

A multiplexed analysis of the transcriptional regulation of yeast pseudohyphal growth recorded the binding of 28 different transcription factors with barcoded transposons. A core set of target genes is identified, and a process of DNA looping at the FLO11 locus that provides transcriptional memory for expression of the gene is described.
Abstract
Pseudohyphal growth is a developmental pathway seen in some strains of yeast in which cells form multicellular filaments in response to environmental stresses. We used multiplexed transposon “Calling Cards” to record the genome-wide binding patterns of 28 transcription factors (TFs) in nitrogen-starved yeast. We identified TF targets relevant for pseudohyphal growth, producing a detailed map of its regulatory network. Using tools from graph theory, we identified 14 TFs that lie at the center of this network, including Flo8, Mss11, and Mfg1, which bind as a complex. Surprisingly, the DNA-binding preferences for these key TFs were unknown. Using Calling Card data, we predicted the in vivo DNA-binding motif for the Flo8-Mss11-Mfg1 complex and validated it using a reporter assay. We found that this complex binds several important targets, including FLO11, at both their promoter and termination sequences. We demonstrated that this binding pattern is the result of DNA looping, which regulates the transcription of these targets and is stabilized by an interaction with the nuclear pore complex. This looping provides yeast cells with a transcriptional memory, enabling them more rapidly to execute the filamentous growth program when nitrogen starved if they had been previously exposed to this condition.
INTRODUCTION
Understanding how signaling pathways activate transcriptional agendas to differentiate cells into a desired fate is a central question of developmental biology. One of the simplest models of cellular differentiation is offered by the budding yeast Saccharomyces cerevisiae. On appropriate stimulation, the normally single-celled organism is capable of undergoing a developmental switch to a multicellular form, a process known as filamentous growth. In response to nutrient deprivation or other stress signals, budding yeast cells grow into elongated cell shapes, or pseudohyphae, that are morphologically and physiologically distinct from the traditional yeast form more commonly associated with budding yeast. Unlike bacteria or the unicellular eukaryotes such as protozoa, yeasts are nonflagellated, nonmotile microbes; thus, faced with nutrient starvation, yeast cells have two options for survival: sporulate or search for nutrients (Vivier et al., 1997). Filamentous growth is believed to allow budding yeast to search for nutrients (Gimeno et al., 1992), as the elongated cells remain associated and in communication with each other. Thus, as the population proliferates, cells can invade solid agar media or move through static liquid media in search of carbon or nitrogen (Honigberg et al., 2011). Filamentous growth is also crucial for virulence in pathogenic yeast such as Candida albicans because it allows cells to adhere to other surfaces and form drug-resistant biofilms (Lo et al., 1997; Verstrepen et al., 2006).
The most commonly used S. cerevisiae strains for the study of filamentous growth are those derived from the Σ1278b background. Based on use of this strain, a large number of signaling pathways have been implicated in a cell's decision to transition to filamentous growth. These include the mitogen-activated protein kinase (MAPK), cyclical adenosine monophosphate (cAMP)–protein kinase A (PKA), and target of rapamycin (TOR) signaling pathways, which are responsible for sensing cell-cycle, nutrient, pH, and osmolarity conditions (Granek et al., 2011). A genome-wide analysis of the effect of single-gene deletions on pseudohyphal growth in the Σ1278b strain identified 691 genes that play a critical role in the cell's ability to execute the filamentation program (Ryan et al., 2012). A smaller set of 550 genes, identified in an overexpression screen (Shively et al., 2013), was found to enhance pseudohyphal growth. Although these studies revealed a large number of genes important for filamentous growth, less is known about the regulatory network that controls and coordinates their expression.
A few of the transcription factors (TFs) that are known to be important for filamentous growth have been analyzed by chromatin immunoprecipitation (ChIP)-chip (Borneman et al., 2006; Bumgarner et al., 2009; Cain et al., 2012). However, given the number and diversity of signals involved in the initiation of filamentous growth and the problem of cross-talk between these pathways (Borneman et al., 2006), our understanding of how these pathways are integrated into a logical response remains limited. The “Calling Card” method represents a way of identifying the genomic binding locations of multiple TFs in a single experiment and recording that binding through development in vivo (Wang et al., 2011, 2012). We used this technology to determine the genome-wide binding patterns of 28 filamentation-related TFs as yeast cells switch to filamentous growth to elucidate the regulatory network governing filamentous growth.
RESULTS
We constructed a set of 28 strains, each with a Sir4-tagged TF relating to pseudohyphal growth (Supplemental Table S1) in a Σ1278b-derived strain of S. cerevisiae. These TFs include MAPK regulators Ste12 and Tec1, PKA regulators Flo8, Sok2, and Phd1, and other TFs spanning the Rim101 and TOR pathways. Each tagged strain consists of the Ty5-targeting domain of Sir4 (YDR227W, amino acids 951–1200) fused to C-terminus of each TF. Each strain was then transfected with a barcoded Ty5 transposon and pooled together for multiplexing through the remainder of the experiment (Figure 1). Pooled cells were then grown on agar plates containing synthetic low–ammonium galactose (SLAG) medium to induce both filamentous growth and Ty5 transposition. The yeast underwent invasive growth into the agar and the barcoded Ty5 transposon inserted into the genome nearby, where its matched Sir4-tagged TF was bound at that time (Wang et al., 2011). After 48 h of induction, the pseudohyphae that had invaded the agar were collected and the insertions sequenced on a HiSeq platform (Illumina, San Diego, CA). The subsequent reads were then aligned to the Σ1278b genome (Dowell et al., 2010). The barcode in each transposon sequence identifies which TF directed its insertion, and the portion of the sequencing read that maps to the yeast genome reveals where the TF was bound.
FIGURE 1:
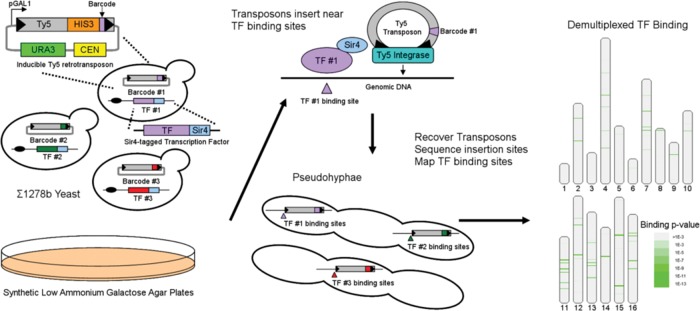
Design of multiplexed experiment. Multiplexed Calling Card analysis begins by transforming 28 barcoded Ty5 transposons into 28 Σ1278b strains in which the genomic copy of a TF is tagged with the Ty5-interacting domain of Sir4. Invasive growth and transposon hopping are simultaneously induced by plating on SLAG plates. Ty5 transposition is directed toward TF binding sites in each tagged strain. After 48 h, cells are collected and transposons are mapped on an Illumina HiSeq with the barcode in each transposon identifying the TF that directed it. Locations are mapped to the Σ1278b genome, and significant clusters are calculated.
Calling Cards accurately identify TF binding in the promoters of essential genes and genes related to pseudohyphal growth in haploid yeast cells
Previous work benchmarked the Calling Card method against ChIP-chip analysis and found it to be accurate and reproducible (Wang et al., 2011). However, that analysis was performed in diploid yeast cells. The study of the haploid invasive growth program of pseudohyphal growth may encounter additional biases that would not be observed in diploid yeast. For example, it is possible that an insertion of the Ty5 transposon in or near the promoter of an essential gene may disrupt its expression, thereby killing the cell and preventing transposon recovery from this promoter. To determine whether such biases exist, we recovered Ty5 insertions from an untagged sir4Δ haploid strain. Because these insertion events were not directed by a TF, they should not be biased to insert near any TF binding sites (Wang et al., 2011). We observed an eightfold decrease in transposon insertions into the coding sequence of essential genes relative to the number expected under the assumption that transposons insert in a uniform distribution into the yeast genome. For nonessential genes, we observed a 0.3-fold decrease for insertions into their coding sequence. However, for insertions into the upstream regulatory regions of essential genes, we observed a 1.7-fold increase over the expected value; for nonessential genes, we observed a 2.5-fold increase (Supplemental Figure S1). The higher-than-expected values can be explained by Ty5’s preference to insert in sequences upstream of genes (Baller et al., 2011; Wang et al., 2011). We also tested the promoters of genes specifically required for the pseudohyphal growth phenotype and observed a 1.5-fold increase over the expected value. From these results, we conclude that the method can recover transposon insertions events in the promoters of essential genes, nonessential genes, and the genes required for pseudohyphal growth in haploid cells.
To further establish that the multiplexed Calling Card is accurately identifying TF targets involved in pseudohyphal growth, we compared the targets determined for three of the TFs in our study (Flo8, Sok2, and Ste12) against those found by ChIP-chip binding data in diploid Σ1278b strains (Borneman et al., 2006). In all three cases, there was a high degree of concordance, as determined by receiver operator characteristic (ROC) curve analysis (Supplemental Figure S2). Differences in binding likely represent physiological differences between the haploid and diploid pseudohyphal growth programs, as these require substantially different sets of necessary genes (Ryan et al., 2012). We next compared the targets bound by at least one of the TFs we tested with the core 61 genes known to be required for all of the filamentous growth development programs (e.g., FLO11, FLO8, TEC1, etc.) and found that 25 of these targets (41%) were bound directly by one of the TFs (p < 1e-17, hypergeometric). This is a high degree of overlap, considering that many of the genes required for filamentous growth will be regulated by factors downstream of our 28 core TFs. Taken together, these results indicate that the multiplexed Calling Card approach is accurately identifying targets related to pseudohyphal growth.
Transcription factor regulation of pseudohyphal growth
The 28 TFs bind a total of 725 targets across the genome (Figure 2A). The binding data for all TFs are listed in Supplemental Table S2. Because many of the TFs included in the experiment have additional regulatory roles beyond their involvement in pseudohyphal growth, it is likely that not all of their targets identified here are involved in the process. To understand better how each TF is regulating filamentous growth, we ranked the TFs based on the percentage of target genes that were either 1) in the 691 genes required for pseudohyphal growth or 2) in the 550 genes that, when overexpressed, enhance pseudohyphal growth (Figure 2B). For any given TF, a minority of its targets were found in one of these sets, with percentages ranging between 0 and 29% for required targets and 0 and 21% for overexpression targets. The majority of the genes in these two classes are not directly bound by one of the 28 TFs. These genes are likely regulated by TFs upstream or downstream of the ones included in this study.
FIGURE 2:

The transcriptional network of pseudohyphal TFs. (A) The 28 TFs bind a total of 725 targets within the filamentous growth transcriptional network. (B) Percentages of targets bound by the 28 TFs that are required for pseudohyphal growth and whose overexpression can enhance pseudohyphal growth. (C) Genes whose promoters are bound by ≥2 of the 28 tested TFs are more likely to be required for or can enhance pseudohyphal growth than genes that are bound by a single TF or none of the TFs. (D) Fourteen of the 28 TFs showed a nonzero betweenness centrality in the transcriptional network of pseudohyphal growth.
Many of the target genes have promoters that are bound by more than one of the 28 TFs analyzed, suggesting combinatorial regulation. For example, the promoters of MGA1 and PHD1 were previously identified as regulated by 6 different TFs in pseudohyphal growth by ChIP-chip (Borneman et al., 2006). These genes are master regulators of the process and can induce pseudohyphal growth in rich media. These examples led us to hypothesize that genes that are regulated by >1 of the 28 TFs analyzed in our study were more likely to be required for filamentous growth or to enhance the phenotype. To test this hypothesis, we divided all genes into three categories: 1) those bound by multiple filamentation-related TFs, 2) those bound by a single TF from the 28 tested, and 3) those not bound by a TF in our study. The targets bound by multiple TFs were more likely both to be required for, and whose overexpression would enhance, pseudohyphal growth compared with the other two sets (p < 0.02, Fisher's exact test; Figure 2C). From this analysis, we conclude that the combinatorial binding of multiple TFs related to pseudohyphal growth makes a gene more likely to play a required role in regulating the process, and these hubs are listed in Supplemental Table S3.
Some of the TFs in our analysis, such as Gcn4 and Fkh1, directly regulate only a small number of targets related to pseudohyphal growth. Because increased binding by multiple TFs correlates with importance in the regulation of the process, we next ordered each TF in the network by its betweenness centrality to determine which are at the core of the transcriptional circuit. In graph theory, betweenness centrality is a count of all the shortest paths through the network that pass through that node and represents a measure of the node's centrality in the network. Of the TFs tested, 14 had a nonzero betweenness centrality score (Figure 2D), suggesting that these TFs are central to the filamentous growth program. Of particular interest were the factors Flo8, Mfg1, and Mss11. These TFs had high centrality scores, bind cooperatively (Shapiro et al., 2012), and are required activators of filamentous growth, as deletion of any of the three factors abolishes the cell's ability to undergo pseudohyphal growth (Ryan et al., 2012).
The in vivo DNA-binding preferences of the Flo8-Mfg1-Mss11 complex
We next analyzed the Calling Card data for all 28 TFs to identify the consensus sequence or motif bound by a TF (Wang et al., 2011). We found significant motifs for 17 of these factors (Figure 3A). Because no consistent consensus binding site has been identified for the Flo8-Mfg1-Mss11 complex (Zhu et al., 2009; Spivak and Stormo, 2012) despite their importance to the filamentous growth program (Shapiro et al., 2012), we focused on their motifs. The most significant motifs found for Flo8, Mfg1, and Mss11 were identical, as would be expected for TFs that bind in a complex. For these TFs, the distribution of Calling Card insertions was centered on this sequence motif, with a mean distances from the center of each distribution less than the length of the motif (Figure 3B), providing further evidence that this motif accurately captures the DNA-binding preferences of the complex. To verify this motif, we created a reporter plasmid with one of our Flo8-Mfg1-Mss11–bound promoters (CUP9) that contains a single putative Flo8-binding site, driving the florescent protein DsRed. CUP9 expression should be activated in the nitrogen starvation condition, and the wild-type (WT) reporter responded as expected, with a 3.6-fold increase in DsRed fluorescence in cells grown in the starved condition (Figure 3C). A mutation in the predicted Flo8-binding site decreased this induction by 82% (p < 1 × 10−4, Student's t test), indicating that this sequence is necessary for full CUP9 activation.
FIGURE 3:

Identification of a novel binding motif for the Flo8-Mfg1-Mss11 complex. (A) Significant DNA-binding motifs for all of the Calling Card tagged TFs in the experiment. (B) The binding distributions of Flo8, Mfg1, and Mss11 are all centered on the same binding motif ±3 base pairs, which is less than the length of the motif. (C) A mutation in the predicted Flo8p-binding site in the CUP9 promoter in a reporter driving DsRed significantly decreases expression in the nitrogen-starved induction condition.
Gene looping at the FLO11 locus
During our investigation into the binding of Flo8, we noticed an unusual pattern of binding at the FLO11 locus. FLO11 encodes a flocculin, a glycosylphosphatidylinositol-anchored cell surface glycoprotein that is necessary for pseudohyphal growth (Lambrechts et al., 1996; Guo et al., 2000) and is a key component of the fibers connecting flocculating yeast cells. We observed a peak of Flo8 binding at the FLO11 promoter centered on a Flo8-Mfg1-Mss11 consensus binding site that is located 1.1 kb upstream from the translation start site (Figure 4A). Of interest, we also observed Flo8 binding in the terminator sequence of FLO11, even though this sequence lacks a consensus binding motif for the Flo8-Mfg1-Mss11 complex. Because the promoter of FLO11 is bound by several of the TFs that were analyzed, we next explored whether similar patterns of binding were observed for any other factors. As expected, the activators Mfg1 and Mss11 both bound FLO11 in a manner that was nearly identical to that observed for Flo8. Both showed ratios of promoter binding to terminator binding of 0.5 (Figure 4B). In contrast, two known repressors of filamentous growth, Sok2 and Sfl1, bound almost exclusively at the FLO11 promoter, both with binding ratios of 0.9. This pattern held true for the eight additional TFs that bind at FLO11, with activators of FLO11 expression bound equally at both the promoter and terminator and repressors of FLO11 expression bound only at the promoter (Figure 4B).
FIGURE 4:

DNA looping occurs at the FLO11 locus. (A) Binding of three activators at FLO11 for three activators (Flo8, Mfg1, and Mss11) shows equal binding in the promoter and terminator. The binding of two repressors (Sok2 and Sfl1) shows higher binding in the promoter. (B) Relative binding of all of the tagged TFs that bind in the promoter of FLO11 correlates with the function of the TF. The y-axis is the phenotypic score for haploid invasive growth for knockouts of each protein, with negative scores meaning that the mutant has a deleterious effect and positive scores meaning that the mutant has an enhanced effect. The x-axis represents the relative binding of the TF in the promoter relative to the terminator. (C) Looping between promoter and terminator will occur when activators are bound and FLO11 is expressed, whereas when repressors are bound, there is no looping and no FLO11 expression. (D) Looping between the FLO11 promoter and terminator depends on nitrogen starvation and is abolished in a Flo8 mutant strain. Error bars are shown as SD among three biological replicates.
These observations are consistent with two possible models. In one model, activators are recruited to the FLO11 promoter by specific cis-acting sequences and are then brought into close proximity to the terminator by DNA looping (Singh and Hampsey, 2007). An alternative model is that the activators have affinity for both the promoter and terminator sequences at FLO11. To distinguish between these two possibilities, we used GOMER, a thermodynamic framework that predicts transcription factor binding based on a position-specific weight matrix (PSWM; Granek and Clarke, 2005). Based on sequence preferences alone, Flo8 was predicted by GOMER to be 18-fold more likely to bind at the FLO11 promoter than the terminator. This prediction is in stark contrast to the observed binding ratio of 1:1. Similar results were observed for the other activators that bind at FLO11 (Supplemental Figure S3). These results suggest that activators are indeed first recruited to the promoter and then brought in proximity to the terminator by DNA looping. In our working model, this loop forms when the FLO11 gene is expressed (i.e., activators are bound), which causes the insertion of TF-directed Calling Cards into both the promoter and terminator; however, when FLO11 is not expressed (i.e., repressors are bound), the loop dissipates, and Calling Card insertions only occur in the promoter (Figure 4C).
To test this model, we performed chromatin conformation capture (3C) analysis (Dekker et al., 2002; Singh et al., 2009) to measure the proximity of the FLO11 promoter and terminator (see Materials and Methods). Cells grown in the low-nitrogen filamentous induction condition displayed a twofold higher 3C signal than cells grown in rich media (p = 3.8 × 10−3, Student's t test; Figure 4D), confirming that looping is correlated with FLO11 expression. Of importance, this 3C signal is dependent on Flo8 binding, as deletion of this required activator decreases the looping signal 10-fold. The 3C signal is also significantly higher at FLO11 than at the corresponding positions in MUB1, a gene of approximately the same length as FLO11 and expressed at approximately the same level as FLO11 during pseudohyphal growth. Together these results support our model in which activator binding at FLO11 causes a loop to form, bringing the promoter and terminator in close proximity.
DNA looping provides transcriptional memory for pseudohyphal growth
Given the presence of DNA looping at this important target relating to pseudohyphal growth, we next sought to explore whether this phenomenon plays a role in regulating the process. One proposed physiological advantage for DNA looping is the ability to recycle RNA polymerase II (RNAP II). In this model, RNAP II and transcriptional components are more quickly returned from the terminator to the promoter, given their much closer proximity, enabling more efficient gene expression (Singh and Hampsey, 2007). Such a model would predict that looped genes should be enriched for those that are more highly transcribed in a nitrogen-starved condition. To test this, we reanalyzed the Calling Card data and scored every target gene for TF binding in the terminator sequence that could not be explained by the PSWM for the TF. This analysis identified nine probable looping events (Supplemental Table S4). Six of the nine genes identified were TFs, and the most significant Gene Ontology term for the genes was flocculation (GO:0000128; p = 3.53e-06), indicating that looping may play a role in the transcriptional regulation of filamentous growth. However, when compared with our RNA-seq data, all nine of the “looping” genes were expressed at levels below the average of all genes (Supplemental Figure S4). This result suggests that the observed looping is not increasing gene expression via RNAP II recycling.
Another possible function for gene looping in filamentous growth is for cellular memory. Gene loops at GAL10 (Laine et al., 2009) and INO1 (Brickner et al., 2007) have been shown to provide yeast cells with a “transcriptional memory” of previous exposure to certain environments. For example, gene loops between the GAL10 promoter and terminator are formed when yeast cells are grown under conditions that induce this gene (i.e., galactose); these loops are maintained after removal of the inducing agent and subsequent GAL10 down-regulation. If these cells are later grown in the presence of galactose, GAL10 expression increases more rapidly than in yeast that were not pretreated. This transcriptional memory requires loop formation at GAL10 (Laine et al., 2009). To test whether FLO11 might also exhibit transcriptional memory, we pretreated yeast in nitrogen-starved media and then moved them back to rich media for 4 h. We then moved those cells back to nitrogen-starved media and compared FLO11 expression with cells that had not been pretreated (Figure 5A). We found that pretreated cells induced FLO11 expression significantly faster than did naive cells, suggesting that yeast have evolved a mechanism for transcriptional memory at the FLO11 locus. If this was due to DNA looping, then we should observe gene looping at FLO11 in rich media if the cells were previously nitrogen starved but not otherwise. We performed 3C as before in this condition and observed gene looping in pretreated cells (Figure 5B), even after expression of FLO11 had returned to basal levels (12-fold decrease). In contrast, little gene looping was observed in yeast cells that were not pretreated. This suggests that gene looping at FLO11 provides yeast cells with a mechanism to “remember” prior nutrient deprivation in order to execute more rapidly the filamentous growth program upon reexposure to these conditions.
FIGURE 5:

DNA looping provides transcriptional memory for activation of pseudohyphal growth. (A) Expression of FLO11 increases more rapidly in cells moved from rich to nitrogen-starved media when they had been pretreated with nitrogen-starved medium 4 h earlier. (B) Looping between the FLO11 promoter and terminator persists in pretreated cells, even after expression of FLO11 has returned to basal expression. (C) Strains with knockouts of nucleoporin protein Nup2 show decreased transcriptional memory for the same pretreatment experiment for FLO11. (D) Looping at FLO11 is inhibited in nitrogen-starved conditions in the mutant Nup2 strain. Error bars are shown as SD among three biological replicates.
To test whether gene looping at FLO11 is necessary for the observed transcriptional memory, we next sought to assess the consequences of disrupting looping. Brickner and colleagues demonstrated that gene looping at INO1 was stabilized by a physical interaction between the looped gene and the nuclear pore complex (NPC), where the complex would remain primed for rapid expression (Ahmed et al., 2010). To test whether the observed looping at the FLO11 locus uses a similar mechanism, we deleted NUP2, which encodes a nucleoporin that facilitates nucleocytoplasmic transport, is involved in maintaining gene loops at the NPC, and whose deletion has negligible effects on all assayed traits of filamentous growth (Ryan et al., 2012). A nup2Δ deletion strain, after pretreatment in nitrogen-starved medium, shows significantly slower activation of FLO11 upon returning to the induction condition (Figure 5C). The same strain shows impaired loop formation at FLO11 by 3C (Figure 5D), indicating that the memory mechanism at FLO11 is dependent on loop formation. The lack of effect of the nup2Δ mutation on the pseudohyphal growth phenotype indicates that this mechanism of looping is not required for FLO11 expression or the broader process of filamentous growth; however, NPC-mediated looping is required for the transcriptional memory of FLO11 expression and likely the other identified looping events.
Taken together, our results demonstrate that yeast cells can more rapidly undergo filamentous growth when starved for nitrogen if they had been previously exposed to this condition. This “memory” is achieved by a mechanism of gene looping at the flocculin FLO11 and likely eight other gene targets involved in the activation of pseudohyphal growth. This enables the yeast to express these genes more rapidly if they need to activate pseudohyphal growth in the future. We also showed that the gene loop at FLO11 is stabilized by an interaction with the NPC, as was previously observed for INO1 and GAL10.
DISCUSSION
Using a multiplexed Calling Card approach, we recorded the binding of 28 TFs during the developmental program of pseudohyphal growth. The complexity of the regulatory network that governs this process is illustrated by the large number of genes required for this process and the large number of TFs that coordinate the expression of these genes. The TFs and signaling pathways studied here are not unique to pseudohyphal growth, and many are involved in other cellular process, such as the cell cycle, mating, and stress response. It is therefore not surprising that only a minority of all targets bound by any of the TFs were directly related to the process. Some of the 28 TFs that we analyzed had a very low percentage of targets that could be functionally tied to pseudohyphal growth and seem to only tangentially regulate these genes. A requirement to be included in our analysis was that the TF should have an effect (positive or negative) on the cell's ability to undergo filamentous growth. Some of these TFs may act indirectly by interfering with nitrogen metabolism or other ways of initiating stress responses. By focusing on the core set of genes that were bound by multiple TFs in the multiplex, we were able to enrich for some of the more important targets, which need to be activated for the process. This allowed us to define 14 TFs that were most central in the filamentous growth network, including the Flo8-Mfg1-Mss11 complex, for which we identify a novel motif describing its DNA-binding preferences.
Although one purpose of this study was to provide a comprehensive map of the regulatory network governing filamentous growth, the FLO11 gene emerged as a particularly intriguing node in this pathway. Thirteen of the 28 TFs screened in this study had significant binding in its promoter, and we also observed substantial gene looping at this locus. The FLO11 promoter is one of the largest promoters in the yeast genome (∼3.2 kb), and this may explain why so many TFs that regulate pseudohyphal growth are able to bind at this locus. Given the high number of regulatory mechanisms already known to function at this gene, further refinements by DNA looping are not surprising. For example, expression of the FLO11 transcript has been observed to demonstrate both bistability and hysteresis upon nitrogen starvation (Vinod et al., 2008), and the looping model may help explain these observations.
Because the theme of looping distal DNA elements to promoters is often observed in transcriptional regulation, it is possible the observed phenomenon could be extrapolated to other pathways and organisms. Yeast looping from promoter to terminator relating to memory has been documented at GAL10 and INO1, which seem to have evolved to provide more rapid responses to galactose- or inositol-starved growth conditions. The process of acquiring resistance in yeast to oxidative stress has also been observed to demonstrate cellular memory in an NPC-dependent manner (Guan et al., 2012), indicating the theme of memory could be involved in more cellular processes. Another interesting observation from this study was that DNA looping generated a strong Calling Card peak 4 kb away from the binding motif for that TF. This phenomenon may help explain the fact that many ChIP-sequencing (ChIP-seq) peaks contain no motif for that TF (Kulakovskiy et al., 2010; Yip et al., 2012). For example, across all of the TFs tested in the ENCODE project, the DNA-binding motif of the tested TF was observed in only 55% of the ChIP-seq peaks (ENCODE Project Consortium et al., 2012). ChIP-seq peaks without a binding motif could be explained by DNA-looping events that juxtapose distal enhancers with other regulatory regions. Given the importance and repetitive use of these mechanisms, high-throughput technologies such as Calling Cards should be useful in elucidating the regulatory architecture of many other systems.
MATERIALS AND METHODS
Calling card analysis
All Calling Card experiments used strains derived from the yRM1013 sir4Δ mat α strain from the Σ1278b background: can1ΔSTE2pr-SP_his5 lyp1ΔSTE3pr-LEU2 his::his3G leu2Δ ura3Δ Δsir4::HygMX. PCR products consisting of the Ty5-targeting domain of Sir4 (amino acids 951–1200) coding sequence and a NatMX (nourseothricin resistance) selectable marker flanked by regions of homology to the 3′ end of the targeted gene were transformed into yRM1013. Strains and primers are listed in Supplemental Table S5. Correct genomic integration was confirmed by PCR, followed by Sanger sequencing both ends.
Transposon induction was carried out as a variant of the original multiplexed Calling Card analysis (Wang et al., 2011). Each TF-Sir4–tagged strain was transformed with a barcoded variant of pRM1001 (which carries a galactose-inducible Ty5 transposon with URA3 as the auxotrophic marker). Cells were then pooled together and grown on agar plates containing SLAG medium (Gimeno et al., 1992). After induction of transposition and the growth of pseudohyphae into the agar, the yeastform cells were replica plated to yeast extract/peptone/dextrose (YPD) plates, and any remaining yeastform cells were washed away with deionized water (Guldal and Broach, 2006). The agar plates with embedded pseudohyphal cells were then adjoined to new YPD plates for 24 h. YPD plates with pseudohyphal cells were grown for an additional day to allow the cells to lose the Ty5 plasmid. Cells were then serially replica plated onto Glu –His, +5-fluoroorotic acid agar plates to select for genomic insertions of the transposon. Libraries for mapping insertion positions were constructed as described in Wang et al. (2011) and aligned to the genomic sequence and annotations for the Σ1278b strain (Dowell et al., 2010). Network figures were generated using Cytoscape (Smoot et al., 2011).
The sequence data from this study have been submitted to the NCBI Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo) under accession number GSE54831.
Flo8p binding-site reporter
The DNA sequence corresponding to bases −1 to −1217 from the transcription start site of CUP9 (YPL177C) was amplified by PCR from Σ1278b genomic DNA and cloned to drive expression of DsRed on a yeast centromere vector pRS314 to create plasmid pRM1145. The Flo8-binding site at −617 to −607 bases from the transcription start site was mutated from the consensus sequence CGGGGTTTTCT to ACACACACAGA to create plasmid pRM1146. Both plasmids were transformed into Σ1278b yeast and grown in synthetic complete–Trp and SLAG-Trp media and imaged on a Zeiss Axiovert200 fluorescent microscope (Carl Zeiss, Jena, Germany).
Reverse transcription-PCR analysis
Total RNA was isolated using TRIzol as described in Rio et al. (2010) from 10-ml yeast cultures grown for the specified times in either YPD or SLAG medium. Primers to amplify FLO11 transcript were ODM1178 and ODM1179 and normalized to UBC6 transcript and were amplified with ODM1120 and ODM1121. Primer sequences are listed in Supplemental Table S6.
3C
DNA looping was measured with 3C protocol optimized for short-range gene loops (Singh et al., 2009). Samples were normalized by control PCRs using a pair of convergent primers within the chromosome V centromere: ODM1191 and ODM1192. Primer sequences are listed in Supplemental Table S6.
Supplementary Material
Acknowledgments
We are grateful to Charlie Boone for generously providing strains and reagents. We thank Victoria Brown-Kennerly, Aimee Dudley, Mark Johnston, Maxim Schillebeeckx, and Francesco Vallania for helpful discussions and comments on the manuscript. Next-generation sequencing was performed at the Washington University Genomic Technology Access Center. This work was supported by National Institutes of Health Grants R21RR023960, 5R01DA025744-02, R01NS076993-02, and T32 HG000045 and a Keck Foundation Research Award.
Abbreviations used:
- 3C
chromatin conformation capture
- ChIP
chromatin immunoprecipitation
- NPC
nuclear pore complex
- PSWM
position-specific weight matrix
- RNAP II
RNA polymerase II
- SLAG
synthetic low-ammonium galactose
- TF
transcription factor
- YPD
yeast extract/peptone/dextrose
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-04-0871) on July 9, 2014.
REFERENCES
- Ahmed S, Brickner DG, Light WH, Cajigas I, McDonough M, Froyshteter AB, Volpe T, Brickner JH. DNA zip codes control an ancient mechanism for gene targeting to the nuclear periphery. Nat Cell Biol. 2010;12:111–118. doi: 10.1038/ncb2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baller JA, Gao J, Voytas DF. Access to DNA establishes a secondary target site bias for the yeast retrotransposon Ty5. Proc Natl Acad Sci USA. 2011;108:20351–20356. doi: 10.1073/pnas.1103665108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borneman AR, Leigh-Bell JA, Yu H, Bertone P, Gerstein M, Snyder M. Target hub proteins serve as master regulators of development in yeast. Genes Dev. 2006;20:435–448. doi: 10.1101/gad.1389306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickner DG, Cajigas I, Fondufe-Mittendorf Y, Ahmed S, Lee P, Widom J, Brickner JH. H2A.Z-mediated localization of genes at the nuclear periphery confers epigenetic memory of previous transcriptional state. PLoS Biol. 2007;5:e81. doi: 10.1371/journal.pbio.0050081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumgarner SL, Dowell RD, Grisafi P, Gifford DK, Fink GR. Toggle involving cis-interfering noncoding RNAs controls variegated gene expression in yeast. Proc Natl Acad Sci USA. 2009;106:18321–18326. doi: 10.1073/pnas.0909641106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain CW, Lohse MB, Homann OR, Sil A, Johnson AD. A conserved transcriptional regulator governs fungal morphology in widely diverged species. Genetics. 2012;190:511–521. doi: 10.1534/genetics.111.134080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- Dowell RD, Ryan O, Jansen A, Cheung D, Agarwala S, Danford T, Bernstein DA, Rolfe PA, Heisler LE, Chin B, et al. Genotype to phenotype: a complex problem. Science. 2010;328:469. doi: 10.1126/science.1189015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium. Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno CJ, Ljungdahl PO, Styles CA, Fink GR. Unipolar cell divisions in the yeast S. cerevisiae lead to filamentous growth: regulation by starvation and RAS. Cell. 1992;68:1077–1090. doi: 10.1016/0092-8674(92)90079-r. [DOI] [PubMed] [Google Scholar]
- Granek JA, Clarke ND. Explicit equilibrium modeling of transcription-factor binding and gene regulation. Genome Biol. 2005;6:R87. doi: 10.1186/gb-2005-6-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granek JA, Kayıkçı Ö, Magwene PM. Pleiotropic signaling pathways orchestrate yeast development. Cur Opin Microbiol. 2011;14:676–681. doi: 10.1016/j.mib.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Q, Haroon S, Bravo DG, Will JL, Gasch AP. Cellular memory of acquired stress resistance in saccharomyces cerevisiae. Genetics. 2012;192:495–505. doi: 10.1534/genetics.112.143016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldal CG, Broach J. Assay for adhesion and agar invasion in S. cerevisiae. J Vis Exp. 2006;1:e64. doi: 10.3791/64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Styles CA, Feng Q, Fink GR. A Saccharomyces gene family involved in invasive growth, cell-cell adhesion, and mating. Proc Natl Acad Sci USA. 2000;97:12158–12163. doi: 10.1073/pnas.220420397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honigberg SM. Cell signals, cell contacts, and the organization of yeast communities. Eukaryot Cell. 2011;10:466–473. doi: 10.1128/EC.00313-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulakovskiy IV, Boeva VA, Favorov AV, Makeev VJ. Deep and wide digging for binding motifs in ChIP-Seq data. Bioinformatics. 2010;26:2622–2623. doi: 10.1093/bioinformatics/btq488. [DOI] [PubMed] [Google Scholar]
- Laine JP, Singh BN, Krishnamurthy S, Hampsey M. A physiological role for gene loops in yeast. Genes Dev. 2009;23:2604–2609. doi: 10.1101/gad.1823609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts MG, Bauer FF, Marmur J, Pretorius IS. Muc1, a mucin-like protein that is regulated by Mss10, is critical for pseudohyphal differentiation in yeast. Proc Natl Acad Sci USA. 1996;93:8419–8424. doi: 10.1073/pnas.93.16.8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo HJ, Kohler JR, DiDomenico B, Loebenberg D, Cacciapuoti A, Fink GR. Nonfilamentous C. albicans mutants are avirulent. Cell. 1997;90:939–949. doi: 10.1016/s0092-8674(00)80358-x. [DOI] [PubMed] [Google Scholar]
- Rio DC, Ares M, Hannon GJ, Nilsen TW. Purification of RNA using TRIzol (TRI Reagent) Cold Spring Harb Protoc. 2010 doi: 10.1101/pdb.prot5439. doi:10.1101/pdb.prot5439. [DOI] [PubMed] [Google Scholar]
- Ryan O, Shapiro RS, Kurat CF, Mayhew D, Baryshnikova A, Chin B, Lin Z, Cox MJ, Vizeacoumar F, Cheung D, et al. Global gene deletion analysis exploring yeast filamentous growth. Science. 2012;337:1353–1356. doi: 10.1126/science.1224339. [DOI] [PubMed] [Google Scholar]
- Shapiro RS, Ryan O, Boone C, Cowen LE. Regulatory circuitry governing morphogenesis in Saccharomyces cerevisiae and Candida albicans. Cell Cycle. 2012;11:4294–4295. doi: 10.4161/cc.22608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shively CA, Eckwahl MJ, Dobry CJ, Mellacheruvu D, Nesvizhskii A, Kumar A. Genetic networks inducing invasive growth in Saccharomyces cerevisiae identified through systematic genome-wide overexpression. Genetics. 2013;193:1297–1310. doi: 10.1534/genetics.112.147876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh BN, Ansari A, Hampsey M. Detection of gene loops by 3C in yeast. Methods. 2009;48:361–367. doi: 10.1016/j.ymeth.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh BN, Hampsey M. A transcription-independent role for TFIIB in gene looping. Mol Cell. 2007;27:806–816. doi: 10.1016/j.molcel.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivak AT, Stormo GD. ScerTF: a comprehensive database of benchmarked positional weight matrices for Saccharomyces species. Nucleic Acids Res. 2012;40:D162–168. doi: 10.1093/nar/gkr1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstrepen KJ, Klis FM. Flocculation, adhesion and biofilm formation in yeasts. Mol Microbiol. 2006;60:5–15. doi: 10.1111/j.1365-2958.2006.05072.x. [DOI] [PubMed] [Google Scholar]
- Vinod PK, Sengupta N, Bhat PJ, Venkatesh KV. Integration of global signaling pathways, cAMP-PKA, MAPK, and TOR in the regulation of FLO11. PLoS One. 2008;3:e1663. doi: 10.1371/journal.pone.0001663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier MA, Lambrechts MG, Pretorius IS. Coregulation of starch degradation and dimorphism in the yeast Saccharomyces cerevisiae. Crit Rev Biochem Mol Biol. 1997;32:405–435. doi: 10.3109/10409239709082675. [DOI] [PubMed] [Google Scholar]
- Wang H, Mayhew D, Chen X, Johnston M, Mitra RD. Calling Cards enable multiplexed identification of the genomic targets of DNA-binding proteins. Genome Res. 2011;21:748–755. doi: 10.1101/gr.114850.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Mayhew D, Chen X, Johnston M, Mitra RD. “Calling cards” for DNA-binding proteins in mammalian cells. Genetics. 2012;190:941–949. doi: 10.1534/genetics.111.137315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip KY, Chen C, Bhardwaj N, Brown JB, Leng J, Kundaje A, Rozowsky J, Birney E, Bickel P, Snyder M, et al. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol. 2012;13:R48. doi: 10.1186/gb-2012-13-9-r48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Byers KJ, McCord RP, Shi Z, Berger MF, Newburger DE, Saulrieta K, Smith Z, Shah MV, Radhakrishnan M, et al. High-resolution DNA-binding specificity analysis of yeast transcription factors. Genome Res. 2009;19:556–566. doi: 10.1101/gr.090233.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.