

Abstract
Chronic mucocutaneous candidiasis is a rare disorder characterized by persistent and recurrent infections by Candida due to changes in cellular immunity and may be associated with autoimmune endocrine disorders. It is refractory to the usual antifungal treatments, which merely control it with imidazole derivatives. This reports the case of a 50-year-old female patient who referred vaginal discharge associated with vulvar ulcerated lesions and whitish plaques on oral and genital mucous membranes of onset in adolescence besides cutaneous horns in nipples. The clinical picture, family history, culture and anatomopathological studies were consistent with chronic infection by candida. Treatment with systemic antifungals obtained partial response of lesions characterizing a clinical picture of Chronic Mucocutaneous Candidiasis.
Keywords: Candidiasis; Candidiasis, chronic mucocutaneous; Autoimmune polyendocrinopathies
INTRODUCTION
Chronic Mucocutaneous Candidiasis (CMC) is a rare clinical syndrome characterized by the development of recurrent and progressive infections caused by yeast of the genus Candida, especially Candida albicans. The characteristic lesions are in whitish plaques, with crusts and ulcers typically found in Candida carriers, with the oral and pharyngeal, gastrointestinal tract and vagina mucosae being the most frequently affected; however, CMC patients progress to recurrence and chronicity of the illness. It starts during infancy for 60% to 80% of the patients, whereas late onset is rare.1,2 The immunodeficiency related to candida may be associated with endocrine and autoimmune disorders of hereditary origin. The diagnosis of the syndrome is based on the clinical aspect of the lesions demonstrably caused by candida, of chronic evolution. It is necessary to rule out other causes of immunodeficiency, usually refractory to antifungal treatments, and the infection is merely controlled with the use of imidazole derivatives, such as ketoconazole, fluconazole and itraconazole.3
CASE REPORT
A 50-year-old, black female patient referred pruriginous and painful ulcerated vulvar lesions, with onset in adolescence associated with vaginal discharge. While breastfeeding her second child hardened projections of progressive growth appeared in her nipples.
Her family history revealed that two of the patient's children had a similar clinical picture, with whitish plaques on the tongue and esophageal mucosa.
At the physical examination she presented adherent whitish plaques on the tongue and vulva, with hyperemia and ulceration (Figures 1 and 2). She also presented 3-centimeter long cornified structures in the nipples, besides hyperkeratotic and verrucous lesions on the nose and fingers (Figure 3).
FIGURE 1.
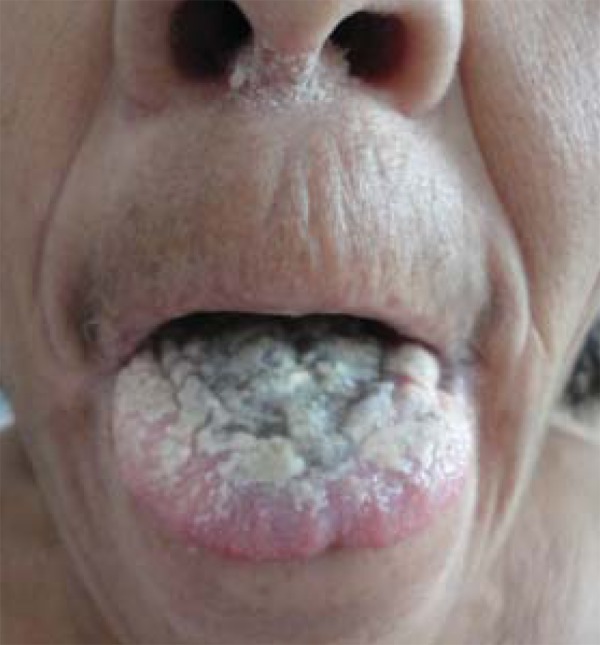
Whitish plaques on tongue
FIGURE 2.
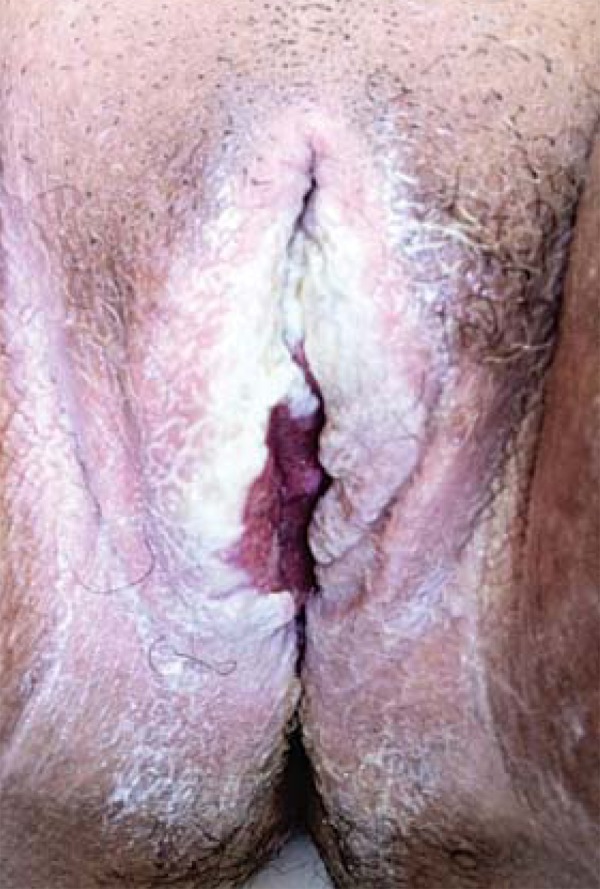
Whitish plaques with hyperemia and ulceration on vulva
FIGURE 3.
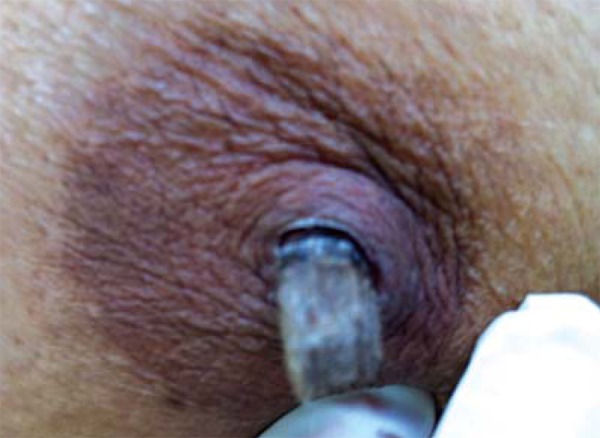
Cornified structures in nipples
Surgical excision of the cutaneous nipple horns and biopsy of the vulva were performed. The histopathological study evidenced pseudoepitheliomatous hyperplasia with intense hyperkeratosis. PAS staining revealed pseudohyphae and spores compatible with candida (Figures 4 e 5). Proofs of immunity like HIV and hepatitis B serology, dosages of immunoglobulins, leukocyte count and differential count showed normal parameters excluding immunodeficiencies. Proof of candidin was not reactive after 48 hours. Upper Digestive Endoscopy evidenced esophageal mucosa covered by whitish plaques; the anatomopathological examination was compatible with candida (Figure 6). Laboratory exams did not demonstrate any endocrine dysfunctions. The treatment was carried out with venous fluconazole 200 mg/day and after 22 days there was improvement of pain with partial regression of lesions.
FIGURE 4.
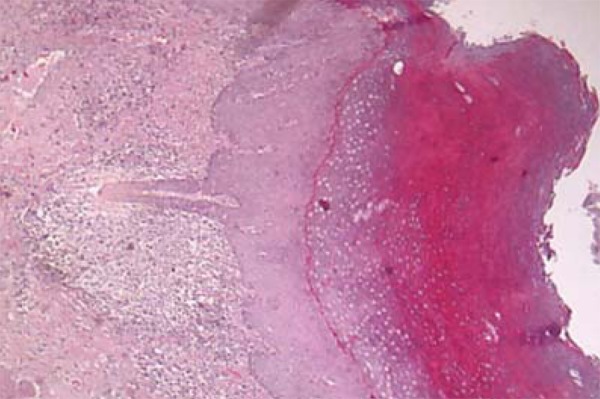
Histopathological examination of vulva evidencing pseudoepitheliomatous hyperplasia with intense hyperkeratosis
FIGURE 5.
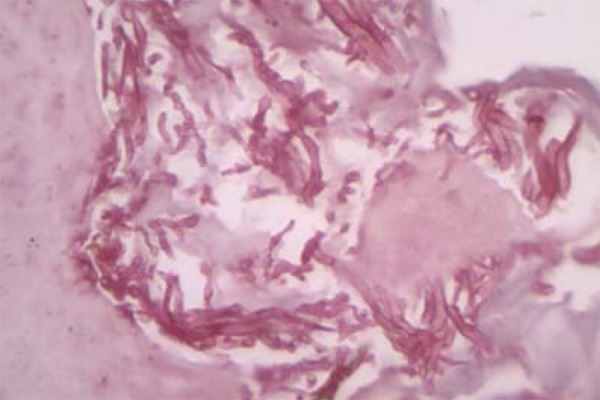
PAS staining revealed pseudohyphae and spores compatible with candida
FIGURE 6.
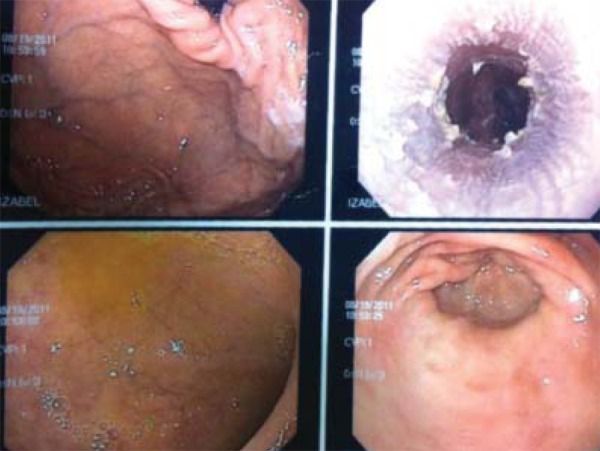
Esophagic mucosa covered by whitish plaques
DISCUSSION
Chronic Mucocutaneous Candidiasis is a rare clinical syndrome characterized by recurrent and progressive infections of the skin, nails and mucosae caused by candida, with onset during infancy in most cases.1 The affected individuals present defects in the acquired cellular immunity, which make an effective response against these microorganisms difficult. The disease may be associated with endocrine and autoimmune disorders of hereditary origin.4 Infections by candida in patients affected by CMC are usually superficial, and systemic candidiasis is rarely observed.2
Patients with CMC present alterations in acquired cellular immunity, specifically by T-cells deficiency, preventing an effective response against Candida. There may be a deficit of cytokines of Th1 response.5 In some patients the deficit is specific for Candida, in others, the response for other organisms is also defective.6 Several patients with CMC present cutaneous allergy by antigens for Candida (Candidin test), even though the innate cellular immunity (especially the quality and quantity of neutrophils) is usually preserved, justifying the low risk of septicemia by Candida in these patients.7,3 The candidin test was considered weak in the clinical case, showing specific immunodeficiency for candida.
CMC affected patients frequently present autoimmune endocrine disorders. An example is the Autoimmune PolyEndocrinopathy Candidiasis Ectodermal Dystrophy syndrome (APECED), also known as autoimmune polyendocrine syndrome type I, a genetic autoimmune disease, autosomal and recessive, with onset in infancy, and that combines chronic mucocutaneous candidiasis and several autoimmune endocrinopathies, more commonly hypoparathyroidism and adrenal insufficiency.8 They may also be associated with other autoimmune diseases, such as diabetes mellitus and hypothyroidism.1
APECED syndrome arises from a mutation of the AIRE gene (autoimmune regulator). In mice, this mutation was shown to avoid deletion of specific T cells in the thymus organ, promoting the development of autoimmune diseases.9
Patient evaluation through laboratory exams did not demonstrate endocrine dysfunctions; glycemia, thyroid, parathyroid and adrenal hormones were within physiological limits. The patients must be evaluated at the moment of diagnosis and later for development of endocrinopathies, as CMC usually precedes endocrine manifestations by several years.8
Clinical manifestations and severity of CMC can be varied. The disease is more frequently manifested with recurrent oral moniliasis with erythematous and scaly cutaneous plaques in intertriginous areas, mucosae and nails. Lesions may evolve until the onset of generalized, hyperkeratotic granulomatous crusty plaques. Nails are thick, dystrophic, loose and brittle, with associated paronychia.1 The patient presented adherent whitish plaques on the tongue and vulva, the latter with hyperemia and ulceration and presence of horns in nipples (Figures 1, 2 and 3). In CMC, besides oral candidiasis other mucosae can be involved, like esophageal, genital and laryngeal mucosae, and may cause stenosis.1 Upper Digestive Endoscopy of patient revealed esophageal mucosa covered by whitish plaques.
The diagnosis consists of observation of the clinical picture of persistent and recurrent lesions compatible with candida of onset in infancy and research of familial history.4 Detection of microorganisms can be done through direct mycological test, culture or histopathological.10 In the clinical case, to rule out lesion malignancies, it was opted for performing biopsies and the anatomopathological examination identified pseudohyphae with pseudoepitheliomatous hyperplasia.
The hypothesis of chronic mucocutaneous candidiasis was raised based on the chronic evolution of the disease with onset in the adolescence, characterized by lesions caused by candida with histopathological, direct mycological and culture proof, familial history and absence of other immunodeficiencies.
The treatment consists of therapy for the infectious disease, endocrinopathies and specific treatment for the immunological deficiency.8 The antifungal therapies used were long-term fluconazole and ketoconazole and as expected, there was partial improvement.
Footnotes
Conflict of interest: None
Financial funding: None
Work performed at Santa Casa de Misericórdia de Vitória - Vitória (ES), Brazil.
How to cite this article: Chambô Filho A, Souza Filho JB, Pignaton CC, Zon I, Fernandes AS, Cardoso LQ. Chronic mucocutaneous candidiasis: a case with cutaneous horns lush in nipples. An Bras Dermatol. 2014;89(4):641-4
REFERENCES
- 1.Bolognia JL, Jorizzo JL, Rapini RP, Callen JP, Horn TD, Mancini AJ, et al. Dermatologia. 2. ed. Rio de Janeiro: Elsevier; 2011. pp. 804–806. [Google Scholar]
- 2.Julián-González RE, Valdebrán-Canales MA, Guidos-Morales HE. Candidiasis mucocutánea crónica. Informe de un caso. Arch Argent Pediatr. 2010;108:e37–e40. doi: 10.1590/S0325-00752010000200015. [DOI] [PubMed] [Google Scholar]
- 3.Collins JR, Sickels JEV. Chronic mucocutaneous candidiasis. J Oral Maxillofac Surg. 1983;41:814–818. doi: 10.1016/s0278-2391(83)80049-4. [DOI] [PubMed] [Google Scholar]
- 4.Fazlollahi MR, Farhoudi A, Movahedi M, Gharagozlou M, Mohammadi K, Rezaei N. Chronic Mucocutaneous Candidiasis; Report of Three Cases with Different Phenotypes. Iran J Allergy Asthma Immunol. 2005;4:39–42. [PubMed] [Google Scholar]
- 5.Kirkpatrick CH. Chronic Mucocutaneous Candidiasis. J Am Acad Dermatol. 1994;31:S14–S17. doi: 10.1016/s0190-9622(08)81260-1. [DOI] [PubMed] [Google Scholar]
- 6.Chilgren RA, Quie PG, Meuwissen HJ, Hong R. Chronic mucocutaneous candidiasis, deficiency of delayed hypersensitivity, and selective local antibody defect. Lancet. 1967;2:688–693. doi: 10.1016/s0140-6736(67)90974-9. [DOI] [PubMed] [Google Scholar]
- 7.Grouhi M, Dalal I, Nisbet-Brown E, Roifman CM. Cerebral vasculitis associated with chronic mucocutaneous candidiasis. J Pediatr. 1998;133:571–574. doi: 10.1016/s0022-3476(98)70072-1. [DOI] [PubMed] [Google Scholar]
- 8.Orpha.net . European Union: Poliendocrinopatia autoimune de tipo 1. 2010. [updated 2010 May 28]. [cited 2012 Jul 9]. [Internet] Available from: https://www.orpha.net/data/patho/Pro/pt/Urgencia_PoliendocrinopatiaAutoimmuneTipo1.pdf. [Google Scholar]
- 9.Ng WF, Tudhope SJ, von Delwig A, Lilic D. Invariant natural killer T (iNKT) cell deficiency in chronic mucocutaneous candidiasis - A consequence or a cause? Immunol Lett. 2011;135:180–183. doi: 10.1016/j.imlet.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 10.Icb.ufmg.br. Instituto de Ciências Biológicas. Universidade Federal de Minas Gerais . Belo Horizonte: Agentes Etiológicos das Leveduroses. 2006. [acesso 9 jul 2012]. [Internet] Disponível em: http://www.icb.ufmg.br/mic/material/agentesetiologicosdasleveduroses.pdf. [Google Scholar]