

Myosin A, an unconventional class XIV myosin of the protozoan parasite Toxoplasma gondii, undergoes calcium-dependent phosphorylation, providing a mechanism by which the parasite can regulate motility-based processes such as escape from the infected host cell at the end of the parasite's lytic cycle.
Abstract
Class XIVa myosins comprise a unique group of myosin motor proteins found in apicomplexan parasites, including those that cause malaria and toxoplasmosis. The founding member of the class XIVa family, Toxoplasma gondii myosin A (TgMyoA), is a monomeric unconventional myosin that functions at the parasite periphery to control gliding motility, host cell invasion, and host cell egress. How the motor activity of TgMyoA is regulated during these critical steps in the parasite's lytic cycle is unknown. We show here that a small-molecule enhancer of T. gondii motility and invasion (compound 130038) causes an increase in parasite intracellular calcium levels, leading to a calcium-dependent increase in TgMyoA phosphorylation. Mutation of the major sites of phosphorylation altered parasite motile behavior upon compound 130038 treatment, and parasites expressing a nonphosphorylatable mutant myosin egressed from host cells more slowly in response to treatment with calcium ionophore. These data demonstrate that TgMyoA undergoes calcium-dependent phosphorylation, which modulates myosin-driven processes in this important human pathogen.
INTRODUCTION
Parasites of the phylum Apicomplexa infect a wide range of hosts and are responsible for severe human and veterinary diseases such as malaria, toxoplasmosis, cryptosporidiosis, and babesiosis. Infection by Toxoplasma gondii, the causative agent of toxoplasmosis, can result in fatal brain defects in the fetus during pregnancy and can be life threatening in immunocompromised individuals (Wong and Remington, 1993). The rapidly dividing T. gondii tachyzoite causes acute infection through repeated cycles of host cell invasion and lysis (Black and Boothroyd, 2000). Invasion is a multistep process that involves attachment to the host cell, reorientation, formation of a “moving junction” (sometimes also referred to as a tight junction), penetration through the moving junction into an induced invagination in the host cell membrane, and pinching off of the invaginated membrane to form a vacuole within which the intracellular parasite subsequently grows and multiplies (Suss-Toby et al., 1996; Mital et al., 2005; Giovannini et al., 2011; Sibley, 2011; Tonkin et al., 2011; Meissner et al., 2013; Sinai, 2014). The parasites then actively disrupt and escape their host cell to disseminate throughout the body, crossing biological barriers and moving between cells by a unique form of substrate-dependent gliding motility that involves no motile appendages such as cilia or flagella and no changes in cell shape (Black and Boothroyd, 2000; Carruthers and Boothroyd, 2007; Sibley, 2004).
The machinery of T. gondii that controls parasite motility is a multisubunit motor complex, at the heart of which is T. gondii myosin A (TgMyoA). Genetic disruption of TgMyoA inhibits motility, invasion, and host cell egress (Meissner et al., 2002; Andenmatten et al., 2013; Egarter et al., 2014). TgMyoA is the founding member of a class of unconventional myosins (class XIVa) present only in apicomplexan parasites and ciliates, the motor domains of which share only 23–34% identity with mammalian myosins (Heintzelman and Schwartzman, 1997). Furthermore, class XIVa myosins do not follow the TEDS rule (Bement and Mooseker, 1995), lack a conserved glycine at the lever arm pivot point, and have very short C-terminal tails, which in other myosins play a critical role in mechanochemical function and regulation of motor activity (Heintzelman and Schwartzman, 1997; Bosch et al., 2007; Frenal et al., 2008). TgMyoA is indirectly anchored at the parasite periphery via binding of its tail to myosin light chain-1 (TgMLC1). TgMLC1 in turn binds to a palmitoylated and myristoylated protein, TgGAP45, which is anchored in the inner membrane complex (IMC), an interconnected series of flattened vesicles beneath the parasite plasma membrane (Porchet and Torpier, 1977; Rabjeau et al., 1994, 1997; Frenal et al., 2010; Supplemental Figure S1). TgMyoA, TgMLC1, and TgGAP45 bind as a complex to TgGAP50, an integral membrane protein that is immobilized within detergent-resistant domains of the IMC, further anchoring the motor complex (Johnson et al., 2007; Frenal et al., 2010). An essential light chain (TgELC1; Nebl et al., 2011) and a polytopic transmembrane protein of unknown function (TgGAP40; Frenal et al., 2010) were recently identified as additional components of the motor complex.
In the linear motor model that has dominated the field for the last decade, TgMyoA walks along short actin filaments that polymerize transiently between the parasite IMC and plasma membrane (Heintzelman and Schwartzman, 1997; Herm-Gotz et al., 2002; Wetzel et al., 2003; Sahoo et al., 2006; Skillman et al., 2011). The actin filaments are bound to the cytosolic tails of transmembrane surface adhesins through a bridging protein (Jewett and Sibley, 2003; Shen and Sibley, 2014; Sheiner et al., 2010). The extracellular domains of the adhesins bind to ligands on the substrate (or host cell) and, because actin is physically connected to the substrate through the surface adhesins, whereas TgMyoA is anchored in the IMC (Supplemental Figure S1), as TgMyoA walks along the actin, the parasite moves relative to the ligands on the substrate. Recent data challenge this model (Munter et al., 2009; Andenmatten et al., 2013; Egarter et al., 2014); rather than producing the force necessary for motility, myosin and actin could instead be required to form and release attachment sites, thereby defining the directionality and mode of parasite motility that is driven by some other mechanism (Meissner et al., 2013; Egarter et al., 2014). Although there is controversy about the precise role of TgMyoA in motility, there is little disagreement about its importance (Meissner et al., 2002; Andenmatten et al., 2013).
Intracellular tachyzoites are immotile, whereas egressing and extracellular tachyzoites are highly motile. Extracellular parasites show three distinct types of myosin-mediated motility on a two-dimensional (2D) surface: circular gliding, helical gliding, and upright twirling (Hakansson et al., 1999). The parasites also move at different speeds and sometimes pause while gliding (Hakansson et al., 1999). Motility within a three-dimensional (3D) model extracellular matrix is even more complex; parasites move along irregular corkscrew-like trajectories, with periodic changes in velocity and in the curvature and torsion of their trajectory (Leung et al., 2014a). How the parasite regulates the activity of its myosin motor to produce these motile behaviors is unknown. Recent phosphoproteomics studies revealed that phosphorylation occurs on most of the components of the TgMyoA motor complex in tachyzoites, including TgMyoA itself (Nebl et al., 2011; Treeck et al., 2011), suggesting that phosphorylation is an important regulatory mechanism. Although phosphorylation regulates the function of many nonmuscle myosin heavy chains (Redowicz, 2001), nothing is known about the biological relevance of class XIVa myosin phosphorylation. Genetic and/or chemical disruption of a variety of apicomplexan protein kinases affects motility-based processes (Donald et al., 2002; Lourido et al., 2010, 2012, 2013; Garrison et al., 2012; McCoy et al., 2012), but whether TgMyoA is a relevant substrate of any of the kinases affected is also unknown.
Pharmacological perturbation is one approach to understanding the regulatory mechanisms governing parasite motility, invasion, and egress. For example, membrane-permeant calcium chelators block all three processes, at least in part through inhibiting the secretion of surface adhesins from apical secretory organelles known as micronemes. Conversely, treatment with calcium ionophore increases microneme secretion and stimulates parasite egress (Endo, 1982; Carruthers et al., 1999; Carruthers and Sibley, 1999; Wetzel et al., 2004). How the intracellular calcium signaling pathways that mediate these processes might operate at the level of the TgMyoA motor complex is unknown. A high-throughput small-molecule screen in our laboratory identified 21 novel inhibitors of parasite motility, one of which was subsequently shown to bind directly to TgMLC1, leading to reduced myosin motor activity (Carey et al., 2004; Heaslip et al., 2010; Leung et al., 2014b). Unexpectedly, the screen also identified six enhancers of parasite motility. Although these compounds have little in common structurally, they all trigger microneme secretion and enhance motility and invasion, suggesting that they act through either common or converging signaling pathways.
We show here that treating parasites with the enhancers causes a significant increase in TgMyoA phosphorylation. Focusing on one of the enhancers, compound 130038, we show that this effect is due to the enhancer's capacity to induce intracellular calcium release in the parasite, and treatment of parasites with calcium ionophore increases TgMyoA phosphorylation even in the absence of the enhancer. These observations provided an opportunity to study the biological function of class XIVa myosin phosphorylation. By mapping and mutating the major sites of TgMyoA phosphorylation, we show that an inability to phosphorylate TgMyoA alters parasite motility in the presence of enhancer and slows the kinetics of host cell egress, demonstrating that TgMyoA phosphorylation modulates the phenotypic output of the calcium signaling pathways that regulate these processes.
RESULTS
Phosphorylation of TgMyoA is increased by a small-molecule enhancer of motility and invasion
The enhancers identified in our earlier high-throughput screen exert their effect on parasite motility within 10 min after compound addition, strongly suggesting posttranslational mechanism(s) of action. Given that TgMyoA is known to be phosphorylated on multiple sites in tachyzoites (Nebl et al., 2011; Treeck et al., 2011), we tested whether the enhancers alter TgMyoA phosphorylation. T. gondii tachyzoites were metabolically labeled with 32P before treatment with either dimethyl sulfoxide (DMSO; vehicle control) or individual enhancers (compounds 153753, 141852, 130038, and 158513; Carey et al., 2004), and the TgMyoA motor complex was then immunoprecipitated with an antibody against TgGAP45. The phosphorylation profile of the isolated motor complex revealed that both GAP45 and TgMyoA are phosphorylated at a basal level in control parasites and that phosphorylation of TgMyoA (but not TgGAP45) is increased by each of the enhancers (Figure 1). Of the four enhancers tested, 130038 consistently enhanced TgMyoA phosphorylation to the greatest extent; it was therefore chosen for subsequent experiments.
FIGURE 1:
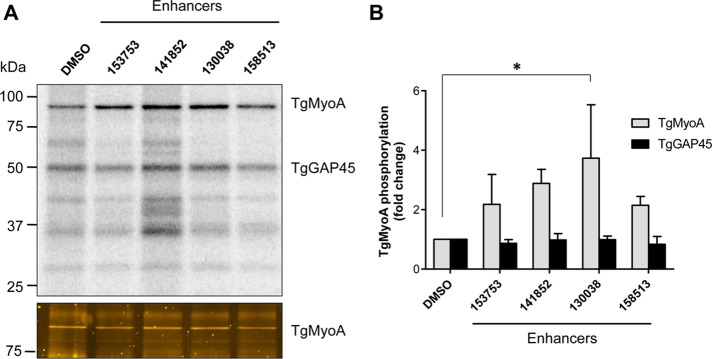
TgMyoA phosphorylation is enhanced upon treatment with small-molecule enhancers of motility and invasion. (A) RH strain T. gondii tachyzoites metabolically labeled with [32P]orthophosphoric acid were exposed to either 100 μM small-molecule enhancers 153753, 141852, 130038, or 158513 (previously referred to as Enhancers 5, 3, 1, and 6, respectively; Carey et al., 2004) or an equivalent amount of DMSO (vehicle control) for 20 min. The TgMyoA motor complex was then isolated by immunoprecipitation using anti-GAP45 antibody and resolved by SDS–PAGE. Two major phosphorylated bands were visualized by phosphorimaging: TgMyoA (∼93 kDa) and TgGAP45 (∼50 kDa; top). The other 32P–labeled bands in the immunoprecipitate likely include TgGAP40 (∼37 kDa) and TgMLC1 (∼30 kDa), both of which are also know to be multiply phosphorylated (Jacot and Soldati-Favre, 2012). The 32P signals associated with TgMyoA and TgGAP45 were quantified and normalized to the relative amount of TgMyoA protein in the immunoprecipitate (bottom, Sypro-Ruby stain; only the relevant portion of the gel is shown). (B) The normalized TgMyoA and TgGAP45 phosphorylation levels for each treatment are shown as fold change relative to DMSO. *p < 0.05 (one-way analysis of variance [ANOVA]). Experiments were repeated at least twice for each compound; bars, mean value; error bars, SD.
To identify sites of phosphorylation on TgMyoA in enhancer-treated parasites, a strain ectopically expressing FLAG-tagged TgMLC1 (Heaslip et al., 2010) was treated with 130038, and the TgMyoA motor complex was immunoaffinity purified and resolved by SDS–PAGE. Liquid chromatography–tandem mass spectrometry analysis of the excised TgMyoA band identified three phosphorylation sites, S20, S21, and S29 (Figure 2A and Supplemental Figure S2, A–C). Quantitative analysis revealed S21 to be the most frequently phosphorylated of the three sites; phosphorylated S20 or S29 was only detected on peptides that were also phosphorylated on S21 (Supplemental Figure S2, B and C, and unpublished data).
FIGURE 2:

Generation of parasites stably expressing TgMyoA phosphorylation-site mutations. (A) Schematic representation of phosphorylation sites on TgMyoA. The pink vertical lines represent phosphorylation sites identified in this study (S29) or in this study and by others (S20, S21; Nebl et al., 2011; Treeck et al., 2011; www.toxodb.org). The different domains of TgMyoA are shown in light purple (N-terminal domain; amino acids [aa] 1–93, uncharacterized function), green (motor domain; aa 94–774), yellow (tail domain; aa 775–831), and blue (IQ motif; aa 808–814). (B) TgMyoA mutations generated in this study and the abbreviated names of the strains expressing them. (C) N-terminally FLAG-tagged TgMyoA (wild type or mutant) was expressed in TgMyoA KO parasites under the control of the endogenous TgMyoA promoter. Lysates from each of the strains were analyzed by SDS–PAGE and Western blotting with anti-TgMyoA (green); anti-TgGRA8 (red) was used as a loading control. The TgMyoA WT6A and TgMyoA SAAA6A samples were from a twice-bleomycin-selected population (>98% FLAG-positive parasites by immunofluorescence), whereas all other parasite lines were clonal. (D) The cellular distributions of the expressed proteins were analyzed by immunofluorescence microscopy using mouse anti-FLAG antibody (red); rabbit anti-TgIMC1 was used to label the inner membrane complex (green). Scale bars, 10 μm.
To test whether these three sites can account for the enhanced phosphorylation seen in response to 130038, we mutated either S21 alone or all three serines (S20, S21, and S29) simultaneously to alanine (TgMyoA S21A and TgMyoA SAAA, respectively; Figure 2B). We also generated a phosphomimetic mutant in which all three serines were mutated to negatively charged amino acids (TgMyoA SEDD; Figure 2B). N-terminally FLAG-tagged constructs encoding either wild-type (WT) TgMyoA or the phosphorylation-site mutations were expressed in a recently developed TgMyoA knockout strain (TgMyoA KO), which is viable in culture but impaired in motility, invasion, and host cell egress (Andenmatten et al., 2013). Western blot and immunofluorescence analysis were used to confirm equivalent FLAG-TgMyoA expression levels and proper localization for each of the expressed proteins (Figure 2, C and D). The expression level of the FLAG-tagged TgMyoA constructs is also comparable to that of TgMyoA in RH strain (Supplemental Figure S3), although parasites expressing FLAG-tagged MyoA grow slightly more slowly (Supplemental Figure S4), presumably due to a minor effect of the N-terminal FLAG tag on TgMyoA activity.
Incorporation of 32P into the expressed TgMyoA was then evaluated with or without enhancer treatment (Figure 3A). The two mutants showed markedly less basal 32P incorporation than TgMyoA WT (on average, 4.9-fold reduction for TgMyoA S21A and 8-fold reduction for TgMyoA SAAA; Figure 3B, DMSO). As with TgMyoA WT, phosphorylation of the mutants increased in response to 130038 treatment, (on average, 4.2-fold for TgMyoA WT, 2.9-fold for TgMyoA S21A, and 2.2-fold for TgMyoA SAAA), but, because basal phosphorylation was lower in the mutants than TgMyoA WT, the final level of 32P incorporation after enhancer treatment was also markedly reduced in the mutants (on average, 7.2- and 15.4-fold less than TgMyoA WT for TgMyoA S21A and TgMyoA SAAA, respectively; Figure 3B, 130038). These three serines, and S21 in particular, are therefore the major determinants of both basal TgMyoA phosphorylation and the enhanced phosphorylation observed upon treatment with 130038.
FIGURE 3:
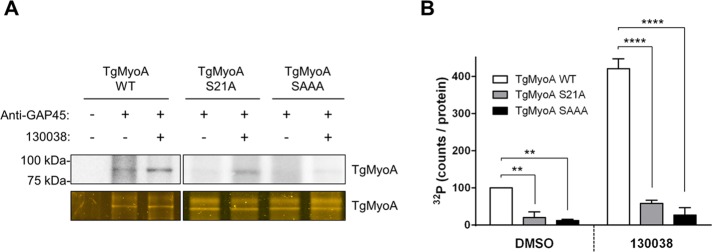
The S21A and SAAA mutations reduce basal and 1300038-enhanced TgMyoA phosphorylation. (A) Tachyzoites of the indicated strains were metabolically labeled with [32P]orthophosphoric acid and exposed to either 100 μM 130038 (+) or DMSO (vehicle; –) for 20 min. The TgMyoA motor complex was then isolated by immunoprecipitation using anti-TgGAP45 antibody and resolved by SDS–PAGE; the 32P signal associated with TgMyoA was analyzed as in Figure 1, except that the relative amount of TgMyoA present in the immunoprecipitate was determined by Oriole (bottom) rather than Sypro Ruby staining. (B) TgMyoA phosphorylation under each condition shown as the intensity of the TgMyoA-associated 32P signal, normalized to the amount TgMyoA protein present in the immunoprecipitate. Values are plotted relative to DMSO-treated TgMyoA WT, which was arbitrarily set to 100. Experiments were repeated twice; error bars, SD. **p < 0.01 and ****p < 0.0001 (two-way ANOVA analysis and Bonferroni multiple comparisons test).
The 130038-enhanced phosphorylation of TgMyoA is calcium dependent
Enhancer 130038 stimulates gliding motility, invasion, and microneme secretion (Carey et al., 2004), which are all processes controlled by calcium-dependent signaling pathways (Billker et al., 2009). To test whether 130038 requires calcium to up-regulate TgMyoA phosphorylation, we treated 32P-labeled parasites with BAPTA-AM, an intracellular calcium chelator, before 130038 treatment. Pretreatment with BAPTA-AM blocked the 130038-enhanced phosphorylation of TgMyoA (Figure 4, A and B), suggesting that the enhanced phosphorylation is indeed calcium dependent.
FIGURE 4:
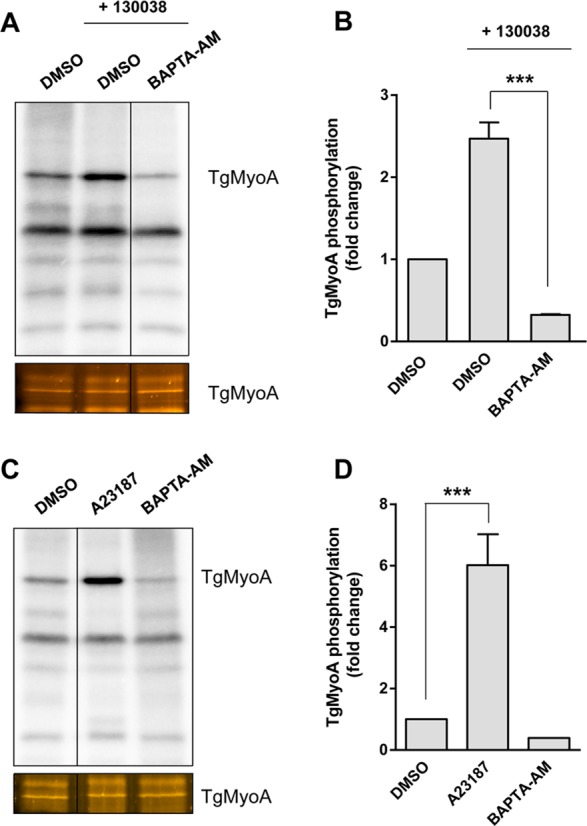
Enhancement of TgMyoA phosphorylation by 130038 is calcium dependent. (A) RH strain tachyzoites metabolically labeled with [32P]orthophosphoric acid were pretreated with BAPTA-AM (20 μM) or vehicle (DMSO) for 20 min. Compound 130038 was then added where indicated to a final concentration of 100 μM, and samples were incubated for an additional 15 min. The TgMyoA motor complex was then isolated by immunoprecipitation using anti-TgGAP45 antibody and resolved by SDS–PAGE; the 32P signal associated with TgMyoA was analyzed as in Figure 3. Images were cropped from selected lanes from the same phosphorimager scan/stained gel; all lanes were processed identically. (B) Normalized TgMyoA phosphorylation level for each treatment in A shown as fold change relative to DMSO, no 130038 treatment. A similar analysis showed no significant change in TgGAP45 phosphorylation after enhancer treatment. (C, D) RH strain tachyzoites were metabolically labeled with [32P]orthophosphoric acid and exposed to DMSO (vehicle control), A23187 (2 μM), or BAPTA-AM (20 μM) for 20 min, immunoprecipitated, and processed as in A and B. Images were cropped from selected lanes from the same phosphorimager scan/stained gel; all lanes were processed identically. All experiments were repeated twice; error bars, SD. ***p < 0.001 (one-way ANOVA).
Next we tested whether increasing intracellular calcium levels enhanced TgMyoA phosphorylation even in the absence of enhancer. When 32P-labeled parasites were treated with the calcium ionophore A23187, TgMyoA phosphorylation increased approximately sixfold compared with treatment with DMSO (Figure 4, C and D). Conversely, BAPTA-AM decreased the basal phosphorylation level of TgMyoA (Figure 4, C and D). As was the case with 130038-enhanced phosphorylation of TgMyoA, the level of 32P incorporation into TgMyoA after treatment with calcium ionophore A23187 was significantly reduced in the TgMyoA S21A and SAAA mutants (Figure 5), with greater inhibition in the triple mutant.
FIGURE 5:
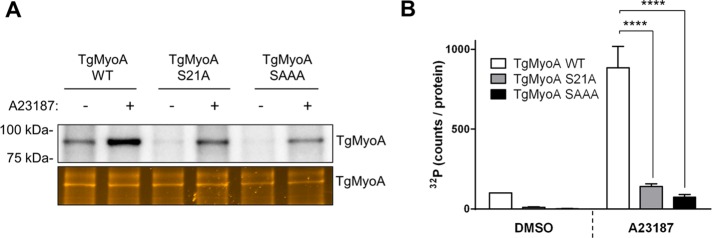
The S21A and SAAA mutations block calcium-induced phosphorylation of TgMyoA. (A) Tachyzoites of the indicated strains were metabolically labeled with [32P]orthophosphoric acid and exposed to either 2 μM A23187 or DMSO (vehicle) for 15 min. The TgMyoA motor complex was then isolated by immunoprecipitation using anti-TgGAP45 antibody and resolved by SDS–PAGE; the 32P signal associated with TgMyoA was analyzed as in Figure 3. (B) TgMyoA phosphorylation under each condition shown as normalized intensity of the TgMyoA-associated 32P signal, as described in Figure 3. Experiments were repeated twice, error bars, SD. ****p < 0.0001 (two-way ANOVA analysis and Bonferroni multiple comparisons test).
In these experiments, the motor complex was isolated from 32P-labeled parasites by immunoprecipitation with an anti-TgGAP45 antibody. To ensure that the differences observed in the TgMyoA-associated32P signal were not due to changes in the interaction between TgMyoA and TgGAP45, TgMyoA WT, TgMyoA SAAA, and TgMyoA SEDD parasites were metabolically labeled with [35S]cysteine/methionine, treated with or without 130038 or A23187. and immunoprecipitated with anti-TgGAP45. The amount of 35S-labeled TgMyoA present in the immunoprecipitates was equivalent (relative to 35S-labeled TgGAP45) under all conditions (Supplemental Figure S5), confirming that neither compound treatment nor the mutations introduced into TgMyoA significantly disrupt the interaction between TgMyoA and TgGAP45.
It remained formally possible that the increased 32P incorporation in TgMyoA was due to a compound-induced change in the specific activity of the intracellular 32P-ATP pool rather than a change in phosphorylation stoichiometry. This seemed unlikely, given the unchanged level of TgGAP45 32P incorporation under the same conditions (Figure 1). However, to definitively rule out this possibility, we used Phos-tag gel electrophoresis to resolve different phosphorylated forms of TgMyoA under the different treatment conditions. TgMyoA SAAA ran as a single band, both from untreated and either A23187- or enhancer-treated parasites; this band presumably corresponds to largely unphosphorylated TgMyoA (Supplemental Figure S6A). In contrast, TgMyoA WT ran as a doublet, the lower band of which comigrated with TgMyoA SAAA. The other band (24.5% of the total TgMyoA) runs more slowly in the Phos-Tag gels, indicative of phosphorylation, and the percentage of the protein in this upper form increased to 45.9 and 69.2% upon treatment with 130038 or A23187, respectively (Supplemental Figure S6, A and B). These results demonstrate that treatment with either 130038 or calcium ionophore A23187 causes an increase in the stoichiometry of TgMyoA phosphorylation; the lack of an upper band in the TgMyoA SAAA mutant even after compound treatment implicates these three residues as the major sites of basal and stimulated TgMyoA phosphorylation.
130038 and the other invasion and motility enhancers stimulate intracellular calcium release
Given that the enhanced phosphorylation of TgMyoA in response to 130038 was calcium dependent and that calcium ionophore A23187 alone could induce the same effect, we tested whether 130038 treatment affects parasite intracellular calcium levels. Compound 130038 does indeed stimulate Ca2+ release into the cytosol from intracellular stores (Figure 6A), and the effect is enhanced in the presence of extracellular Ca2+ (Figure 6B and Supplemental Figure S7). Similar results were observed with two of the other three enhancers tested (153753 and 158513); the third (141852) could not be evaluated because of a quenching effect on the fluorescence of the calcium indicator. Supplemental Figure S7B shows a comparison of the rate of Ca2+ release stimulated by 20 μM 130038, 153753, and 158513.
FIGURE 6:
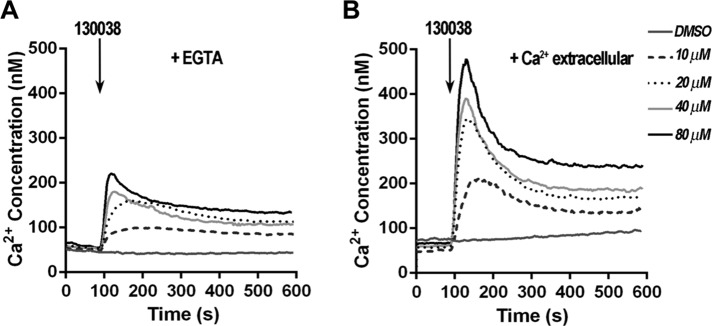
Compound 130038 stimulates Ca2+ release into the cytosol of T. gondii tachyzoites. (A) The addition of various concentrations of 130038 to Fura 2-AM–labeled parasites at 100 s demonstrates that 130038 stimulates a dose-dependent increase in cytosolic Ca2+. The extracellular buffer was Ringer containing 100 μM ethylene glycol tetraacetic acid. The control trace shows the effect of adding DMSO at the maximum volume used (5 μl or 0.2%). (B) Experimental conditions were similar to A, but the extracellular buffer contained 1 mM Ca2+.
The contribution of other TgMyoA phosphorylation sites to 130038-enhanced phosphorylation
It was previously reported that S21 of TgMyoA shows only slightly increased phosphorylation in response to calcium stimulation (Nebl et al., 2011). In contrast, our data demonstrate that treatment with calcium ionophore causes a 5- to 10-fold increase in 32P incorporation into TgMyoA, and much of this increase is abrogated by the S21A mutation. The block is even more complete when the S21A mutation is coupled with the S20A and S29A mutations. It is possible that phosphorylation on S20, S21, and/or S29 enables the phosphorylation of other sites on TgMyoA in response to the enhancer, rather than S20, S21, and/or S29 being the major sites of enhancer-induced phosphorylation themselves. To test this possibility, we determined the extent to which TgMyoA containing the triple SEDD phosphomimetic mutation was phosphorylated after enhancer or calcium ionophore treatment. Only a low basal level of TgMyoA phosphorylation was observed in the SEDD parasites (Figure 7, A and B). Incorporation of 32P into TgMyoA SEDD increased upon treatment with 130038 or calcium ionophore by approximately fivefold, similar to the fold increase seen for TgMyoA WT, but the final levels of enhanced 32P incorporation were significantly lower in TgMyoA SEDD than with TgMyoA WT (Figure 7, A and B). Thus S20, S21, and S29 are likely themselves the major sites of both basal and enhanced TgMyoA phosphorylation, but additional phosphorylation sites make a detectable, enhancer- and calcium ionophore–sensitive contribution to overall phosphorylation of TgMyoA.
FIGURE 7:
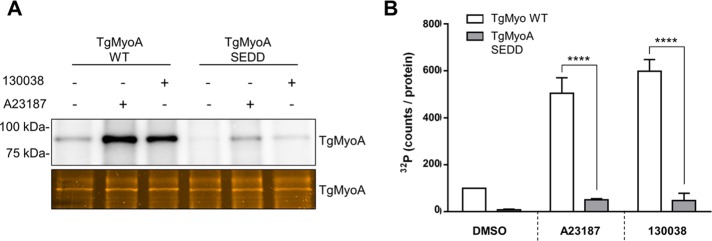
The SEDD phosphomimetic mutations do not enhance phosphorylation of other sites on TgMyoA. (A) Tachyzoites of the indicated strains were metabolically labeled with [32P]orthophosphoric acid and exposed to DMSO (vehicle control), 100 μM 130038, or 2 μM A23187 for 20 min. The TgMyoA motor complex was then isolated by immunoprecipitation using anti-TgGAP45 antibody and resolved by SDS–PAGE; the 32P signal associated with TgMyoA was analyzed as in Figure 3. (B) TgMyoA phosphorylation under each condition shown as normalized intensity of the TgMyoA-associated 32P signal, as described in Figure 3. Experiments were repeated twice; error bars, SD. Statistically significant differences were observed between the two strains after treatment with either 130038 or A23187 (****p < 0.0001; two-way ANOVA analysis and Bonferroni multiple comparisons test); no significant difference was observed in TgMyoA SEDD parasites between treatments.
A recent phosphoproteomics study (Treeck et al., 2011; see also ToxoDB [www.toxodb.org]) identified six phosphorylation sites on TgMyoA in addition to S20 and 21: S7, S517, S518, T561, S748, and S823 (Figure 2A). To quantify the relative contribution of these other sites to phosphorylation enhancement, we generated two additional TgMyoA mutants in the TgMyoA KO background. One left S20, S21, and S29 unchanged but contained alanine mutations at the six other phosphorylation sites (TgMyoA WT6A), whereas the second mutant had serine-to-alanine mutations at all nine phosphorylation sites (TgMyoA SAAA6A; Figure 2B). The 32P labeling of TgMyoA WT6A showed a strong basal level of phosphorylation and robust enhancement of TgMyoA phosphorylation in response to A23187 and 130038. On the other hand, TgMyoA SAAA6A showed no detectable labeling in untreated parasites and significantly less 32P incorporation than TgMyoA WT6A after treatment with either A23187 or 130038 (Supplemental Figure S8). These data once again implicate S20, S21, and S29 as the primary sites of both basal and enhanced TgMyoA phosphorylation; the other six sites are phosphorylated in untreated and enhancer-treated parasites, but to a much lesser extent. The observation that the SAAA6A mutant shows a detectable level of 32P labeling after treatment with 130038 or A23187 demonstrates the existence of yet other phosphorylation sites on TgMyoA, beyond the nine that were collectively identified here and in earlier phosphoproteomic analyses (Nebl et al., 2011; Treeck et al., 2011).
Phenotypic consequences of TgMyoA phosphorylation
Compound 130038 was originally identified as an enhancer of T. gondii invasion and motility. To determine whether TgMyoA phosphorylation contributes to the enhancement of invasion by 130038, we tested the capacity of compound-treated WT and SAAA mutant parasites to invade host cells using a quantitative laser scanning cytometry (LSC)–based invasion assay (Mital et al., 2006). Both the WT and SAAA mutant parasites showed ∼2.5-fold enhancement of invasion in response to 130038; however, the basal level of invasion was lower in the mutant than the WT, so the final enhanced level was also lower (Figure 8A). The effect was modest (23% decrease) but statistically significant. The phosphomimetic SEDD mutant partially restored the enhanced invasion levels (Figure 8A). Phosphorylation of TgMyoA on S20, 21, and/or 29 may therefore be required for maximally efficient invasion but does not appear to play a role in the enhancement of invasion by 130038, suggesting some other calcium-dependent mechanism of enhancement. In the absence of TgMyoA, invasion is reduced by ∼90% but not abolished, confirming the existence of TgMyoA-independent invasion mechanism(s) (Andenmatten et al., 2013). Addition of the enhancer has no effect on the residual invasion of the TgMyoA KO parasites, confirming that enhanced invasion, whatever the mechanism, requires the presence of TgMyoA (Figure 8A).
FIGURE 8:
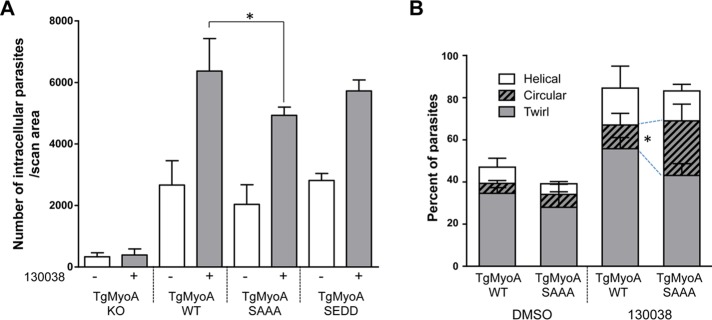
The effect of TgMyoA phosphorylation site mutations on parasite invasion and motility. (A) The ability of TgMyoA KO and TgMyoA WT, TgMyoA S21A, and TgMyoA SAAA complemented strains to invade host cells in the presence or absence of 100 μM 130038 was determined using the laser scanning cytometry–based invasion assay. Mean number of intracellular parasites per scan area for each condition. Experiments were repeated three times; error bars, SD. *p < 0.05 (two-way ANOVA analysis and Bonferroni multiple comparisons test). (B) Quantification of parasite motility by 2D live videomicroscopy. Parasite motile behavior is plotted as the mean percentage of total parasites in the observed fields exhibiting twirling, circular, or helical motility. Including motile and nonmotile parasites, n = 350–650 parasites were analyzed per treatment for each parasite line; the experiment was performed twice; error bars, SD. Two-way ANOVA was performed on each type of motility; to preserve statistical power, an uncorrected Fisher least significant difference test was performed on each type of motility for multiple comparisons between strains and treatments. *p < 0.05.
To test for a more specific effect of TgMyoA phosphorylation on motility, we treated WT and SAAA mutant parasites with and without 130038 and monitored their motility on a protein-coated glass surface by live videomicroscopy. The percentages of untreated WT and SAAA parasites moving during the 60-s observation period were similar, and exposure to 130038 roughly doubled the number of parasites moving in both parasite lines (from 48 to 84.5% in WT and from 39.6 to 81.6% in SAAA; Figure 8B). Three distinctly different types of motility can be observed in these assays, as previously reported: twirling and circular and helical gliding (Hakansson et al., 1999). There was no significant difference in the types of motility exhibited by WT and SAAA parasites in the absence of 130038, but after enhancer treatment, more than twice as many SAAA parasites were seen undergoing circular gliding than were similarly treated WT parasites (25.9 vs. 11.3%, respectively, p < 0.05; Figure 8B). Although the physiological relevance of circular gliding on a glass coverslip is unknown, these data demonstrate that an inability to phosphorylate TgMyoA on S20, 21, and/or 29 alters the motile behavior of 130038-treated parasites.
Because TgMyoA phosphorylation can be induced by calcium ionophore alone, we tested whether another key calcium- and TgMyoA-dependent step in the parasite life cycle—host cell egress (Meissner et al., 2002; Garrison et al., 2012; Lourido et al., 2012; Andenmatten et al., 2013; Egarter et al., 2014)—was also altered in the phosphorylation-defective mutants. Host cells infected with WT or SAAA mutant parasites were treated with low levels of calcium ionophore to experimentally induce egress, and the number of cells from which parasites had egressed was determined over the subsequent 30 min (Figure 9). The SAAA mutant showed significantly delayed egress, whereas the SEDD mutant was indistinguishable from wild type. Treatment of infected cells with 130038 did not induce egress on its own, even after prolonged incubation (unpublished data), perhaps because the compound cannot penetrate the membranes of both the host cell and parasitophorous vacuole to access the intracellular parasite. However, Figure 9 shows clearly that TgMyoA phosphorylation accelerates the process of calcium-induced host cell egress.
FIGURE 9:
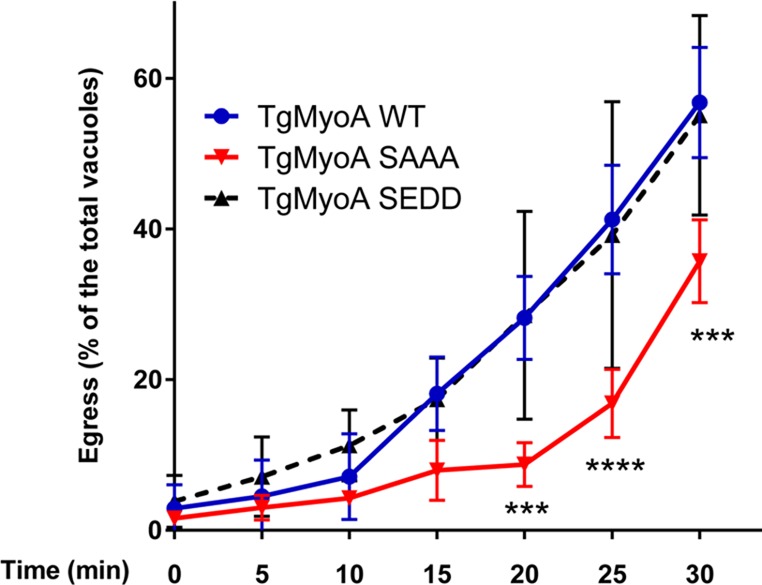
Parasites expressing the SAAA TgMyoA mutation show a delay in calcium-induced host cell egress. HFF cells were infected with TgMyoA WT, TgMyoA SAAA, or TgMyoA SEDD parasites and exposed 36–48 h postinfection to 0.1 μM calcium ionophore A23187 for the times indicated. The number of vacuoles from which parasites had egressed was determined by immunofluorescence microscopy and is plotted as the mean percentage of the total number of vacuoles (egressed plus intact) observed. Experiments were repeated three times; error bars, SD. Statistically significant differences were observed between A23187-treated TgMyoA WT and TgMyoA SAAA parasites at the time points indicated. ***p < 0.001, ****p < 0.0001 (two-way ANOVA with Bonferroni multiple comparison test).
DISCUSSION
Apicomplexan parasites use substrate-dependent motility to invade and escape from host cells, to cross biological barriers such as the placenta and blood–brain barrier, and to disseminate throughout the infected host (reviewed in Sibley, 2004). The data presented here suggest that the signaling pathways regulating motility-based processes in T. gondii can do so, at least in part, through calcium-mediated phosphorylation of parasite myosin A.
Phosphorylation of the myosin heavy chain affects motor function in different ways in different systems, including altering the catalytic cycle, changing how the heavy chains interact with each other and disrupting tail interaction with cargo (Rogers et al., 1999; Redowicz, 2001; Liu et al., 2013a, b). Class XIVa myosins are different from other myosins in several respects (Heintzelman and Schwartzman, 1997; Hettmann et al., 2000; Foth et al., 2006) and therefore may be regulated by different mechanisms. For example, class XIVa myosins do not follow the TEDS rule, that is, they do not contain a conserved phosphorylatable or acidic residue within the motor domain that is important in many other nonmuscle myosins for enzymatic activity (Bement and Mooseker, 1995; Brzeska and Korn, 1996; Redowicz, 2001). The TgMyoA motor domain shares only 23–34% identity with the motor domains of other myosins (Heintzelman and Schwartzman, 1997), making it difficult to predict the biophysical or biochemical consequences of phosphorylation. Nonetheless, the importance of TgMyoA in the parasite life cycle (Meissner et al., 2002; Andenmatten et al., 2013; Egarter et al., 2014) and the significant differences between TgMyoA and other mammalian myosins highlight TgMyoA and the mechanisms that regulate its activity as promising potential antiparasitic drug targets.
In a previous mass spectrometry–based study, S21 was the only TgMyoA phosphorylation site detected, and its phosphorylation increased only slightly, if at all, in response to increased intracellular calcium (Nebl et al., 2011). In contrast, our 32P-labeling results suggest a relatively large increase in TgMyoA phosphorylation after calcium ionophore treatment (Figures 4, 5, and 7 and Supplemental Figures S6 and S8). In both studies the motor complex was isolated for analysis by anti-TgGAP45 immunoprecipitation. The discrepancy is unlikely to be explained by either a calcium-induced increase in the specific activity of the ATP pool in our experiments or increased turnover of 32P on TgMyoA that had not yet been labeled to steady state, since the Phos-tag results strongly suggest a significant increase in phosphorylation stoichiometry. Another possibility is that basal phosphorylation on a primary site (e.g., S21) promotes or enables enhanced phosphorylation on other sites, but this also seems unlikely since our triple phosphomimetic SEDD mutant failed to achieve a highly phosphorylated state. Perhaps to achieve the enhanced phosphorylation of TgMyoA, phosphorylation on the primary sites needs to be dynamic rather than constitutive (as simulated by the phosphomimetic mutations), or some particular combination of phosphorylated residues or ordered phosphorylation is necessary, as observed in other systems (Gingras et al., 2001; Huang et al., 2007; Ko et al., 2010). Of interest, a recent study in which three of the nine phosphorylation sites on TgMLC1 were mutated to nonphosphorylatable alanines resulted in increased phosphorylation on a fourth site (Jacot et al., 2014), suggesting that cross-talk occurs between the phosphorylation sites on TgMLC1.
We also did not detect the calcium-induced increase in TgGAP45 phosphorylation reported previously (Nebl et al., 2011). However, this increase occurred on only two or three of the 13 known sites of TgGAP45 phosphorylation (Gilk et al., 2009; Nebl et al., 2011; Treeck et al., 2011). If phosphorylation on some of the other sites were to either decrease or simply be present at a higher stoichiometry, then an increase in phosphorylation on these two or three sites, although of potential functional importance, might have little effect on the overall phosphorylation state of the protein. Given the total number of phosphorylation sites present on TgMyoA and other members of the motor complex (at least 35; Nebl et al., 2011; Treeck et al., 2011; Jacot and Soldati-Favre, 2012), the generation of parasites containing all possible combinations of phosphorylation site mutations is not experimentally feasible. However, this same combinatorial complexity provides the parasite with a potentially extraordinary level of posttranslational control of TgMyoA motor complex activity.
The finding that all of the invasion and motility enhancers tested stimulate an increase in intracellular calcium release was unexpected. Our earlier experiments did not detect this effect (Carey et al., 2004); however, these earlier experiments were nonquantitative, used a different, nonratiometric calcium indicator, and were confounded by spectral overlap between the compounds and the indicator. The discovery that the different enhancers all stimulate intracellular calcium release provides a rationale for how this group of structurally diverse compounds can cause the same set of phenotypes, that is, enhanced invasion, microneme secretion, and motility (Carey et al., 2004). It can also explain why the TgMyoA mutations only partially abrogate the effects of the compounds; that is, these mutations block the calcium-induced effects on MyoA phosphorylation but do not affect other targets such as microneme secretion that could contribute to the calcium-induced phenotypes. Indeed, we found no evidence that myosin phosphorylation is involved in the enhancement of invasion by 130038; this particular enhancer-induced phenotype may be mediated through other calcium effectors. Although intracellular calcium is known to play an important role in motility (Lovett and Sibley, 2003), invasion (Vieira and Moreno, 2000), and egress (Garrison et al., 2012; Lourido et al., 2012; McCoy et al., 2012), it may be that calcium oscillations, rather than sustained changes in calcium levels, are what is required for optimal invasion (Wetzel et al., 2004). This provides a cautionary note for mutations that introduce a constitutive negative charge onto a potential phosphorylation substrate (e.g., the TgMyoA SEDD mutant), since phosphomimetic mutations do not allow for dynamic cycles of phosphorylation/dephosphorylation.
The kinase(s) responsible for TgMyoA phosphorylation are not known. The dominant calcium-responsive kinases in apicomplexan parasites are the calcium-dependent protein kinases (CDPKs), characterized by a protein kinase domain fused to a calmodulin-like domain (Nagamune and Sibley, 2006). Both TgCDPK1 and TgCDPK3 appear to play a role in regulating motility (Lourido et al., 2012), but the substrate specificity and the identity of the targets these kinases phosphorylate are only just beginning to emerge (Green et al., 2008; Thomas et al., 2012; Lourido et al., 2013). Intriguingly, TgMyoA S21 (which we show here to be a major site of calcium-induced phosphorylation) lies within a sequence (TALKKRSS21D) that could potentially serve as a TgCDPK1 substrate (Lourido et al., 2013). Although the substrate preference of TgCDPK3 is unknown, the delayed-egress phenotype of parasites expressing the S21A mutation reported here (Figure 9) is strikingly similar to the phenotype of parasites containing a disruption in TgCDPK3 (Garrison et al., 2012; see also Lourido et al., 2012; McCoy et al., 2012). On the other hand, in the malaria parasite Plasmodium falciparum, S19 of PfMyoA, which may be functionally equivalent to S21 in TgMyoA, is phosphorylated by protein kinase A (Lasonder et al., 2012). Identification of the kinase(s)/phosphatases(s) that regulate the function of the class XIVa myosin motor complex in apicomplexan parasites will be an important subject for future investigation.
MATERIALS AND METHODS
Parasite culture
Parasites were maintained by continuous passage in confluent human foreskin fibroblast (HFF) monolayers cultured in DMEM (HyClone, South Logan, UT), supplemented with 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.6 (Life Technologies, Grand Island, NY), 1% (vol/vol) fetal bovine serum (FBS; Life Technologies), and 100 U penicillin plus 100 μg streptomycin/ml (Life Technologies) as previously described (Roos et al., 1994).
Metabolic labeling of parasites
For 32P labeling, freshly released extracellular parasites were filtered through 3-μm Nuclepore track-etch membranes (Whatman, Bound Brook, NJ), washed (4 min, 1050 × g), and resuspended in phosphate-free DMEM (Life Technologies) supplemented with 1% dialyzed FBS (Life Technologies). A 1-mCi amount of [32P]H3PO4 in H2O (MP Biomedical, Solon, OH) was added to every 108 parasites suspended in 160 μl of medium, which were then incubated at 37°C for 35–45 min. Labeled parasites were then washed with phosphate-free medium twice (4 min, 1000 × g) before compound treatment. Labeling of parasites with [35S]cysteine/methionine was performed as previously described (Gaskins et al., 2004).
Immunoprecipitation of the TgMyoA motor complex
Immunoprecipitation was performed as described by Gilk et al. (2009), with the following modifications. Filtered parasites, 5 × 107, were pelleted (4 min, 1050 × g) and lysed in 1 ml of ice-cold TX-100 IP buffer (1% [vol/vol] Triton X-100, 150 mM NaCl, 5 mM EDTA, 50 mM Tris, pH 8, 1:100 [vol/vol] protease inhibitor cocktail [P8340; Sigma-Aldrich, St. Louis, MO], 20 mM β-glycerophosphate, 30 mM sodium fluoride, and 5 mM sodium pyrophosphate). The lysate was incubated on ice for 15 min and cleared (15 min, 21,130 × g, 4°C). Rabbit anti-TgGAP45 antiserum (1:1000; Gaskins et al., 2004) was added to the supernatant, and the mixture was gently agitated at 4°C for 1 h, followed by incubation with rec-protein A–Sepharose 4B beads (Invitrogen, Frederick, MD) at 4°C for 1 h. The beads were washed four to six times with ice-cold TX-100 IP buffer, and the bound proteins were eluted by boiling at 100°C in SDS–PAGE sample buffer containing 5% (vol/vol) β-mercaptoethanol.
Gels were stained either with Sypro Ruby (Figure 1A) or Oriole (all other figures) according to manufacturer's instruction (Bio-Rad, Hercules, CA), and the TgMyoA bands were quantified with Quantity One software (Bio-Rad). The stained gels were dried onto filter paper and exposed to a phosphorimager screen for 4 d before being scanned with a Bio-Rad FX imager. The 32P or 35S incorporation into individual bands was quantified using Quantity One software.
Generation of the FLAG-tagged TgMyoA construct
Primers P1 and P2 (see Table 1) were used to amplify FLAGTgMyoA from an intermediate plasmid containing N-terminal FLAG-Tagged TgMyoA, which had been originally amplified from T. gondii cDNA (RH). The PCR product was subsequently ligated into the AvrII and XmaI sites of plasmid RPS13(IV)-YFP-IMC13/sagCAT (a generous gift from Marc-Jan Gubbels, Boston College), a plasmid modified from ptub-YFP–IMCx/sagCAT (Anderson-White et al., 2010) by substitution of ptub with the RPS13(IV) promoter (van Poppel et al., 2006). The TgMyoA endogenous promoter was amplified from RH T. gondii genomic DNA using primers P3 and P4 and cloned upstream of FLAG-TgMyoA using PmeI and AvrII. The sagCAT cassette from the resulting plasmid was replaced with the graBle fragment excised from GRA1-Ble vector (Messina et al., 1995) using the PacI and HindIII sites. The DHFR 3′ untranslated region was amplified from RPS13(IV)-YFP-IMC13/sagCAT using primers P5 and P6 and ligated downstream of TgMyoA sequence using XmaI and NotI. The TgMyoA open reading frame (ORF) of the final construct pmyoA-FLAGTgMyoA-WT/graBle was confirmed by sequencing. TgMyoA mutations were generated in pmyoA-FLAGTgMyoA-WT/graBle using either the QuikChangeII XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) with the sense and antisense primer pairs listed in Table 1 or by “round-the-horn” mutagenesis (http://openwetware.org/wiki/%27Round-the-horn_site-directed_mutagenesis) with the phosphorylated primer pairs listed in Table 1.
TABLE 1:
Primers used in this study.
| Primer name | Sequence 5′-3′ |
|---|---|
| P1 | CCACCTAGGAAAATGGACTACAAGGACGATGACGAC |
| P2 | CACCCCGGGTTACTAGAACGCCGGCTGAACAG |
| P3 | CAGGTTTAAACATTCGTCGACTCCTCAAGAGCGGTGGATTC |
| P4 | CAGCCTAGGGCTTCAGAGAGAGATAACACGAGGAAAGAGGTA |
| P5 | CAGCCCGGGATATCGAGCTCTAACC |
| P6 | CCTCTTCGCTATTACGCCAGCTG |
| P7 | GGGGCCATGGGCCATCATCACCATCACCACATGGCG |
| P8 | GGGGCACCATCACCACATGGCGAGCAAGACCACGTCTG |
| P9 | GGGGGGATCCCTACTACTAGAACGCCGGCTGAACAGTC |
| S7A sense | GCGAGCAAGACCACGGCTGAGGAGCTGAAAA |
| S7A antisense | TTTTCAGCTCCTCAGCCGTGGTCTTGCTCGC |
| S20A sense | CGCTGAAGAAGAGGGCGGCCGATGTCCAC |
| S20A antisense | ATGGACATCGGCCGCCCTCTTCTTCAGAG |
| S20E sense | GCGCTGAAGAAGAGGGAGGACGATGTCCACGC |
| S20E antisense | GCGTGGACATCGTCCTCCCTCTTCTTCAGCGC |
| S21A sense | TGAAGAAGAGGTCGGCCGATGTCCACGCG |
| S21A antisense | CGCGTGGACATCGGCCGACCTCTTCTTCA |
| S21D sense | GCTGAAGAAGAGGTCGGACGATGTCCACGCGGTC |
| S21D antisense | GACCGCGTGGACATCGTCCGACCTCTTCTTCAGC |
| S29A sense | CGCGGTCGACCACGCCGGCAATGTGTA |
| S29A antisense | TACACATTGCCGGCGTGGTCGACCGCG |
| S29D sense | CCACGCGGTCGACCACGACGGCAATGTGTACAAA |
| S29D antisense | TTTGTACACATTGCCGTCGTGGTCGACCGCGTGG |
| S517/S518 AA sense | ATCGTGACGAGGGTGTCGCCGCCAAGGAGTTGATTTTC |
| S517/S518 AA antisense | GAAAATCAACTCCTTGGCGGCGACACCCTCGTCACGAT |
| T561A sense | GAAAAGTTCCTCTCGGCCTGCAAGAACGCGC |
| T561A antisense | GCGCGTTCTTGCAGGCCGAGAGGAACTTTTC |
| S748 sense | AAAGCAGCAAACTGCCCGCCGAAGAATACCAGCTCG |
| S748 antisense | CGAGCTGGTATTCTTCGGCGGGCAGTTTGCTGCTTT |
| Phospho-S823A Fr | P-GCCCCCGCGACTGTTCAGCC |
| Phospho-S823A Rv | P-GACGTTGTTGTCCACCAGGTGTC |
P indicates that the primer is phosphorylated at its 5′ end.
Parasite transfection and selection
Filtered parasites, 2 × 107, were pelleted (4 min, 1050 × g) and resuspended in 300 μl of electroporation cytomix solution (120 mM KCl, 0.15 mM CaCl2, 10 mM potassium phosphate, pH 7.6, 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES]–KOH, pH 7.6, 2 mM EDTA, 5 mM MgCl2) supplemented with 2 mM ATP and 5 mM glutathione. Plasmid DNA (100 μg) was ethanol precipitated and dissolved in 100 μl of cytomix solution. The dissolved plasmid and the parasites were gently mixed and transferred to a cuvette for electroporation (1500 V, 25 Ω, 25 μF) in a Genetronics Precision Pulse ECM630 electroporator (BTX-Harvard Apparatus, Holliston, MA). Transfected parasites were used to infect an HFF monolayer and grown without selection until they fully lysed the monolayer. For bleomycin selection (Messina et al., 1995), 5 × 106 parasites were treated extracellularly with 50 μg/ml bleomycin in DMEM for 4 h at 37°C and used to infect an HFF monolayer, which was then cultured in the presence of 5 μg/ml bleomycin until the host cells fully lysed. The selection procedure was repeated, and the bleomycin resistant parasites were cloned by limiting dilution as previously described (Roos et al., 1994).
Generation of TgMyoA antibody
Hexahistidine (His6)-TgMyoA was amplified through nested PCR with forward primers P7 and P8 and reverse primer P9 from RH tachyzoite cDNA. The PCR product was subcloned into pCR II-TOPO (Invitrogen) according to the manufacturer's instructions. The His6-TgMyoA fragment was then digested with NcoI and BamHI from the TOPO vector and ligated into pET28a(+) (Novagen, Darmstadt, Germany). The TgMyoA ORF in the final construct was confirmed by sequencing and used to transform BL21-CodonPlus(DE)-RIPL Escherichia coli strain. Recombinant His6-TgMyoA expression was induced by culturing with 1 mM isopropyl-β-d-thiogalactoside in Luria-Bertani medium for 4 h at 37°C. The bacterial culture was then spun down and resuspended in ice-cold lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, and 2 mM EDTA supplemented with 1 mg/ml lysozyme) and sonicated six times on ice before adding 5 μg/ml DNase/RNase (Sigma). The lysate was then centrifuged at 13,000 × g for 30 min at 4°C to collect the pellet, which was subsequently dislodged, resuspended in solubilizing buffer (20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 10 mM imidazole, and 8 M urea) at 23°C for 1 h with constant gentle agitation and centrifuged again at 13,000 × g for 4 min at 4°C. The supernatant was then mixed with 4× SDS–PAGE sample buffer without boiling to avoid protein carbamylation. The mixture was resolved by SDS–PAGE and stained with colloidal Coomassie blue. The His6-TgMyoA gel fraction was excised and sent to Cocalico Biologicals (Reamstown, PA) for generation of rabbit anti-TgMyoA polyclonal antiserum. The specificity of the anti-TgMyoA serum was confirmed by Western blot.
Immunofluorescence
Infected HFF monolayers were fixed in phosphate-buffered saline (PBS) containing 4% (vol/vol) paraformaldehyde for 15–30 min, washed three times in PBS, permeabilized with PBS containing 0.25% (vol/vol) Triton X-100 for 30 min, and blocked with PBS containing 1% FBS for 10 min. Samples were incubated for 1 h with mouse anti-FLAG (Sigma-Aldrich) and rabbit anti-TgIMC1 (a generous gift from Con Beckers, University of North Carolina) diluted 1:5000 (vol/vol) and 1:1000 (vol/vol), respectively, in PBS containing 1% (vol/vol) FBS, followed by 45-min incubation with Alexa 488–conjugated goat anti-rabbit immunoglobulin G (IgG) and Alexa 546–conjugated goat anti-mouse IgG (Invitrogen), each diluted 1:1000 (vol/vol) in PBS with 1% FBS. All incubations were carried out at 25°C.
Fluorescently stained parasites were imaged using a 60× PlanApo objective on Nikon Eclipse TE300 epifluorescence microscope. Images were captured using an iXon 885 camera (Andor Technology, Belfast, Ireland) cooled to −70°C and driven by NIS Elements, version 3.20 software (Nikon Instruments). Final images were processed by Photoshop (Adobe, San Jose, CA).
Western immunoblotting
Protein samples from 107 parasites were resolved by SDS–PAGE and transferred to polyvinylidene fluoride (PVDF) as previously described (Carey et al., 2004). Blots were probed with rabbit anti-TgMyoA antiserum at 1:1000, mouse anti-TgGRA8 at 1 μg/ml, and mouse anti-FLAG antibody (Sigma-Aldrich) at 1:7500 in PBS containing 0.5% (vol/vol) bovine serum albumin (BSA), followed by incubation with IRDye 680–conjugated anti-rabbit IgG, IRDye 800–conjugated anti-mouse IgG, and IRDye 680–conjugated goat anti-mouse IgG1 at (1:20,000; LI-COR Biosciences, Lincoln, NE), each at 1:20,000 in PBS containing 0.5% BSA. Blots were then washed in PBS and scanned using an Odyssey CLx Infrared Imaging System (LI-COR Biosciences). Images were processed using Image Studio software (LI-COR Biosciences).
Phos-tag PAGE
Phos-tag PAGE was performed according to manufacturer's instructions (Phos-tag AAL-107; Nard Institute, Amagasaki, Japan). To separate TgMyoA, 100 μM Phos-tag and 400 μM MnCl2 were added to a conventional SDS–PAGE separating gel mix containing 6% (wt/vol) acrylamide. Lysates from 1.5 × 107 parasites treated with compound or the vehicle control (DMSO) were boiled in SDS sample buffer containing β-mercaptoethanol and resolved at 4°C overnight under constant voltage. The gel was washed three times in SDS–PAGE running buffer containing 10 mM EDTA and once in running buffer alone before transferring to PVDF membrane for Western immunoblotting.
Calcium measurements
Parasites were loaded with Fura 2-AM as described (Moreno and Zhong, 1996). After the parasites were harvested and purified, they were washed twice at 500 × g for 10 min at room temperature in Ringer buffer, which contained 155 mM NaCl, 3 mM KCl, 1 mM MgCl2, 3 mM NaH2PO4H2O, 10 mM HEPES, pH 7.3, and 10 mM glucose. Cells were resuspended to a final density of 1 × l09 cells/ml in loading buffer, which consisted of Ringer buffer plus 1.5% (wt/vol) sucrose and 5 μM Fura 2-AM. The suspensions were incubated for 30 min in a 26°C water bath with mild agitation. Subsequently, the cells were washed twice with Ringer buffer to remove extracellular dye. Cells were resuspended to a final density of 1 × 109 cells/ml in Ringer buffer and kept on ice. Parasites were viable for several hours under these conditions. For fluorescence measurements, a 50-μl aliquot of the cell suspension was diluted into 2.5 ml of Ringer buffer (2 × l07 cells/ml final density) in a cuvette placed in a Hitachi F-4500 spectrofluorometer. Excitation was at 340 and 380 nm, and emission was at 510 nm. The Fura 2 fluorescence response to intracellular Ca2+ concentration ([Ca2+]i) was calibrated from the ratio of 340/380 nm fluorescence values after subtraction of the background fluorescence of the cells at 340 and 380 nm as previously described (Grynkiewicz et al., 1985). Calcium release rate is the difference in Ca2+ concentration in the first 20 s after drug addition. The slope was determined as the linear regression of concentration versus time. Slope average of at least three independent experiments was used for quantifications. Compounds 130038, 153753, and 158513 were dissolved in DMSO, and their excitation spectra showed no peaks between 300 and 400 nm.
LSC invasion assay
Parasites, 1.5 × 106, were pretreated at 25°C for 15 min with 100 μM compound 130038 or the equivalent volume of DMSO (vehicle control) in Hank's buffer supplemented with 10 mM HEPES, pH 7.6, and used to infect a 25-mm2 coverslip of confluent HFF cells, which was then incubated at 37°C for 50 min. Coverslips were then washed with PBS and fixed with PBS containing 3.1% (vol/vol) paraformaldehyde and 0.06% (vol/vol) glutaraldehyde for 30 min at 23°C. All subsequent immunofluorescence staining and laser scanning cytometry procedures were performed as described previously (Mital et al., 2006).
Egress assay
Parasites were used to infect confluent HFF monolayers on coverslips in 12-well plates (4 × 104 parasite/well; multiplicity of infection, ∼0.1). The infected cells were incubated for 36 h at 37°C and then treated with 0.1 μM A23187 for 0–30 min and fixed in PBS containing 4% (vol/vol) paraformaldehyde. The monolayer was then blocked with PBS containing 1% (vol/vol) FBS for 10 min at 25°C. Extracellular parasites were stained with mouse monoclonal anti-SAG1 (Argene, North Massapequa, NY) at 1:1000 in PBS, followed by incubation with Alexa 546–conjugated goat anti-mouse IgG (Invitrogen) at 1:1000. The cells were then permeabilized with PBS containing 0.25% (vol/vol) Triton X-100 for 30 min before staining with rabbit anti-TgIMC1 at 1:1000, followed by Alexa 488–conjugated goat anti-rabbit IgG (Invitrogen) at 1:1000. Experiments in which there was >5% natural egress at 0 min were discarded. From 300 to 400 vacuoles were counted in each sample per time point.
Two-dimensional video microscopy of parasite motility
An IbiTreat Ibidi chamber μ-Slide VI0.4 (Ibidi GmbH, Munich, Germany) was precoated with 50 μg/ml BSA in H2O at 37°C for 30 min and then washed three times with PBS. Parasites freshly released from HFF cells were filtered through a 3-μm Nuclepore membrane and resuspended at 3 × 107/ml in motility buffer (Hank's buffered salt solution supplemented with 10 mM HEPES, pH 7.6, and 10 mM glucose). The parasite suspension, 50 μl, was gently mixed with 50 μl of motility buffer containing 200 μM 130038 or an equal volume of DMSO and 5 μl of 400 μg/ml Hoechst 33342 (EMD Millipore, Billerica, MA). The mixture was perfused into the Ibidi chamber and allowed to settle at 25°C for 5 min in the dark before imaging.
Fluorescent parasite nuclei were imaged using a 20× PlanApo objective (numerical aperture 0.75) on a preheated (36 ± 1°C) Nikon Eclipse TE300 epifluorescence microscope with three tandem neutral density filters: ND 4, ND 8 (Nikon Instruments), and ND OD = 1.0 (Thorlabs, Newton, NJ). Time-lapse images were captured every second for 60 s using an iXon 885 camera (Andor Technology) cooled to −70°C and driven by NIS Elements, version 3.20 software (Nikon Instruments). A maximum intensity projection (Max-IP) was prepared from each image stack using NIS Elements, and motility was scored manually. Each Max-IP image contained 150–350 tracks.
Plaque assay
Confluent HFF monolayers in a 12-well plate were infected with 50 parasites/well and left undisturbed at 37°C for 8–9 d. The monolayer was then fixed with 100% ethanol, stained with PBS containing 2% (wt/vol) crystal violet and 20% (vol/vol) methanol for 10 min, washed three times with PBS, air dried, and imaged.
In-gel digestion and mass spectrometry analysis
Colloidal Coomassie-stained gel bands of TgMyoA were sliced into 1-mm cubes and destained with destain solution (50% [vol/vol] acetonitrile (MeCN) and 50% [vol/vol] 50 mM ammonium bicarbonate) at 37°C until colorless and then dehydrated with 100% MeCN. The dehydrated gel cubes were hydrated in 50 mM ammonium bicarbonate containing 25 mM dithiothreitol (DTT) at 55°C for 30 min, and the excess liquid was removed. The gel cubes were dehydrated with 100% MeCN, rehydrated in 50 mM ammonium bicarbonate containing10 mM iodoacetamide, and left for 30–60 min at 25°C in the dark. The alkylated samples were then incubated with destain solution twice and dehydrated with 100% MeCN. The dried gel cubes were then hydrated on ice in 50 mM ammonium bicarbonate containing 6 ng/μl sequencing-grade modified trypsin (Promega, Madison, WI) and incubated at 37°C for 16–18 h. Peptides were extracted once with 50% (vol/vol) MeCN and 2.5% (vol/vol) formic acid (FA) in H2O. The extraction was performed for 15 min while spinning at 15,000 × g. A second extraction was performed with 100% MeCN for 10 min without spinning. The two extracts were combined and dried using a SpeedVac. The dry peptides were dissolved in H2O containing 2.5% MeCN and 2.5% FA for mass spectrometry (MS) analysis.
Samples were loaded using a Micro AS autosampler (Thermo Electron, Waltham, MA) and a Surveyor MS Pump Plus (Thermo Electron) onto a nano-electrospray microcapillary column packed with 12 cm of reverse-phase MagicC18 material (5 mm, 200 Å; Michrom Bioresources, Auburn, CA). Elution was performed with a 5–35% MeCN (0.15% FA) gradient over 45 min, after a 15-min isocratic loading at 2.5% MeCN/0.15% FA. Solvent A was 2.5% MeCN/0.15% FA, and Solvent B was 99.85% MeCN/0.15% FA. Mass spectra were acquired in an LTQ XL linear ion trap mass spectrometer (Thermo Electron). Throughout the entire run a precursor survey (MS1) scan was followed by 10 data-dependent MS/MS scans on the most abundant ions (dynamic exclusion repeat count, 1; duration, 180 s). Mass spectral data were searched against the sequence of TgMyoA (GT1 strain; www.toxodb.org) using Turbo SEQUEST v27, Revision 12 (Thermo Electron) requiring no enzyme specificity, a 2-Da precursor mass tolerance, and a 1-Da fragment ion mass tolerance. Cysteine residues were required to have a static increase of 57.0 Da for carbamidomethylation. Differential modifications of 16.0 Da on methionine and 80.0 Da on serine, threonine, and tyrosine were permitted.
Supplementary Material
Acknowledgments
DNA sequencing was performed in the Vermont Cancer Center DNA Analysis Facility. We thank Alan Howard for advice on statistical analysis and Aimee Shen, Mary Tierney, Jacqueline Leung, and Pramod Rompikuntal for critically reading the manuscript. TgMyoA sequence and phosphorylation-site information were obtained from the Toxoplasma Genome Database (ToxoDB.org). ToxoDB is a component of the Eukaryotic Pathogen Genomics Resource (EuPathDB.org), a Bioinformatics Resource Center supported by the National Institute of Allergy and Infectious Diseases; we gratefully acknowledge the staff responsible for developing and maintaining this resource. This work was supported by U.S. Public Health Service grants AI054961 (G.E.W.) and AI096836 (S.J.N.M.); funding for the mass spectrometry analysis was provided by Vermont Genetics Network/National Institutes of Health Grant 8P20GM103449 from the IDeA Network of Biomedical Research Excellence Program of the National Institute of General Medical Sciences.
Abbreviations used:
- BSA
bovine serum albumin
- FBS
fetal bovine serum
- IMC
inner membrane complex
- PBS
phosphate-buffered saline
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-11-0648) on July 2, 2014.
REFERENCES
- Andenmatten N, Egarter S, Jackson AJ, Jullien N, Herman J-P, Meissner M. Conditional genome engineering in Toxoplasma gondii uncovers alternative invasion mechanisms. Nat Methods. 2013;10:125–127. doi: 10.1038/nmeth.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson-White BR, Ivey FD, Cheng K, Szatanek T, Lorestani A, Beckers CJ, Ferguson DJ, Sahoo N, Gubbels MJ. A family of intermediate filament-like proteins is sequentially assembled into the cytoskeleton of Toxoplasma gondii. Cell Microbiol. 2010;13:18–31. doi: 10.1111/j.1462-5822.2010.01514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bement WM, Mooseker MS. TEDS rule: a molecular rationale for differential regulation of myosins by phosphorylation of the heavy chain head. Cell Motil Cytoskeleton. 1995;31:87–92. doi: 10.1002/cm.970310202. [DOI] [PubMed] [Google Scholar]
- Billker O, Lourido S, Sibley LD. Calcium-dependent signaling and kinases in apicomplexan parasites. Cell Host Microbe. 2009;5:612–622. doi: 10.1016/j.chom.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black MW, Boothroyd JC. Lytic cycle of Toxoplasma gondii. Microbiol Mol Biol Rev. 2000;64:607–623. doi: 10.1128/mmbr.64.3.607-623.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch J, Turley S, Roach CM, Daly TM, Bergman LW, Hol WG. The closed MTIP-myosin A-tail complex from the malaria parasite invasion machinery. J Mol Biol. 2007;372:77–88. doi: 10.1016/j.jmb.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzeska H, Korn ED. Regulation of class I and class II myosins by heavy chain phosphorylation. J Biol Chem. 1996;271:16983–16986. doi: 10.1074/jbc.271.29.16983. [DOI] [PubMed] [Google Scholar]
- Carey KL, Westwood NJ, Mitchison TJ, Ward GE. A small-molecule approach to studying invasive mechanisms of Toxoplasma gondii. Proc Natl Acad Sci USA. 2004;101:7433–7438. doi: 10.1073/pnas.0307769101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carruthers V, Boothroyd JC. Pulling together: an integrated model of Toxoplasma cell invasion. Curr Opin Microbiol. 2007;10:83–89. doi: 10.1016/j.mib.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Carruthers VB, Giddings OK, Sibley LD. Secretion of micronemal proteins is associated with Toxoplasma invasion of host cells. Cell Microbiol. 1999;1:225–235. doi: 10.1046/j.1462-5822.1999.00023.x. [DOI] [PubMed] [Google Scholar]
- Carruthers VB, Sibley LD. Mobilization of intracellular calcium stimulates microneme discharge in Toxoplasma gondii. Mol Microbiol. 1999;31:421–428. doi: 10.1046/j.1365-2958.1999.01174.x. [DOI] [PubMed] [Google Scholar]
- Donald RG, Allocco J, Singh SB, Nare B, Salowe SP, Wiltsie J, Liberator PA. Toxoplasma gondii cyclic GMP-dependent kinase: chemotherapeutic targeting of an essential parasite protein kinase. Eukaryot Cell. 2002;1:317–328. doi: 10.1128/EC.1.3.317-328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egarter S, Andenmatten N, Jackson AJ, Whitelaw JA, Pall G, Black JA, Ferguson DJP, Tardieux I, Mogilner A, Meissner M. The Toxoplasma acto-MyoA motor complex is important but not essential for gliding motility and host cell invasion. PLoS One. 2014;9:e91819. doi: 10.1371/journal.pone.0091819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo T, Sethi KK, Piekarski G. Toxoplasma gondii: calcium ionophore A23187-mediated exit of trophozoites from infected murine macrophages. Exp Parasitol. 1982;53:179–188. doi: 10.1016/0014-4894(82)90059-5. [DOI] [PubMed] [Google Scholar]
- Foth BJ, Goedecke MC, Soldati D. New insights into myosin evolution and classification. Proc Natl Acad Sci USA. 2006;103:3681–3686. doi: 10.1073/pnas.0506307103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenal K, Foth B, Soldati D. In: Myosins: A Superfamily of Molecular Motors. ed. L Collucio, Dordrecht, Netherlands: Springer, 421–440; 2008. Myosin class XIV and other myosins in protists. [Google Scholar]
- Frenal K, Polonais V, Marq JB, Stratmann R, Limenitakis J, Soldati-Favre D. Functional dissection of the apicomplexan glideosome molecular architecture. Cell Host Microbe. 2010;8:343–357. doi: 10.1016/j.chom.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Garrison E, Treeck M, Ehret E, Butz H, Garbuz T, Oswald BP, Settles M, Boothroyd J, Arrizabalaga G. A forward genetic screen reveals that calcium-dependent protein kinase 3 regulates egress in Toxoplasma. PLoS Pathog. 2012;8:e1003049. doi: 10.1371/journal.ppat.1003049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaskins E, Gilk S, DeVore N, Mann T, Ward G, Beckers C. Identification of the membrane receptor of a class XIV myosin in Toxoplasma gondii. J Cell Biol. 2004;165:383–393. doi: 10.1083/jcb.200311137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilk SD, Gaskins E, Ward GE, Beckers CJ. GAP45 phosphorylation controls assembly of the Toxoplasma myosin XIV complex. Eukaryot Cell. 2009;8:190–196. doi: 10.1128/EC.00201-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Gygi SP, Niedzwiecka A, Miron M, Burley SK, Polakiewicz RD, Wyslouch-Cieszynska A, Aebersold R, Sonenberg N. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001;15:2852–2864. doi: 10.1101/gad.912401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannini D, Spath S, Lacroix C, Perazzi A, Bargieri D, Lagal V, Lebugle C, Combe A, Thiberge S, Baldacci P, et al. Independent roles of apical membrane antigen 1 and rhoptry neck proteins during host cell invasion by apicomplexa. Cell Host Microbe. 2011;10:591–602. doi: 10.1016/j.chom.2011.10.012. [DOI] [PubMed] [Google Scholar]
- Green JL, Rees-Channer RR, Howell SA, Martin SR, Knuepfer E, Taylor HM, Grainger M, Holder AA. The motor complex of Plasmodium falciparum: phosphorylation by a calcium-dependent protein kinase. J Biol Chem. 2008;283:30980–30989. doi: 10.1074/jbc.M803129200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hakansson S, Morisaki H, Heuser J, Sibley LD. Time-lapse video microscopy of gliding motility in Toxoplasma gondii reveals a novel, biphasic mechanism of cell locomotion. Mol Biol Cell. 1999;10:3539–3547. doi: 10.1091/mbc.10.11.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaslip AT, Leung JM, Carey KL, Catti F, Warshaw DM, Westwood NJ, Ballif BA, Ward GE. A small-molecule inhibitor of T. gondii motility induces the posttranslational modification of myosin light chain-1 and inhibits myosin motor activity. PLoS Pathog. 2010;6:e1000720. doi: 10.1371/journal.ppat.1000720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzelman MB, Schwartzman JD. A novel class of unconventional myosins from Toxoplasma gondii. J Mol Biol. 1997;271:139–146. doi: 10.1006/jmbi.1997.1167. [DOI] [PubMed] [Google Scholar]
- Herm-Gotz A, Weiss S, Stratmann R, Fujita-Becker S, Ruff C, Meyhofer E, Soldati T, Manstein DJ, Geeves MA, Soldati D. Toxoplasma gondii myosin A and its light chain: a fast, single-headed, plus-end-directed motor. EMBO J. 2002;21:2149–2158. doi: 10.1093/emboj/21.9.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettmann C, Herm A, Geiter A, Frank B, Schwarz E, Soldati T, Soldati D. A dibasic motif in the tail of a class XIV apicomplexan myosin is an essential determinant of plasma membrane localization. Mol Biol Cell. 2000;11:1385–1400. doi: 10.1091/mbc.11.4.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Chen S, Li S, Cha J, Long C, Li L, He Q, Liu Y. Protein kinase A and casein kinases mediate sequential phosphorylation events in the circadian negative feedback loop. Gene Dev. 2007;21:3283–3295. doi: 10.1101/gad.1610207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacot D, Frenal K, Marq JB, Sharma P, Soldati-Favre D. Assessment of phosphorylation in Toxoplasma glideosome assembly and function. Cell Microbiol. 2014 doi: 10.1111/cmi.12307. DOI: 10.1111/cmi.12307. [DOI] [PubMed] [Google Scholar]
- Jacot D, Soldati-Favre D. Does protein phosphorylation govern host cell entry and egress by the Apicomplexa. Int J Med Microbiol. 2012;302:195–202. doi: 10.1016/j.ijmm.2012.07.012. [DOI] [PubMed] [Google Scholar]
- Jewett TJ, Sibley LD. Aldolase forms a bridge between cell surface adhesins and the actin cytoskeleton in apicomplexan parasites. Mol Cell. 2003;11:885–894. doi: 10.1016/s1097-2765(03)00113-8. [DOI] [PubMed] [Google Scholar]
- Johnson TM, Rajfur Z, Jacobson K, Beckers CJ. Immobilization of the type XIV myosin complex in Toxoplasma gondii. Mol Biol Cell. 2007;18:3039–3046. doi: 10.1091/mbc.E07-01-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko HW, Kim EY, Chiu J, Vanselow JT, Kramer A, Edery I. A hierarchical phosphorylation cascade that regulates the timing of PERIOD nuclear entry reveals novel roles for proline-directed kinases and GSK-3β/SGG in circadian clocks. J Neurosci. 2010;30:12664–12675. doi: 10.1523/JNEUROSCI.1586-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasonder E, Green JL, Camarda G, Talabani H, Holder AA, Langsley G, Alano P. The Plasmodium falciparum schizont phosphoproteome reveals extensive phosphatidylinositol and cAMP-protein kinase A signaling. J Proteome Res. 2012;11:5323–5337. doi: 10.1021/pr300557m. [DOI] [PubMed] [Google Scholar]
- Leung JM, Rould MA, Konradt C, Hunter CA, Ward GE. Disruption of TgPHIL1 alters specific parameters of Toxoplasma gondii motility measured in a quantitative, three-dimensional live motility assay. PLoS One. 2014a;9:e85763. doi: 10.1371/journal.pone.0085763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung JM, Tran F, Pathak RB, Poupart S, Heaslip AT, Ballif BA, Westwood NJ, Ward GE. Identification of T. gondii myosin light chain-1 as a direct target of tachyplegina-2, a small-molecule inhibitor of parasite motility and invasion. PLoS One. 2014b;9:e98056. doi: 10.1371/journal.pone.0098056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Hong M-S, Shu S, Yu S, Korn ED. Regulation of the filament structure and assembly of Acanthamoeba myosin II by phosphorylation of serines in the heavy-chain nonhelical tailpiece. Proc Natl Acad Sci USA. 2013a;110:E33–E40. doi: 10.1073/pnas.1219727110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Lee D-Y, Cai S, Yu S, Shu S, Levine RL, Korn ED. Regulation of the actin-activated MgATPase activity of Acanthamoeba myosin II by phosphorylation of serine 639 in motor domain loop 2. Proc Natl Acad Sci USA. 2013b;110:E23–E32. doi: 10.1073/pnas.1219713110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourido S, Jeschke GR, Turk BE, Sibley LD. Exploiting the unique ATP-binding pocket of Toxoplasma calcium-dependent protein kinase 1 to identify its substrates. ACS Chem Biol. 2013;8:1155–1162. doi: 10.1021/cb400115y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourido S, Shuman J, Zhang C, Shokat KM, Hui R, Sibley LD. Calcium-dependent protein kinase 1 is an essential regulator of exocytosis in Toxoplasma. Nature. 2010;465:359–362. doi: 10.1038/nature09022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourido S, Tang K, Sibley LD. Distinct signalling pathways control Toxoplasma egress and host-cell invasion. EMBO J. 2012;31:4524–4534. doi: 10.1038/emboj.2012.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett JL, Sibley LD. Intracellular calcium stores in Toxoplasma gondii govern invasion of host cells. J Cell Sci. 2003;116:3009–3016. doi: 10.1242/jcs.00596. [DOI] [PubMed] [Google Scholar]
- McCoy JM, Whitehead L, van Dooren GG, Tonkin CJ. TgCDPK3 regulates calcium-dependent egress of Toxoplasma gondii from host cells. PLoS Pathog. 2012;8:e1003066. doi: 10.1371/journal.ppat.1003066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner M, Ferguson DJP, Frischknecht F. Invasion factors of apicomplexan parasites: essential or redundant. Curr Opin Microbiol. 2013;16:438–444. doi: 10.1016/j.mib.2013.05.002. [DOI] [PubMed] [Google Scholar]
- Meissner M, Schluter D, Soldati D. Role of Toxoplasma gondii myosin A in powering parasite gliding and host cell invasion. Science. 2002;298:837–840. doi: 10.1126/science.1074553. [DOI] [PubMed] [Google Scholar]
- Messina M, Niesman I, Mercier C, Sibley LD. Stable DNA transformation of Toxoplasma gondii using phleomycin selection. Gene. 1995;165:213–217. doi: 10.1016/0378-1119(95)00548-k. [DOI] [PubMed] [Google Scholar]
- Mital J, Meissner M, Soldati D, Ward GE. Conditional expression of Toxoplasma gondii apical membrane antigen-1 (TgAMA1) demonstrates that TgAMA1 plays a critical role in host cell invasion. Mol Biol Cell. 2005;16:4341–4349. doi: 10.1091/mbc.E05-04-0281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mital J, Schwarz J, Taatjes DJ, Ward GE. Laser scanning cytometer-based assays for measuring host cell attachment and invasion by the human pathogen Toxoplasma gondii. Cytometry A. 2006;69:13–19. doi: 10.1002/cyto.a.20202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno SN, Zhong L. Acidocalcisomes in Toxoplasma gondii tachyzoites. Biochem J. 1996;31:655–659. doi: 10.1042/bj3130655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munter S, Sabass B, Selhuber-Unkel C, Kudryashev M, Hegge S, Engel U, Spatz JP, Matuschewski K, Schwarz US, Frischknecht F. Plasmodium sporozoite motility is modulated by the turnover of discrete adhesion sites. Cell Host Microbe. 2009;6:551–562. doi: 10.1016/j.chom.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Nagamune K, Sibley LD. Comparative genomic and phylogenetic analyses of calcium ATPases and calcium-regulated proteins in the apicomplexa. Mol Biol Evol. 2006;23:1613–1627. doi: 10.1093/molbev/msl026. [DOI] [PubMed] [Google Scholar]
- Nebl T, Prieto JH, Kapp E, Smith BJ, Williams MJ, Yates JR, 3rd, Cowman AF, Tonkin CJ. Quantitative in vivo analyses reveal calcium-dependent phosphorylation sites and identifies a novel component of the Toxoplasma invasion motor complex. PLoS Pathog. 2011;7:e1002222. doi: 10.1371/journal.ppat.1002222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porchet E, Torpier G. Freeze fracture study of Toxoplasma and Sarcocystis infective stages [in French] Z Parasitenkd. 1977;54:101–124. doi: 10.1007/BF00380795. [DOI] [PubMed] [Google Scholar]
- Rabjeau A, Foussard F, Mauras G. The membrane complex of Toxoplasma gondii: ultrastructural study of its constituents [in French] Bull Assoc Anat (Nancy) 1994;78:29–31. [PubMed] [Google Scholar]
- Rabjeau A, Foussard F, Mauras G, Dubremetz JF. Enrichment and biochemical characterization of Toxoplasma gondii tachyzoite plasmalemma. Parasitology. 1997;114:421–426. doi: 10.1017/s0031182096008670. [DOI] [PubMed] [Google Scholar]
- Redowicz MJ. Regulation of nonmuscle myosins by heavy chain phosphorylation. J Muscle Res Cell Motil. 2001;22:163–173. doi: 10.1023/a:1010552929028. [DOI] [PubMed] [Google Scholar]
- Rogers SL, Karcher RL, Roland JT, Minin AA, Steffen W, Gelfand VI. Regulation of melanosome movement in the cell cycle by reversible association with myosin V. J Cell Biol. 1999;146:1265–1276. doi: 10.1083/jcb.146.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos DS, Donald RG, Morrissette NS, Moulton AL. Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell Biol. 1994;45:27–63. doi: 10.1016/s0091-679x(08)61845-2. [DOI] [PubMed] [Google Scholar]
- Sahoo N, Beatty W, Heuser J, Sept D, Sibley LD. Unusual kinetic and structural properties control rapid assembly and turnover of actin in the parasite Toxoplasma gondii. Mol Biol Cell. 2006;17:895–906. doi: 10.1091/mbc.E05-06-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheiner L, Santos JM, Klages N, Parussini F, Jemmely N, Friedrich N, Ward GE, Soldati-Favre D. Toxoplasma gondii transmembrane microneme proteins and their modular design. Mol Microbiol. 2010;77:912–929. doi: 10.1111/j.1365-2958.2010.07255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Sibley LD. Toxoplasma aldolase is required for metabolism but dispensable for host-cell invasion. Proc Natl Acad Sci USA. 2014;111:3567–3572. doi: 10.1073/pnas.1315156111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley LD. Intracellular parasite invasion strategies. Science. 2004;304:248–253. doi: 10.1126/science.1094717. [DOI] [PubMed] [Google Scholar]
- Sibley LD. Invasion and intracellular survival by protozoan parasites. Immunol Rev. 2011;240:72–91. doi: 10.1111/j.1600-065X.2010.00990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinai AP. The Toxoplasma gondii parasitophorous vacuole membrane: a multifuntional organelle in the infected cell. In: Weiss LM, Kim K, editors. In: Toxoplasma gondii—The Model Apicomplexan: Perspectives and Methods. London: Academic Press; 2014. pp. 375–387. [Google Scholar]
- Skillman KM, Diraviyam K, Khan A, Tang K, Sept D, Sibley LD. Evolutionarily divergent, unstable filamentous actin is essential for gliding motility in apicomplexan parasites. PLoS Pathog. 2011;7:e1002280. doi: 10.1371/journal.ppat.1002280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suss-Toby E, Zimmerberg J, Ward GE. Toxoplasma invasion: the parasitophorous vacuole is formed from host cell plasma membrane and pinches off via a fission pore. Proc Natl Acad Sci USA. 1996;93:8413–8418. doi: 10.1073/pnas.93.16.8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DC, Ahmed A, Gilberger TW, Sharma P. Regulation of Plasmodium falciparum glideosome associated protein 45 (PfGAP45) phosphorylation. PLoS One. 2012;7:e35855. doi: 10.1371/journal.pone.0035855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonkin ML, Roques M, Lamarque MH, Pugnière M, Douguet D, Crawford J, Lebrun M, Boulanger MJ. Host cell invasion by apicomplexan parasites: insights from the co-structure of AMA1 with a RON2 peptide. Science. 2011;333:463–467. doi: 10.1126/science.1204988. [DOI] [PubMed] [Google Scholar]
- Treeck M, Sanders JL, Elias JE, Boothroyd JC. The phosphoproteomes of Plasmodium falciparum and Toxoplasma gondii reveal unusual adaptations within and beyond the parasites’ boundaries. Cell Host Microbe. 2011;10:410–419. doi: 10.1016/j.chom.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Poppel NF, Welagen J, Duisters RF, Vermeulen AN, Schaap D. Tight control of transcription in Toxoplasma gondii using an alternative tet repressor. Int J Parasitol. 2006;36:443–452. doi: 10.1016/j.ijpara.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Vieira MC, Moreno SN. Mobilization of intracellular calcium upon attachment of Toxoplasma gondii tachyzoites to human fibroblasts is required for invasion. Mol Biochem Parasitol. 2000;106:157–162. doi: 10.1016/s0166-6851(99)00182-6. [DOI] [PubMed] [Google Scholar]
- Wetzel DM, Chen LA, Ruiz FA, Moreno SN, Sibley LD. Calcium-mediated protein secretion potentiates motility in Toxoplasma gondii. J Cell Sci. 2004;117:5739–5748. doi: 10.1242/jcs.01495. [DOI] [PubMed] [Google Scholar]
- Wetzel DM, Hakansson S, Hu K, Roos D, Sibley LD. Actin filament polymerization regulates gliding motility by apicomplexan parasites. Mol Biol Cell. 2003;14:396–406. doi: 10.1091/mbc.E02-08-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SY, Remington JS. Biology of Toxoplasma gondii. AIDS. 1993;7:299–316. doi: 10.1097/00002030-199303000-00001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.