1F32GM095311-01 and National Institute of Neurological Disorders and Stroke 1K99NS082376-01A1. S.S.W.H. was supported by a Clinician Development Award from the American Academy of Neurology/ALS Association and the Harvard Catalyst KL2 Medical Research Investigator Training Program. T.R.C. is an investigator and K.C.E. is an Early Career Scientist of the Howard Hughes Medical Institute.
Abbreviations used:
- ALS
amyotrophic lateral sclerosis
- bp
base pairs
- ChIP-seq
chromatin immunoprecipitation followed by high-throughput sequencing
- CTD
carboxy-terminal domain of RNA Pol II
- DAPI
4’,6-diamidino-2-phenylindole
- DTT
dithiothreitol
- EWSR1
Ewing sarcoma RNA-binding protein 1
- FUS
Fused in Sarcoma
- IF
immunofluorescence
- LC
low complexity
- LDS
lithium dodecyl sulfate
- NLS
nuclear localization signal
- PBS
phosphate-buffered saline
- RNA Pol II
RNA polymerase II
- RPM
reads per million
- Ser2p
CTD phosphorylated at Ser-2
- Ser5p
CTD phosphorylated at Ser-5
- siFUS
small interfering RNA knockdown of FUS
- TSS
transcription start site
- WC
whole cell
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-05-1007) on July 9, 2014.
REFERENCES
Boldface names denote co–first authors.
- Alliegro MC, Alliegro MA. A nuclear protein regulated during the transition from active to quiescent phenotype in cultured endothelial cells. Dev Biol. 1996;174:288–297. doi: 10.1006/dbio.1996.0074. [DOI] [PubMed] [Google Scholar]
- Bachmaier R, Aryee DN, Jug G, Kauer M, Kreppel M, Lee KA, Kovar H. O-GlcNAcylation is involved in the transcriptional activity of EWS-FLI1 in Ewing's sarcoma. Oncogene. 2009;28:1280–1284. doi: 10.1038/onc.2008.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ, Jr, Sapp P, McKenna-Yasek D, Brown RH, Jr, Hayward LJ. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet. 2010;19:4160–4175. doi: 10.1093/hmg/ddq335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvio C, Neubauer G, Mann M, Lamond AI. Identification of hnRNP P2 as TLS/FUS using electrospray mass spectrometry. RNA. 1995;1:724–733. [PMC free article] [PubMed] [Google Scholar]
- Cisse II, Izeddin I, Causse SZ, Boudarene L, Senecal A, Muresan L, Dugast-Darzacq C, Hajj B, Dahan M, Darzacq X. Real-time dynamics of RNA polymerase II clustering in live human cells. Science. 2013;341:664–667. doi: 10.1126/science.1239053. [DOI] [PubMed] [Google Scholar]
- Crozat A, Aman P, Mandahl N, Ron D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature. 1993;363:640–644. doi: 10.1038/363640a0. [DOI] [PubMed] [Google Scholar]
- Das R, Yu J, Zhang Z, Gygi MP, Krainer AR, Gygi SP, Reed R. SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol Cell. 2007;26:867–881. doi: 10.1016/j.molcel.2007.05.036. [DOI] [PubMed] [Google Scholar]
- Fujii R, Okabe S, Urushido T, Inoue K, Yoshimura A, Tachibana T, Nishikawa T, Hicks GG, Takumi T. The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr Biol. 2005;15:587–593. doi: 10.1016/j.cub.2005.01.058. [DOI] [PubMed] [Google Scholar]
- Fujii R, Takumi T. TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J Cell Sci. 2005;118:5755–5765. doi: 10.1242/jcs.02692. [DOI] [PubMed] [Google Scholar]
- Glover-Cutter K, Kim S, Espinosa J, Bentley DL. RNA polymerase II pauses and associates with pre-mRNA processing factors at both ends of genes. Nat Struct Mol Biol. 2008;15:71–78. doi: 10.1038/nsmb1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu B, Eick D, Bensaude O. CTD serine-2 plays a critical role in splicing and termination factor recruitment to RNA polymerase II in vivo. Nucleic Acids Res. 2013;41:1591–1603. doi: 10.1093/nar/gks1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackl W, Luhrmann R. Molecular cloning and subcellular localisation of the snRNP-associated protein 69KD, a structural homologue of the proto-oncoproteins TLS and EWS with RNA and DNA-binding properties. J Mol Biol. 1996;264:843–851. doi: 10.1006/jmbi.1996.0681. [DOI] [PubMed] [Google Scholar]
- Halliday G, Bigio EH, Cairns NJ, Neumann M, Mackenzie IR, Mann DM. Mechanisms of disease in frontotemporal lobar degeneration: gain of function versus loss of function effects. Acta Neuropathol. 2012;124:373–382. doi: 10.1007/s00401-012-1030-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallier M, Lerga A, Barnache S, Tavitian A, Moreau-Gachelin F. The transcription factor Spi-1/PU.1 interacts with the potential splicing factor TLS. J Biol Chem. 1998;273:4838–4842. doi: 10.1074/jbc.273.9.4838. [DOI] [PubMed] [Google Scholar]
- Hoell JI, Larsson E, Runge S, Nusbaum JD, Duggimpudi S, Farazi TA, Hafner M, Borkhardt A, Sander C, Tuschl T. RNA targets of wild-type and mutant FET family proteins. Nat Struct Mol Biol. 2011;18:1428–1431. doi: 10.1038/nsmb.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iko Y, Kodama TS, Kasai N, Oyama T, Morita EH, Muto T, Okumura M, Fujii R, Takumi T, Tate S, et al. Domain architectures and characterization of an RNA-binding protein, TLS. J Biol Chem. 2004;279:44834–44840. doi: 10.1074/jbc.M408552200. [DOI] [PubMed] [Google Scholar]
- Immanuel D, Zinszner H, Ron D. Association of SARFH (sarcoma-associated RNA-binding fly homolog) with regions of chromatin transcribed by RNA polymerase II. Mol Cell Biol. 1995;15:4562–4571. doi: 10.1128/mcb.15.8.4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigaki S, Masuda A, Fujioka Y, Iguchi Y, Katsuno M, Shibata A, Urano F, Sobue G, Ohno K. Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci Rep. 2012;2:529. doi: 10.1038/srep00529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Bercier V, Lissouba A, Liao M, Brustein E, Rouleau GA, Drapeau P. FUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet. 2011;7:e1002214. doi: 10.1371/journal.pgen.1002214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–767. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Erickson B, Luo W, Seward D, Graber JH, Pollock DD, Megee PC, Bentley DL. Gene-specific RNA polymerase II phosphorylation and the CTD code. Nat Struct Mol Biol. 2010;17:1279–1286. doi: 10.1038/nsmb.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kino Y, Washizu C, Aquilanti E, Okuno M, Kurosawa M, Yamada M, Doi H, Nukina N. Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucleic Acids Res. 2010;39:2781–2798. doi: 10.1093/nar/gkq1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda M, Sok J, Webb L, Baechtold H, Urano F, Yin Y, Chung P, de Rooij DG, Akhmedov A, Ashley T, et al. Male sterility and enhanced radiation sensitivity in TLS(-/-) mice. EMBO J. 2000;19:453–462. doi: 10.1093/emboj/19.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Kwon I, Kato M, Xiang S, Wu L, Theodoropoulos P, Mirzaei H, Han T, Xie S, Corden JL, McKnight SL. Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell. 2013;155:1049–1060. doi: 10.1016/j.cell.2013.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling SC, Liang TY, Mazur C, et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci. 2012;15:1488–1497. doi: 10.1038/nn.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat. 2013;34:812–826. doi: 10.1002/humu.22319. [DOI] [PubMed] [Google Scholar]
- Lerga A, Hallier M, Delva L, Orvain C, Gallais I, Marie J, Moreau-Gachelin F. Identification of an RNA binding specificity for the potential splicing factor TLS. J Biol Chem. 2001;276:6807–6816. doi: 10.1074/jbc.M008304200. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Munoz DG, Kusaka H, Yokota O, Ishihara K, Roeber S, Kretzschmar HA, Cairns NJ, Neumann M. Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol. 2011;121:207–218. doi: 10.1007/s00401-010-0764-0. [DOI] [PubMed] [Google Scholar]
- Mayer A, Lidschreiber M, Siebert M, Leike K, Soding J, Cramer P. Uniform transitions of the general RNA polymerase II transcription complex. Nat Struct Mol Biol. 2010;17:1272–1278. doi: 10.1038/nsmb.1903. [DOI] [PubMed] [Google Scholar]
- Meissner M, Lopato S, Gotzmann J, Sauermann G, Barta A. Proto-oncoprotein TLS/FUS is associated to the nuclear matrix and complexed with splicing factors PTB, SRm160, and SR proteins. Exp Cell Res. 2003;283:184–195. doi: 10.1016/s0014-4827(02)00046-0. [DOI] [PubMed] [Google Scholar]
- Mo X, Dynan WS. Subnuclear localization of Ku protein: functional association with RNA polymerase II elongation sites. Mol Cell Biol. 2002;22:8088–8099. doi: 10.1128/MCB.22.22.8088-8099.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz MJ, Perez Santangelo MS, Paronetto MP, de la Mata M, Pelisch F, Boireau S, Glover-Cutter K, Ben-Dov C, Blaustein M, Lozano JJ, et al. DNA damage regulates alternative splicing through inhibition of RNA polymerase II elongation. Cell. 2009;137:708–720. doi: 10.1016/j.cell.2009.03.010. [DOI] [PubMed] [Google Scholar]
- Powers CA, Mathur M, Raaka BM, Ron D, Samuels HH. TLS (translocated-in-liposarcoma) is a high-affinity interactor for steroid, thyroid hormone, and retinoid receptors. Mol Endocrinol. 1998;12:4–18. doi: 10.1210/mend.12.1.0043. [DOI] [PubMed] [Google Scholar]
- Scaramuzzino C, Monaghan J, Milioto C, Lanson NA, Jr, Maltare A, Aggarwal T, Casci I, Fackelmayer FO, Pennuto M, Pandey UB. Protein arginine methyltransferase 1 and 8 interact with FUS to modify its sub-cellular distribution and toxicity in vitro and in vivo. PLoS One. 2013;8:e61576. doi: 10.1371/journal.pone.0061576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JC, Ebmeier CC, Podell ER, Heimiller J, Taatjes DJ, Cech TR. FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes Dev. 2012;26:2690–2695. doi: 10.1101/gad.204602.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JC, Wang X, Podell ER, Cech TR. RNA seeds higher-order assembly of FUS protein. Cell Rep. 2013;5:918–925. doi: 10.1016/j.celrep.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Diaz Z, Fang X, Hart MP, Chesi A, Shorter J, Gitler AD. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 2011;9:e1000614. doi: 10.1371/journal.pbio.1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AY, Manley JL. TLS inhibits RNA polymerase III transcription. Mol Cell Biol. 2010;30:186–196. doi: 10.1128/MCB.00884-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AY, Riley TR, Coady T, Bussemaker HJ, Manley JL. TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proc Natl Acad Sci USA. 2012;109:6030–6035. doi: 10.1073/pnas.1203028109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tradewell ML, Yu Z, Tibshirani M, Boulanger MC, Durham HD, Richard S. Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum Mol Genet. 2012;21:136–149. doi: 10.1093/hmg/ddr448. [DOI] [PubMed] [Google Scholar]
- Tsuiji H, Iguchi Y, Furuya A, Kataoka A, Hatsuta H, Atsuta N, Tanaka F, Hashizume Y, Akatsu H, Murayama S, et al. Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol Med. 2013;5:221–234. doi: 10.1002/emmm.201202303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Scotter EL, Nishimura AL, Troakes C, Mitchell JC, Kathe C, Urwin H, Manser C, Miller CC, Hortobagyi T, et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum Mol Genet. 2013;22:2676–2688. doi: 10.1093/hmg/ddt117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Arai S, Song X, Reichart D, Du K, Pascual G, Tempst P, Rosenfeld MG, Glass CK, Kurokawa R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454:126–130. doi: 10.1038/nature06992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi A, Kitajo K. The effect of PRMT1-mediated arginine methylation on the subcellular localization, stress granules, and detergent-insoluble aggregates of FUS/TLS. PLoS One. 2012;7:e49267. doi: 10.1371/journal.pone.0049267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Chen S, Yu Y, Yan B, Haertlein TC, Carrasco MA, Tapia JC, Zhai B, Das R, Lalancette-Hebert M, et al. FUS-SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep. 2012;2:799–806. doi: 10.1016/j.celrep.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Chansky HA, Hickstein DD. EWS.Fli-1 fusion protein interacts with hyperphosphorylated RNA polymerase II and interferes with serine-arginine protein-mediated RNA splicing. J Biol Chem. 2000;275:37612–37618. doi: 10.1074/jbc.M005739200. [DOI] [PubMed] [Google Scholar]
- Zhu X, Zeng X, Huang B, Hao S. Actin is closely associated with RNA polymerase II and involved in activation of gene transcription. Biochem Biophys Res Commun. 2004;321:623–630. doi: 10.1016/j.bbrc.2004.05.229. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Albalat R, Ron D. A novel effector domain from the RNA-binding protein TLS or EWS is required for oncogenic transformation by CHOP. Genes Dev. 1994;8:2513–2526. doi: 10.1101/gad.8.21.2513. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Sok J, Immanuel D, Yin Y, Ron D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci. 1997;110:1741–1750. doi: 10.1242/jcs.110.15.1741. [DOI] [PubMed] [Google Scholar]