

Abstract
Hyperactivation of the amygdala following chronic stress is believed to be one of the primary mechanisms underlying the increased propensity for anxiety-like behaviors and pathological states; however, the mechanisms by which chronic stress modulates amygdalar function are not well characterized. The aim of the current study was to determine the extent to which the endocannabinoid system, which is known to regulate emotional behavior and neuroplasticity, contributes to changes in amygdalar structure and function following chronic stress. To examine the hypothesis, we have exposed C57/Bl6 mice to chronic restraint stress which results in an increase in fatty acid amide hydrolase (FAAH) activity and a reduction in the concentration of the endocannabinoid N-arachidonylethanolamine (AEA) within the amygdala. Chronic restraint stress also increased dendritic arborization, complexity and spine density of pyramidal neurons in the basolateral nucleus of the amygdala (BLA) and increased anxiety-like behavior in wild-type mice. All of the stress-induced changes in amygdalar structure and function were absent in mice deficient in FAAH. Further, the anti-anxiety effect of FAAH deletion was recapitulated in rats treated orally with a novel pharmacological inhibitor of FAAH, JNJ5003 (50 mg/kg/day), during exposure to chronic stress. These studies suggest that FAAH is required for chronic stress to induce hyperactivity and structural remodeling of the amygdala. Collectively, these studies indicate that FAAH-mediated decreases in AEA occur following chronic stress and that this loss of AEA signaling is functionally relevant to the effects of chronic stress. These data support the hypothesis that inhibition of FAAH has therapeutic potential in the treatment of anxiety disorders, possibly by maintaining normal amygdalar function in the face of chronic stress.
Introduction
The amygdala is a central node within the corticolimbic circuitry involved in the processing of external sensory information from the environment and establishing emotional valence to these stimuli (1, 2). The basolateral nucleus of the amygdala (BLA) acts as the primary interface for this processing, being the recipient of inputs from both thalamocortical sensory centers as well as from higher order centers, such as the medial prefrontal cortex (1, 3, 4). If external stimuli are interpreted as negative or aversive, the BLA activates efferent pathways to neighbouring amygdalar nuclei (central and medial nuclei of the amygdala) and local limbic circuits (primarily the bed nucleus of the stria terminalis) to initiate a stress response. The stress response includes a cluster of behavioral, physiological and endocrine responses classically typified as heightened anxiety and vigilance as well as increased sympathetic tone and glucocorticoid secretion (1, 3, 5, 6). Therefore, the sensitivity of the pyramidal neurons within the BLA (which are the primary output neurons of this structure) to neuronal activation is an important determinant of the emotional state of an organism (5, 7, 8).
Pharmacological studies have revealed that suppression of excitation within the BLA reduces anxiety and is a primary mechanism of action of classical anxiolytics, such as benzodiazepines (9–12). In accord with these data, facilitation of excitation within the BLA can produce a state of hypervigilance and anxiety, even in the absence of an impending threat (13–15). In addition, imaging studies of clinical populations have demonstrated that hyperactivity of the amygdala is a hallmark of an array of mood and anxiety disorders, and normalization of activity is a marker of disease remission (1, 16, 17).
Chronic exposure to stress contributes to the development of mood and anxiety disorders (18). Preclinical studies have demonstrated that chronic stress can alter amygdalar function; in particular, chronic stress increases the intrinsic excitability, sensitivity to afferent stimulation and activation of pyramidal neurons within the BLA (8, 19–21). The mechanism of this sensitization is likely mediated by structural remodeling of these neurons as chronic stress exposure increases dendritic arborization, induces the formation of new spines and facilitates the formation of excitatory inputs along pyramidal neurons in the BLA, which together will increase amygdalar sensitivity to sensory inputs (22–26). The mechanisms underlying the effects of stress to change synaptic morphology within the BLA are not well characterized, but are thought to be triggered by the repeated activation of excitatory inputs into the BLA which, in a feed forward process, shifts the balance of excitation and inhibition within the BLA to increased excitation (22, 26). This sensitization of the BLA to excitation is believed to be a primary mechanism underlying the shift from normal vigilance to a state of hypervigilance and anxiety following chronic stress (5, 7, 27). Specifically, any genetic, experiential or hormonal manipulation which increases anxiety-like behaviors correspondingly increases dendritic branching or spine density within pyramidal neurons of the BLA (24, 26, 28–30), suggesting that strengthening the structural basis of synaptic connectivity in the BLA contributes to the development of an anxious state.
The endocannabinoid (eCB) system is a highly plastic, neuromodulatory system in which post-synaptically derived ligands (N-arachidonylethanolamine [anandamide; AEA] and 2-arachidonoylglycerol [2-AG]) suppress presynaptic transmitter release through the activation of cannabinoid CB1 receptors, which are localized to axon terminals of both glutamatergic and GABAergic fibers (31). The eCB system within the BLA (32), particularly AEA and the activity of its primary metabolic enzyme fatty acid amide hydrolase (FAAH), is known to be modulated by stress (33–37) and has been found to contribute to several forms of synaptic plasticity subserving the regulation of emotional behavior (38–44). However, the role of the FAAH activity in regulating changes in amygdalar structure and function following chronic stress has yet to be characterized. Accordingly, the current study was designed to determine whether modulation of amygdalar FAAH activity following chronic stress contributes to the structural remodeling of pyramidal neurons in the BLA and the subsequent shifts in anxiety-like behavior.
Methods
Subjects
For studies examining the effects of chronic stress on the endocannabinoid system, male C57/Bl6 mice (obtained from Charles River, Wilmington, MA), 3 months of age and housed in cages of 5 mice per cage, were used. To examine the effects the role of FAAH in chronic stress, wild-type and FAAH deficient (FAAH−/−) mice, 3–5 months of age and housed in cages of 3–5 mice per cage, were used. These mice were bred on a C57/Bl6 background (more than 8 backcrosses) and were descendants of a previously characterized line (45). FAAH−/− mice and wild type controls were obtained from heterozygous breeding pairs. Genotypes were determined by PCR using DNA isolated from ear tissue obtained at weaning. For all studies, mice were maintained on a 12 hour light:dark cycle with lights on at 0900h. Food and water were available ad libitum. For the pharmacological arm of this study, 70 day male Sprague-Dawley rats were employed. Rats were singly housed for the duration of the study and all environmental conditions were identical to those of the mice. All procedures carried out with mice were approved by the Institutional Animal Use and Care Committees of the Rockefeller University and the Medical College of Wisconsin.
Biochemical Studies
For biochemical analysis of the effects of chronic stress on endocannabinoid biochemistry, wild-type C57/Bl6 mice were divided into two conditions, stress and non-stress. Mice in the stress condition were subjected to 6 h of restraint stress per day for 21 days. Restraint stress took place each day between 1000h–1600h. Mice were placed into wire mesh restrainers (5″ × 4.5″) which were sealed with binder clips, and the restrainers were placed in the home cage for the duration of the stressor. Non-stressed mice were left undisturbed for the duration of the experiment, except for cage changing, which occurred twice weekly. For tissue collection, mice were killed 24 h following the final stressor (or a comparable point in the circadian cycle for non-stressed mice) and the amygdalae were dissected and frozen on dry ice within 5 min of decapitation. The amygdala was dissected bilaterally along the barriers of the external capsule, thus it was primarily composed of the lateral and basolateral amygdala. Two cohorts of animals were generated for biochemical analysis. One cohort (n = 9–10) was used to determine brain regional eCB contents. Another cohort (n = 4) was used to generate membrane fractions, which were used for CB1 receptor radioligand binding and FAAH activity.
Membrane Preparation
Membranes were collected from isolated brain regions by homogenization of frozen tissue in 10 volumes of TME buffer (50 mM Tris HCl, pH 7.4; 1 mM EDTA and 3 mM MgCl2). Homogenates were centrifuged at 18,000 x g for 20 min and the resulting pellet, which contains a crude membrane fraction, was resuspended in 10 volumes of TME buffer. Protein concentrations were determined by the Bradford method (Bio-Rad, Hercules, CA, USA).
CB1 Receptor Radioligand Binding Assay
CB1 receptor agonist binding parameters were determined using radioligand binding using a Multiscreen Filtration System with Durapore 1.2-μM filters (Millipore, Bedford, MA). Incubations (total volume = 0.2 mL) were carried out using TME buffer containing 1 mg/mL bovine serum albumin (TME/BSA). Membranes (10 μg protein per incubate) were added to the wells containing 0.1, 0.25, 0.5, 1.0, 1.5 or 2.5 nM [3H]CP 55,940, a cannabinoid CB1 receptor agonist. Ten μM Δ9-tetrahydrocannabinol was used to determine non-specific binding. KD and Bmax values were determined by nonlinear curve fitting of specific binding data to the single site binding equation using GraphPad Prism (San Diego, CA, USA).
Fatty Acid Amide Hydrolase (FAAH) Activity Assay
FAAH activity was measured as the conversion of AEA labeled with [3H] in the ethanolamine portion of the molecule ([3H]AEA; (46)) to [3H]ethanolamine as reported previously (47). Membranes were incubated in a final volume of 0.5 ml of TME buffer (50 mM Tris-HCl, 3.0 mM MgCl2, and 1.0 mM EDTA, pH 7.4) containing 1.0 mg/ml fatty acid-free bovine serum albumin and 0.2 nM [3H]AEA. Isotherms were constructed using eight concentrations of AEA between 10 nM and 10 μM. Incubations were carried out at 37°C and were stopped with the addition of 2 ml of chloroform/methanol (1:2). After standing at ambient temperature for 30 min, 0.67 ml of chloroform and 0.6 ml of water were added. Aqueous and organic phases were separated by centrifugation at 1,000 rpm for 10 min. The amount of [3H] in 1 ml of the aqueous phase was determined by liquid scintillation counting and the conversion of [3H]AEA to [3H]ethanolamine was calculated. The binding affinity of AEA for FAAH (Km) and maximal hydrolytic activity of FAAH (Vmax) values for this conversion were determined by fitting the data to the Michaelis-Menten equation using GraphPad Prism.
Endocannabinoid Extraction and Analysis
Brain regions were subjected to a lipid extraction process as described previously (35). Tissue samples were weighed and placed into borosilicate glass culture tubes containing two ml of acetonitrile with 84 pmol of [2H8]anandamide and 186 pmol of [2H8]2-AG. Tissue was homogenized with a glass rod and sonicated for 30 min. Samples were incubated overnight at −20°C to precipitate proteins, then centrifuged at 1,500 x g to remove particulates. The supernatants were removed to a new glass tube and evaporated to dryness under N2 gas. The samples were resuspended in 300 μl of methanol to recapture any lipids adhering to the glass tube, and dried again under N2 gas. Final lipid extracts were suspended in 20 μl of methanol, and stored at −80°C until analysis. The contents of the two primary endocannabinoids AEA and 2-AG within lipid extracts in methanol from brain tissue were determined using isotope-dilution, liquid chromatography–mass spectrometry as described previously (48).
Chronic Stress in FAAH−/− mice
To examine the effects of chronic stress in FAAH deficient mice, both wild type (n = 10) and FAAH−/− mice (n = 10) were divided into two conditions, non-stress or stress. The chronic stress procedure was identical to that described above.
Behavioral testing
Twenty four hours following the final stressor, all subjects were examined for anxiety-like behavior using the elevated plus maze. Testing occurred within the colony room, so no habituation to the testing room was required. The elevated plus maze (San Diego Instruments, San Diego, CA) was constructed from beige plastic and consisted of two open arms (30 × 5 cm) and two enclosed arms (30 × 5 × 15 cm) that extended from a central platform (5 × 5 cm). The maze was elevated 40 cm above the floor. All testing occurred under dim light conditions. Each animal was placed in the central quadrant of the elevated plus maze facing an open arm and entries into open and closed arms, and time spent in the open arm were scored over a 5 min trial by a blind observer. Entries into the open arm were calculated as a full body entry to the open arm, such that the entire body up to the base of the tail was on the open arm. At the conclusion of each test, the elevated plus maze was wiped clean with a 10% ethanol solution to remove urine and scent.
A second cohort of FAAH−/− mice was exposed to chronic stress after which they were tested in an open field arena for motor behavior. The open field arena (38.1 × 43.2 cm) was constructed from opaque PVC lined with a beige interior. All testing occurred under dim light conditions. Each animal was placed in the central quadrant of the open field chamber and tracked for 5 min. Tracking and scoring were performed using Ethovision Software (Ethovision v8, Noldus). One hour after the OFT, these mice were euthanized, brains were removed and amygdala dissected, as described above. Amygdalar AEA contents were determined, to establish the role of FAAH in the effects of stress on amygdalar AEA contents, using the extraction and quantification methods described above.
Morphological Analysis
Immediately following testing in the elevated plus maze, mice were rapidly decapitated. Brains were quickly removed, washed in distilled water and were processed for staining of individual neurons following the manufacturer’s instructions for the Rapid Golgi Kit (FD Neurotech, Ellicot City, MD). Golgi-stained tissue containing the BLA was sliced (120 μm), mounted with coverslip and used for morphological analysis of dendritic arborization and spine-density. Neurons were reconstructed in 3 dimensions using a Nikon Eclipse microscope (40x) and the Neurolucida software package (MicroBrightField, Williston, VT).
For morphological analysis of the dendritic tree, 5–6 neurons were reconstructed per animal by a trained experimenter blind to the conditions. Pyramidal neurons within the BLA were examined for dendritic architecture as this class of neurons has been repeatedly characterized as those which exhibit remodeling in response to chronic stress (22–24, 49, 50). Pyramidal neurons within this region were identified based on previously established criteria (22, 50, 51), which requires the presence of a single, clearly defined apical dendrite and at least two intact basilar dendrites extending from the base of the cell body. Further, as previous studies have identified that it is the spiny pyramidal neurons in the BLA which exhibit architectural changes in response to stress (22–24, 49, 50), our analysis was restricted to the spiny pyramidal-like neurons. Neurons within the BLA used for analysis were chosen from slices which were between −1.0–2.0 mm posterior to bregma, and the boundaries of the BLA were clearly defined within Golgi impregnated material by the fiber tracts from the external capsule which defined the dorsal, medial and lateral boundaries of the BLA. For analysis, cells were chosen which met criteria described in earlier reports: 1) cell bodies within the mid section of the tissue to minimize dendritic truncation; 2) relative isolation of the cell body from neighboring impregnated neurons; 3) cell bodies exist within the defined boundaries of the BLA; and 4) the presence of an intact primary, secondary and tertiary dendrites (as evidenced by well defined endings) at least 50 μm from the cell body. Based on the analysis performed in previous studies, we analyzed all dendritic material in both the apical and basilar trees collapsed together as one variable. As such, neurons required the presence of at least two, and no more than five, basilar dendritic trees to be quantified and each animal was balanced so that an equal number of basilar dendritic trees was represented across the six neurons quantified for each animal. For analysis, the total dendritic length and total number of branch points along the dendritic tree were quantified in a total of 28–30 neurons per experimental condition.
Analysis of Dendritic Spine Density
Using the NeuroLucida system (100×, 1.3 numerical aperture, Olympus BX61), all protrusions, irrespective of their morphological characteristics, were counted as spines if they were in direct continuity with the dendritic shaft. For the purpose of this study, an apical dendrite originating directly from the cell soma were classified as the primary apical dendritic shaft, and those originating from this dendrite were classified as secondary dendrites. Further, secondary dendrites originating within 30–40 μm of the cell soma were chosen and only one such dendrite per neuron was used for spine-density analysis. As described earlier (24, 26, 30), starting from the origin of the branch, and continuing away from the cell soma, spines were counted along an 100 μm stretch of the dendrite. This total length of 100 μm was further subjected to a detailed segmental analysis, which consisted of counting the number of spines in successive steps of 20 μm each, for a total of 5 steps. The values for number of spines from each 20 μm segment, at a given distance from the origin of the branch, were then averaged across all neurons in a particular experimental group. For spine density analysis, spines along 30 dendritic branches per experimental condition were quantified. Our analysis, like all those involving Golgi staining, is likely to lead to a systematic underestimation of spine density because it is not possible to visualize spines pointing directly toward the surface or extending beneath the dendrite (52–54). No correction was made for these hidden spines because of previously reported validation of the use of visible spine counts for comparison between different experimental conditions (55).
Pharmacological Inhibition of FAAH and Chronic Stress-induced Anxiety
We examined the effects of pharmacological inhibition of FAAH in adult rats subjected to chronic stress on anxiety-like behavior. Like mice, rats exposed to chronic stress (ranging from nine to twenty one days) have been found to exhibit reductions in AEA content within the amygdala (36, 56). Rats were employed for this arm of the study because we were using a non-invasive means of drug administration through incorporation into the chow that we have previously validated in rats (Hill et al., in preparation). This approach was taken because administration of the FAAH inhibitor via daily injections constitutes significant stress and can mask the effects of chronic stress on amygdala structure and function (29).
Similar to the experimental design for the FAAH−/− mice, rats were divided into two groups – one exposed to chronic stress (6 h/day of restraint stress for 21 days; n = 16) and the other as cage controls (n = 16). Each of these experimental groups was divided into those who had normal chow and those who had 50 mg/kg of the FAAH inhibitor JNJ-40355003 (JNJ-5003; see supplementary information for synthesis and characterization of compound) incorporated into their chow. The dose of JNJ-5003 was based on previous studies in our lab demonstrating that this dose produced a sustained and complete inhibition of FAAH within the limbic forebrain and did not modulate daily eating patterns or body weight gain (Hill et al., in preparation).
Restraint stress was administered each day between 1000h–1600h. Rats were placed into wire mesh restrainers (8″ × 6.5″) that were sealed with binder clips, and the restrainers were placed in the home cage for the duration of the stressor. Non-stressed rats were left undisturbed for the duration of the experiment, except for cage changing, which occurred twice weekly. Behavioral testing in the elevated plus maze done 24 h following the final stressor. All behavioral testing occurred between 8:30–12:00. On the testing day, rats were habituated to the testing room for 30 minutes. Rats were placed in the center of the elevated plus maze and allowed to explore freely for five minutes. Movement, including distance traveled, time spent immobile, and time and distance in the open arms, closed arms and central quadrant was recorded and analyzed by Ethovision XT tracking software (Noldus Information Technology, Leesburg, VA). At the conclusion of each test, the elevated plus maze was wiped clean with a 10% ethanol solution to remove urine and scent.
Statistics
Biochemical analysis of the effects of chronic stress on various parameters of the eCB system was analyzed using an independent t-test. Anxiety-like behavior, dendritic length and branch points as well as dendritic spine counts were all analyzed using a two way analysis of variance (ANOVA), with stress and genotype as fixed factors. Bonferroni post hoc tests were used to determine specific differences between experimental groups. Data generated from segmental analyses of dendritic branches were analyzed using a repeated measure ANOVA, with distance from soma acting as a within subject factor and stress and genotype acting as between subject factors. Corrected Bonferroni post hoc tests were used to determine specific differences between experimental groups at different segments from the soma. Significance for all tests was established at a p < 0.05. All data presented in the figures is listed as mean values +/− standard error.
Results
Chronic Stress Increases FAAH Activity and Reduces NAE Contents in the Amygdala
In the first set of studies, we examined the effects of chronic stress on FAAH activity and N-acylethanolamine (NAE) contents in the amygdala. Previous studies have demonstrated a complete loss of AEA hydrolysis by membranes isolated from FAAH null mice (48). Therefore, AEA hydrolysis is used as an assay of FAAH activity. Chronic stress was found to result in a significant increase in the Vmax for FAAH to hydrolyze AEA in membranes isolated from the amygdala of stressed mice compared to nonstressed controls [t (6) = 2.73, p < 0.04; Fig. 1]. Stress exposure had no significant effect on the Km of AEA for FAAH [t (6) = 0.52, p > 0.05; Fig. 1]. Consistent with the increase in maximal FAAH activity within the stress-exposed amygdala, there was a significant reduction in the tissue content of AEA within the amygdala following chronic stress [t (17) = 2.96, p < 0.01; Fig. 1], as well as a reduction in the tissue content of both palmitoylethanolamide (PEA)[t (17) = 2.33, p < 0.04; control: 142.0 +/− 7.1 vs. chronic stress 117.2 +/− 8.1 pmol/g tissue] and oleoylethanolamide (OEA)[t (17) = 2.52, p < 0.03; control 88.8 +/− 5.9 vs. chronic stress 67.8 +/− 5.9 pmol/g tissue], NAEs that are metabolized by FAAH but do not activate CB1 receptors. There was no effect of chronic stress on the tissue content of the other endocannabinoid, 2-AG [t (17) = 0.79, p > 0.05; Fig. 1]. There was no difference in either the binding site density [t (6) = 0.25, p > 0.05; Fig. 1] or the binding affinity of the CB1 receptor agonist CP55,940 for the cannabinoid CB1 receptor within the amygdala between the control and chronically stressed groups [t (6) = 0.47, p > 0.05; Fig. 1].
Figure 1. Characterization of the amygdalar endocannabinoid system following chronic stress.

Exposure of C57/Bl6 mice to chronic stress (21 days of 6 h/day restraint stress) increased the maximal velocity (Vmax) of AEA hydrolysis by fatty acid amide hydrolase (FAAH) within the amygdala (Panel A), although there was no difference in the binding affinity of AEA for FAAH (Km; Panel B). Consistent with this increase in FAAH activity, chronic stress resulted in a reduction in the tissue levels of the endocannabinoid anandamide (AEA) within the amygdala (Panel C); there was, however, no effect of chronic stress on amygdalar 2-arachidonoylglycerol (2-AG; Panel D). There was no effect of chronic stress on either the maximal binding site density (Bmax; Panel E) or the binding affinity (Kd; Panel F) of the cannabinoid CB1 receptor within the amygdala. Data are presented as means +/− SEM. * denotes significant differences (p < .05) between control and chronically stressed mice.
FAAH−/− mice do not exhibit changes in anxiety-like behavior or structural remodeling in the amygdala following chronic stress
To determine if the increase in FAAH activity and reduction in NAE content within the amygdala contribute to structural and functional changes which occur in response to chronic stress, we utilized mice in which the gene for FAAH has been eliminated (FAAH−/−; (45)). Our rationale for this study is that if a change in FAAH activity is required for an effect of chronic stress, then chronic stress will not produce that particular effect in FAAH−/− mice.
Our hypothesis predicts that chronic stress will not affect AEA concentrations in the amygdala in the absence of FAAH. We tested this prediction in groups of wild type and FAAH−/− mice that were unstressed or exposed to chronic stress. As expected, there was a highly significant main effect of genotype [F (1, 16) = 156.4, p < 0.001; Table 1] because the FAAH−/− mice had significantly higher AEA concentrations than wild types. There was no main effect of stress on AEA content [F (1, 16) = 0.10, p > 0.05; Table 1] and no significant interaction between genotype and stress in the regulation of AEA content in the amygdala [F (1, 16) = 0.43, p > 0.05; Table 1]. We were concerned that the effect of genotype on AEA concentration was so large that it might mask a relatively small effect of stress. Therefore, we calculated the ratio of AEA concentrations in stressed to unstressed mice in each genotype then compared the ratios using an unpaired t-test. This analysis revealed a significant difference in the ratio of AEA content within the amygdala in wild type mice vs. FAAH−/− mice in response to chronic stress [t (9) = 3.49, p < 0.01]. In particular, the stressed wild type mice exhibited a 38.1 (+/− 6.8) % decrease in AEA content in the amygdala in response to chronic stress, while FAAH−/− mice exhibited a 2.0 (+/− 8.7) % increase in AEA content in the amygdala in response to chronic stress. With respect to amygdala 2-AG content, the interaction between stress and genotype was not significant [F (1, 16) = 0.25, p > 0.05; Table 1], and the main effects of both stress [F (1, 16) = 1.46, p > 0.05] and genotype [F (1, 16) = 0.01, p > 0.05] were also not significant.
Table 1. The Effects of Chronic Stress on Endocannabinoid Content in the Amygdala of Wild-type Mice and those Deficient in Fatty Acid Amide Hydrolase.
Effects of three weeks exposure of restraint stress to wild-type C57/Bl6 mice, or those lacking fatty acid amide hydrolase (FAAH−/−), on the tissue content of anandamide (AEA) and 2-arachidonoylglycerol (2-AG) within the amygdala.
| Control | Chronic Stress | |
|---|---|---|
| AEA Content (pmol/g tissue) | ||
| Wild type | 18.64 +/− 5.33 | 10.89 +/− 1.02 |
| FAAH−/− | 101.8 +/− 11.03* | 104.2 +/− 7.73* |
| 2-AG Content (nmol/g tissue) | ||
| Wild type | 5.66 +/− 0.58 | 4.58 +/− 0.73 |
| FAAH−/− | 5.27 +/− 0.70 | 4.82 +/− 0.71 |
Data are presented as mean values +/− SEM (n = 5–6 / condition).
denotes significant differences between wild type and FAAH−/− mice (p < 0.001).
There was a significant interaction between stress exposure and genotype with respect to total dendritic length of pyramidal neurons within the BLA [F (1, 102) = 8.53, p < 0.005; Fig 2]. Post hoc analysis revealed that in wild-type mice, chronic stress resulted in a significant increase in dendritic length of BLA pyramidal neurons (p < 0.05); there was, no difference in dendritic length of BLA neurons between non-stressed and chronically stressed FAAH−/− mice (p > 0.05). There was also no difference in dendritic length between non-stressed wild-type or FAAH−/− mice (p > 0.05), but chronically stressed FAAH−/− mice had significantly less dendritic material than chronically stressed wild-type mice (p < 0.05).
Figure 2. Deficiency in fatty acid amide hydrolase prevents the ability of chronic stress to promote dendritic arborization and complexity in pyramidal neurons of the basolateral amygdala.

(A) Representative tracings of the total dendritic tree of pyramidal neurons from fatty acid amide hydrolase deficient mice (FAAH KO), or their wild type counterparts (WT), under conditions of no stress (CONTROL) or following 21 days of 6 h/day restraint stress (CHRONIC STRESS).
(B) Morphological analysis of pyramidal neurons in the basolateral amygdala in FAAH KO, or their WT counterparts, under conditions of no stress (CONTROL) or following 21 days of 6 h/day restraint stress (CHRONIC STRESS). Analysis encompassed dendritic length (upper panel) and branch points (bottom panel) of pyramidal neurons. Data are displayed as mean ± SEM. * denotes significant differences (p < 0.05) between chronically stressed WT mice and all other treatment conditions.
There was a significant interaction between stress exposure and genotype on the branch points of pyramidal neurons in the BLA [F (1, 102) = 4.344, p < 0.04; Fig. 2]. Post-hoc analyses demonstrated that chronically stressed, wild-type mice exhibited an increase in branch points along the dendritic trees of BLA pyramidal neurons, compared to non-stressed wild-type mice (p < 0.05). There was no difference in the number of dendritic tree branch points of BLA neurons between non-stressed and stressed FAAH−/− mice (p > 0.05), nor between non-stressed wild-type and FAAH−/− mice (p > 0.05). However, chronically stressed, FAAH−/− mice exhibited significantly less dendritic branching than chronically stressed, wild-type mice (p < 0.05). These data demonstrate that FAAH−/− mice do not exhibit an expansion in dendritic arborization following chronic stress.
To further analyze the role of FAAH in stress-induced morphological changes in BLA pyramidal neurons, we examined the effect of chronic stress on BLA pyramidal neuron dendritic spines. This analysis was restricted to the first secondary dendritic branch on pyramidal neurons in the BLA (see Fig. 3 for representative illustration), as spines along this dendritic branch exhibit changes in response to chronic stress and their number correlates with changes in anxiety-like behavior (24, 26, 30). Analysis revealed that there was an interaction between stress exposure and genotype in determining the number of spines along the secondary dendritic arbor on pyramidal neurons of the BLA [F (1, 107) = 5.74, p < 0.02; Fig. 3]. Post hoc analysis demonstrated that chronic stress increased dendritic spine number in wild-type mice relative to their non-stressed counterparts (p < 0.001). There was no effect of chronic stress on dendritic spines in FAAH−/− mice relative to non-stressed FAAH−/− mice (p > 0.05). The number of dendritic spines was not different in chronically stressed, FAAH−/− mice and non-stressed, wild-type mice (p > 0.05). However, the number of dendritic spines was significantly less in chronically stressed, FAAH−/− mice than in chronically stressed, wild-type mice (p < 0.05).
Figure 3. Deficiency in fatty acid amide hydrolase prevents the ability of chronic stress to promote dendritic spinogenesis in pyramidal neurons of the basolateral amygdala.

(A) Low-power photomicrograph of a Golgi stain-impregnated medium spiny neuron in the BLA (Scale bar 50μm.) (Inset) Image of spines on a dendritic segment from the same neuron.
(B) Representative photomicrograph of a dendritic spine densities on a secondary dendritic branch on a pyramidal neuron in the basolateral amygdala from fatty acid amide hydrolase deficient mice (FAAH KO), or their wild type counterparts (WT), under conditions of no stress (Con) or following 21 days of 6 h/day restraint stress (Stress). Scale bars (5 μm) are seen in white in the photomicrograph.
(C) Analysis of total dendritic spines on a secondary branch of pyramidal neurons in the basolateral amygdala in FAAH KO, or their WT counterparts, under conditions of no stress (CONTROL) or following 21 days of 6 h/day restraint stress (CHRONIC STRESS). Data are displayed as mean ± SEM. * denotes significant differences (p < 0.05) between chronically stressed WT mice and all other treatment conditions.
(D) Segmental analysis of dendritic spine density along a secondary dendritic branch of pyramidal neurons in the basolateral amygdala from FAAH KO mice, or their WT counterparts, under conditions of no stress (CONTROL) or following 21 days of 6 h/day restraint stress (CHRONIC STRESS). Total number of dendritic spines along the dendritic branch are expressed for 20 μm segments starting from the origin of the branch up to 100 μm from the origin. Data are displayed as mean ± SEM. * denotes significant differences (p < 0.05) between chronically stressed WT mice and all other treatment conditions.
To further investigate the spatial changes in dendritic spinogenesis along the dendritic branch, we performed a segmental analysis of the secondary branch to determine where the changes in dendritic spines were occurring. Analyses revealed a significant interaction among stress, genotype and distance along the dendritic branch on spine density [F (12, 428) = 2.59, p < 0.005; Fig. 3]. Subsequent analysis of treatment conditions by distance along the dendritic branch revealed that in wild-type mice, chronic stress resulted in an increase in dendritic spines at more distal regions of dendritic branch (p < 0.001; at segments which were 60–80 μm and 80–100 μm distance from origin of branch). There were no differences in dendritic spines at any segment along the dendritic branch between non-stressed and chronically stressed FAAH−/− mice (p > 0.05), or between wild-type and FAAH−/− mice that were not stressed (p > 0.05). FAAH−/− mice exposed to chronic stress exhibited far fewer dendritic spines than their chronically stressed wild-type counterparts at multiple segments along the dendritic branch (p < 0.01 at segments which were 0–20 μm, 60–80 μm and 80–100 μm from the origin of the branch).
To identify if these structural changes in pyramidal neurons of the BLA were associated with concomitant changes in emotional behavior, we examined the effects of chronic stress on anxiety-like behavior in the elevated plus maze of wild-type and FAAH−/− mice. Consistent with the effects on neuronal morphology, there was a significant interaction between stress and genotype on percentage of time mice spent in the open arms of the elevated plus maze [F (1, 16) = 4.61, p < 0.05; Fig. 4]. Post-hoc analyses demonstrated that wild-type mice exposed to chronic stress exhibit a lower percentage of time spent in the open arm relative to all other groups (p < 0.05 for all comparisons). There was no significant difference between non-stressed wild-type and FAAH−/− mice in percent time spent in the open arm (p > 0.05). Regarding closed arm entries, while there was no significant interaction between genotype and stress [F (1, 16) = 0.23, p > 0.05; data not shown], there was a significant main effect of stress [F (1, 16) = 9.40, p < 0.01; data not shown], such that chronic stress exposure increased closed arm entries, regardless of genotype. Since these data could be explained as an effect of chronic stress to affect locomotor activity rather than anxiety-like behaviors, we examined open arm entries as a percent of total entries. There was a significant interaction between stress and genotype on percent of entries into the open arm [F (1, 16) = 4.95, p < 0.05; Fig. 4], such that chronically stressed, wild-type mice exhibited a significantly lower percentage of entries made into the open arm than all other treatment conditions (p < 0.05). There was no significant difference between non-stressed wild-type and FAAH−/− mice in this measure (p > 0.05).
Figure 4. Deficiency in fatty acid amide hydrolase prevents the ability of chronic stress to promote anxiety-like behavior.
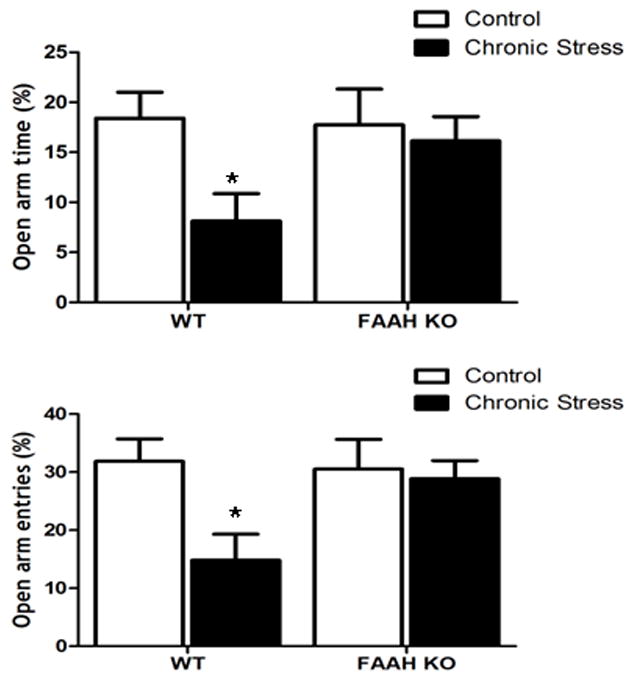
Behavioral analysis of fatty acid amide hydrolase deficient (FAAH KO), or their wildtype counterparts (WT), under conditions of no stress (CONTROL) or following 21 days of 6 h/day restraint stress (CHRONIC STRESS), in the elevated plus maze. Analysis encompassed both the (A) percentage of time spent in and (B) the percentage of entires that the mice made into the open arms. Data are displayed as mean ± SEM. * denotes significant differences (p < 0.05) between chronically stressed WT mice and all other treatment conditions.
To verify that the effects seen in the elevated plus maze were not confounded by differences in locomotor function, we examined the effects of chronic stress on motor activity in an open field arena (Table 2). There was no significant interaction between genotype and stress [F (1, 13) = 0.05, p > 0.05; Table 2]; and neither stress [F (1, 13) = 0.73, p > 0.05] nor genotype [F (1, 13) = 0.17, p > 0.05] exerted significant main effects on total distance travelled in the open field arena. Similarly, there was no significant interaction between stress and genotype [F (1, 13) = 0.35, p > 0.05; Table 2]; or significant main effects of either stress [F (1, 13) = 2.56, p > 0.05] or genotype [F (1, 13) = 0.13, p > 0.05] on velocity of movement.
Table 2. The Effects of Chronic Stress on Locomotor Activity of Wild-type Mice and those Deficient in Fatty Acid Amide Hydrolase.
Three weeks exposure of restraint stress to wild-type C57/Bl6 mice, or those lacking fatty acid amide hydrolase (FAAH−/−), had no effect on locomotor activity in an open field arena.
| Control | Chronic Stress | |
|---|---|---|
| Total Distance Travelled (cm) | ||
| Wild type | 1896 +/− 138 | 2035 +/− 171 |
| FAAH−/− | 1870 +/− 50 | 1954 +/− 114 |
| Velocity (cm/s) | ||
| Wild type | 5.77 +/− 0.93 | 6.92 +/− 0.63 |
| FAAH−/− | 6.26 +/− 0.17 | 6.80 +/− 0.35 |
Data are presented as mean values +/− SEM (n = 4–6 / condition).
Pharmacological inhibition of FAAH similarly confers an anti-anxiety phenotype in rats exposed to chronic stress
The aim of this arm of the study was to verify that inhibition of FAAH itself, and not a neurodevelopmental compensation that could have developed in the FAAH−/− mice, was the principal factor in preventing the development of anxiety-like behaviour following chronic stress. It was also necessary to show that FAAH inhibition in adult life had similar effects to the knock-out in another species, and we thus used rats. To this extent, rats were exposed to normal chow, or chow in which the FAAH inhibitor JNJ-5003 was incorporated at a dose of 50 mg/kg/day for twenty ones days. Both of these treatment conditions were then divided into cage controls and those exposed to twenty-one days of 6h/day restraint stress, in line with the protocol employed in the FAAH−/− mice. Consistent with the behavioural data generated in the FAAH−/− mice, there was a significant interaction between stress and FAAH inhibition on time spent in the open arms of the elevated plus maze [F (1, 26) = 4.63, p < 0.05; Table 3]. Post-hoc analyses demonstrated that normal chow fed rats exposed to chronic stress exhibit a lower amount of time spent in the open arm relative to all other groups (p < 0.05 for all comparisons), demonstrating a protective effect of JNJ-5003 administration in the rat chow during chronic stress on changes in anxiety-like behavior. As another measure of anxiety, we also examined the latency for the rats to enter the open arm, with longer latencies associating with higher levels of anxiety. In this measure, no significant interaction was found between stress exposure and treatment with JNJ-5003 [F (1, 26) = 2.032, p > 0.05; Table 3]. However, based on the data generated with the FAAH−/− mice demonstrating a resistance to the effects of chronic stress, our a priori hypothesis was that the FAAH inhibitor would abrogate the effects of chronic stress on anxiety. Using this hypothesis to guide our analysis, we compared all of the treatment conditions with a Bonferroni test and found that normal chow fed rats exposed to chronic stress exhibited a significant increase in latency to enter the open arms relative to normal chow fed, non-stressed rats (p < 0.01). Consistent with our previous findings, there was no difference between chronically stressed rats treated with JNJ-5003 in their chow versus normal chow fed, non-stressed rats (p > 0.01), indicating that administration of the FAAH inhibitor in the rat chow during chronic stress prevented stress-induced changes in anxiety-like behavior.
Table 3. Fatty acid amide hydrolase inhibitor JNJ-5003 prevents the effects of chronic stress on anxiety-like behavior in the elevated plus maze.<.
br>Three weeks exposure of restraint stress (6 h/day) to rats fed normal rat chow (control) increased indices of anxiety in the elevated plus maze (time spent in the open arm and latency to enter the open arm). The FAAH inhibitor JNJ-5003 was incorporated into the diet and which was fed to rats for the twenty-one day duration of the chronic stress regimen, resulting in an estimated dose of 50 mg/kg/day. Administration of JNJ-5003 during chronic stress mitigated the increased indices of anxiety seen in the rats fed control chow.
| Control | Chronic Stress | |
|---|---|---|
| Time Spent in the Open Arms (s) | ||
| Control | 112.6 +/− 10.1 | 55.3 +/− 7.5* |
| JNJ-5003 | 106.6 +/− 11.4 | 93.4 +/− 10.8 |
| Latency to Enter the Open Arms (s) | ||
| Control | 9.9 +/− 2.6 | 27.4 +/− 6.3✦ |
| JNJ-5003 | 12.1 +/− 2.9 | 18.8 +/− 2.4 |
Data are presented as mean values +/− SEM (n = 7–8 / condition).
denotes significant differences (p < 0.05) between chronically stressed rats fed normal chow and all other treatment conditions.
denotes significant difference (p < 0.05) between chronically stress rats fed normal chow and unstressed rats fed normal chow.
Discussion
In this study, we demonstrated that exposure of mice to chronic stress resulted in an increase in the Vmax of AEA hydrolysis in the amygdala. Since previous studies have demonstrated that the enzyme FAAH is the only mechanism in brain for AEA hydrolysis (45), these data indicate that the hydrolytic activity of FAAH within the amygdala is increased by chronic stress. The amygdala of mice exposed to chronic stress also had significantly reduced amygdalar contents of AEA and related fatty acid amides, which is consistent with increased FAAH activity; further support for this hypothesis comes from the finding that AEA contents in FAAH−/− mice were not affected by chronic stress. Chronic stress exposure neither affected the amygdalar content of 2-AG nor altered the agonist binding parameters to the CB1 receptor in the amygdala. Chronic-stress induced increase in FAAH activity within the amygdala likely contributes to stress-induced morphological alterations within pyramidal neurons in the BLA and increases in anxiety-like behaviors, since mice lacking FAAH expression did not exhibit either of these changes following chronic stress. Similarly, rats treated orally with JNJ-5003, a pharmacological inhibitor of FAAH, during exposure to chronic stress did not develop any significant increases in anxiety-like behavior, consistent with the data obtained using the FAAH−/− mice. These data identify FAAH as a contributor to stress-induced neuronal plasticity within the amygdala and emotional behavior and support the potential therapeutic value of FAAH inhibitors in the context of stress-related anxiety disorders. The determination of whether the loss of FAAH activity can confer resilience against other aspects of chronic stress, including structural remodeling in other brain areas such as the prefrontal cortex or hippocampus, has yet to be determined, but is a focus of future research for our group.
The finding that exposure to chronic stress resulted in an increase in dendritic arborization, as well as spinogenesis, of pyramidal neurons in the BLA is consistent with previous reports (22–24, 26, 30, 49, 50). The mechanism by which stress produces these changes is not completely clear. The predominant hypothesis is that the increase in excitatory transmission within the amygdala that occurs in response to stress engages a feed-forward process which induces spinogenesis and dendritic branching on pyramidal neurons, possibly through increased intracellular calcium signaling and the activation of transduction cascades which induce cytoskeletal remodeling (22, 24, 26). This hypothesis is supported by findings that mice deficient in the NR2A subunit of NMDA receptors have reduced pyramidal neuronal dendritic spines in the BLA under steady-state conditions (57). Similarly, genetic overexpression of the SK2 potassium channel in pyramidal neurons of the BLA, which reduces the intrinsic excitability of these neurons, decreases dendritic arborization in both control and stressed rodents (28). These studies lead to the hypothesis that the dendritic complexity of pyramidal neurons in the BLA is positively regulated by excitation and the structural remodeling of pyramidal dendritic spines following chronic stress is a direct consequence of increased excitatory transmission.
The present results demonstrate that chronic stress results in increased activity of FAAH and that loss of FAAH activity prevented the neuronal remodeling of BLA pyramidal neurons in response to chronic stress. An explanation of these findings is that increased FAAH activity in response to chronic stress couples the effects of stress to increased excitatory neurotransmission in the BLA. FAAH is known to regulate the metabolism of multiple fatty acid ethanolamides, such as AEA, OEA and PEA. All of these molecules are elevated in FAAH−/− mice (48) and reduced following chronic stress, suggesting that any of these could mediate the ability of FAAH activity to couple stress to neuronal plasticity within the amygdala. Outside of one recent report demonstrating that peripheral OEA signaling can influence amygdalar function through a regulation of autonomic fibers and noradrenergic transmission through the nucleus of the solitary tract (58), there is no evidence of a functional role of PEA or OEA signaling within the amygdala. However, the possibility exists that the ability of FAAH activity to modulate the effects of chronic stress on amygdalar function may involve changes in peripheral OEA concentrations.
AEA, which is an endocannabinoid and is also a substrate of FAAH, has been demonstrated to have significant effects within the amygdala. AEA, via activation of the CB1 cannabinoid receptor, inhibits the release of both GABA and glutamate within the BLA (39, 42). Interestingly, despite the presence of CB1 receptors on both GABAergic and glutamatergic populations (31), there is some evidence that endogenous AEA signaling could preferentially target CB1 receptors localized on glutamatergic terminals (59–62). Within the BLA, FAAH expression is restricted to pyramidal neurons (63) and CB1 receptors are known to gate excitatory inputs to pyramidal cells (40, 42, 64, 65). Thus, FAAH activity could modulate presynaptic glutamate release onto pyramidal neurons through the regulation of AEA, which, in turn, would regulate CB1 receptor activation. We hypothesize that chronic stress increases FAAH activity, resulting in a reduction of the ability of AEA to gate excitatory inputs to BLA pyramidal neurons, resulting in disinhibition of glutamate release within the BLA that promotes dendritic remodeling and shifts in emotional behavior.
Consistent with the increased complexity and architecture of pyramidal neurons in the BLA, chronic stress also increased indices of anxiety in the elevated plus maze. Anxiety-like behavior can result from direct changes in the excitability of the BLA. Local disruption of GABAergic signaling within the BLA can produce an increased state of anxiety in rodents, and pharmacological suppression of excitation within the BLA reliably produces a state of anxiolysis (see (5)). The increase in dendritic complexity and spinogenesis following chronic stress is believed to result in a robust increase in excitatory synaptic contacts along pyramidal neurons in the BLA (24), rendering them exquisitely sensitive to excitation. This shift in excitatory sensitivity of the BLA following chronic stress is thought favor a state of hypervigilance and heightened sensitivity to environmental stimuli that manifests as anxiety (5, 8). In line with the ability of FAAH deletion to prevent dendritic expansion and spinogenesis following chronic stress, FAAH knockout mice did not develop stress-induced anxiety. Interestingly, several previous reports have demonstrated that the ability of FAAH inhibition to modulate emotional behavior is entirely dependent upon the aversive nature of the environmental context (60, 66–71). That is, disruption of FAAH activity has little to no effect on anxiety-like behavior in conditions of low aversion, such as in low light testing or in familiar environments (66, 67). Our current data support this, as we did not detect any difference in anxiety-like behavior in non-stressed FAAH knockout mice under our testing conditions (low-light conditions in a familiar testing environment). However, pharmacological or genetic disruption of FAAH can produce a reliable reduction in anxiety-like behavior in aversive environments (60, 66–68, 71). Similarly, we reveal the anxiolytic phenotype of the FAAH deficient mice after exposure to chronic stress, wherein wild type mice develop a characteristic increase in anxiety-like behavior that is absent in the FAAH knockout mice. As such, it has previously been hypothesized that FAAH inhibition may not produce generalized anxiolytic effects, as other agents such as benzodiazepines do, but simply counter stress-induced anxiety (66, 67, 72). The current data provide the first experimental evidence to support this contention by directly demonstrating that a loss of FAAH activity can prevent stress-induced anxiety.
Based on the ability of FAAH inhibition to selectively modulate emotional behavior in highly aversive contexts, and the fact that stress exposure can increase FAAH activity, we propose the following model. Exposure to stress produces a rapid induction of FAAH activity within the amygdala, which results in a reduction in AEA signaling. This loss of AEA signaling disinhibits glutamatergic release within the BLA and increases the firing activity and sensitivity of pyramidal neurons, resulting in an increase in anxiety and vigilance. In the short term, this shift in behavioral anxiety and arousal is likely an adaptive response to ensure survival in a threatening environment, and is transient in nature, returning to normative function once the threat is removed. Under conditions of chronic stress, however, it appears that the steady-state activity of FAAH goes up, resulting in a loss of tonic AEA signaling. Over time, this reduction in AEA signaling results in a progressive excitation of the BLA which then engages a feed-forward response and promotes dendritic expansion and spinogenesis. These morphological changes in pyramidal neurons within the BLA would render them more sensitive to stimuli and thus increasing the basal anxiety state of the organism. Thus, stress-induced FAAH activity regulates anxiety in response to both acute and chronic stress through related, but possibly distinct pathways. That is, in the short term, FAAH inhibition counters stress-induced anxiety, possibly by preventing an increase in glutamatergic transmission and excitation within the BLA. Under conditions of chronic stress, however, a continuous disruption of FAAH activity may prevent stress-induced anxiety by suppressing the architectural remodeling which occurs in pyramidal neurons of the BLA, possibly through an attenuation of excitatory transmission.
We did not demonstrate directly that the effects of FAAH are mediated by changes in CB1 receptor activity. However, data that CB1 receptor deficient mice exhibit the opposite phenotype (increased anxiety and dendritic branching in the BLA under non-stress conditions and a further increase in branching following chronic stress (50)), and that the ability of FAAH inhibition to reduce stress-induced anxiety is blocked by CB1 receptor antagonism (66) support this contention. We were not able to examine whether effects seen in the FAAH−/− mice were reversible by CB1 receptor antagonist administration due to the stressful nature of chronic injections. Specifically, it has previously been demonstrated that repeated injections are sufficiently stress-inducing to reduce limbic AEA content (S. Amen and C.J. Hillard, unpublished data) and induce dendritic hypertrophy within pyramidal neurons of the BLA (29). As such, the possibility exists that these effects could be due to either a non-CB1 receptor target of AEA (such as TRPV1; (73) or a non-AEA fatty acid ethanolamide substrate of FAAH (such as OEA or PEA, as discussed above).
A very important open question is whether comparable changes in FAAH activity and AEA signaling also occur in humans exposed to chronic stress. There is circumstantial evidence to support a role for FAAH in stress-induced anxiety in humans. First, we have previously demonstrated that anxiety levels in individuals with major depression are inversely correlated to circulating levels of AEA, such that people with low levels of circulating AEA exhibit much higher levels of both cognitive and somatic anxiety (74). Second, carriers of the 385A polymorphism of FAAH, which reduces enzyme activity through increased proteolytic degradation and increases levels of AEA (75), exhibit a blunted activation of the amygdala in response to threatening stimuli (76), which is consistent with the hypothesis that reduced FAAH activity suppresses excitation of the amygdala.
In summary, these data indicate that FAAH activity and AEA signaling within the amygdala are important modulators of stress-induced anxiety and structural plasticity. Further, these data highlight FAAH inhibition as a promising target for anxiety disorders, especially those involving hyperactivation of the amygdala, such as post-traumatic stress disorder or generalized anxiety disorder (77, 78).
Supplementary Material
Acknowledgments
The authors would like to thank Katharine McCarthy, Ilia Karatsoreos and Melinda Miller for their technical assistance on the execution of these studies. This research was supported by operating grants from the National Institutes of Health grants MH41256 (BSM), DA09155 and DA026996 (CJH) and the Lightfighter Trust (BSM), as well as an unrestricted research grant from Johnson and Johnson (BSM). SC and SAK are supported by funds from the National Centre for Biological Sciences (NCBS). MNH was the recipient of a postdoctoral fellowship from the Canadian Institutes of Health Research.
Footnotes
Conflict of Interest
This research was supported, in part, by an unrestricted operating grant to BSM from Johnson and Johnson Pharmaceuticals. JMK is an employee of Janssen Research and Development L.L.C, of Johnson and Johnson Pharmaceuticals. The funding body had no role in the design of this study, collection or analysis of the data or decision to publish, outside of the development of the pharmacological compound, JNJ-5003, which was developed and used in a portion of this study.
References
- 1.Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005;48(2):175–187. doi: 10.1016/j.neuron.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 2.van Marle HJ, Hermans EJ, Qin S, Fernandez G. From specificity to sensitivity: how acute stress affects amygdala processing of biologically salient stimuli. Biol Psychiatry. 2009;66(7):649–655. doi: 10.1016/j.biopsych.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 3.Pitkanen A, Savander V, LeDoux JE. Organization of intra-amygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci. 1997;20(11):517–523. doi: 10.1016/s0166-2236(97)01125-9. [DOI] [PubMed] [Google Scholar]
- 4.McDonald AJ. Cortical pathways to the mammalian amygdala. Prog Neurobiol. 1998;55(3):257–332. doi: 10.1016/s0301-0082(98)00003-3. [DOI] [PubMed] [Google Scholar]
- 5.Shekhar A, Truitt W, Rainnie D, Sajdyk T. Role of stress, corticotrophin releasing factor (CRF) and amygdala plasticity in chronic anxiety. Stress. 2005;8(4):209–219. doi: 10.1080/10253890500504557. [DOI] [PubMed] [Google Scholar]
- 6.Joels M, Baram TZ. The neuro-symphony of stress. Nat Rev Neurosci. 2009;10 (6):459–466. doi: 10.1038/nrn2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rainnie DG, Bergeron R, Sajdyk TJ, Patil M, Gehlert DR, Shekhar A. Corticotrophin releasing factor-induced synaptic plasticity in the amygdala translates stress into emotional disorders. J Neurosci. 2004;24(14):3471–3479. doi: 10.1523/JNEUROSCI.5740-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenkranz JA, Venheim ER, Padival M. Chronic stress causes amygdala hyperexcitability in rodents. Biol Psychiatry. 2010;67(12):1128–1136. doi: 10.1016/j.biopsych.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanders SK, Shekhar A. Anxiolytic effects of chlordiazepoxide blocked by injection of GABAA and benzodiazepine receptor antagonists in the region of the anterior basolateral amygdala of rats. Biol Psychiatry. 1995;37(7):473–476. doi: 10.1016/0006-3223(94)00183-4. [DOI] [PubMed] [Google Scholar]
- 10.Petersen EN, Braestrup C, Scheel-Kruger J. Evidence that the anticonflict effect of midazolam in amygdala is mediated by the specific benzodiazepine receptors. Neurosci Lett. 1985;53(3):285–288. doi: 10.1016/0304-3940(85)90552-x. [DOI] [PubMed] [Google Scholar]
- 11.Pesold C, Treit D. The central and basolateral amygdala differentially mediate the anxiolytic effects of benzodiazepines. Brain Res. 1995;671(2):213–221. doi: 10.1016/0006-8993(94)01318-c. [DOI] [PubMed] [Google Scholar]
- 12.Green S, Vale AL. Role of amygdaloid nuclei in the anxiolytic effects of benzodiazepines in rats. Behav Pharmacol. 1992;3(3):261–264. [PubMed] [Google Scholar]
- 13.Sajdyk TJ, Shekhar A. Sodium lactate elicits anxiety in rats after repeated GABA receptor blockade in the basolateral amygdala. Eur J Pharmacol. 2000;394(2–3):265–273. doi: 10.1016/s0014-2999(00)00128-x. [DOI] [PubMed] [Google Scholar]
- 14.Sanders SK, Shekhar A. Regulation of anxiety by GABAA receptors in the rat amygdala. Pharmacol Biochem Behav. 1995;52(4):701–706. doi: 10.1016/0091-3057(95)00153-n. [DOI] [PubMed] [Google Scholar]
- 15.Sajdyk TJ, Shekhar A. Excitatory amino acid receptors in the basolateral amygdala regulate anxiety responses in the social interaction test. Brain Res. 1997;764(1–2):262–264. doi: 10.1016/s0006-8993(97)00594-5. [DOI] [PubMed] [Google Scholar]
- 16.Price JL, Drevets WC. Neurocircuitry of mood disorders. Neuropsychopharmacology. 2010;35(1):192–216. doi: 10.1038/npp.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shin LM, Liberzon I. The neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharmacology. 2010;35(1):169–191. doi: 10.1038/npp.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87(3):873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- 19.Buffalari DM, Grace AA. Chronic cold stress increases excitatory effects of norepinephrine on spontaneous and evoked activity of basolateral amygdala neurons. Int J Neuropsychopharmacol. 2009;12(1):95–107. doi: 10.1017/S1461145708009140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Correll CM, Rosenkranz JA, Grace AA. Chronic cold stress alters prefrontal cortical modulation of amygdala neuronal activity in rats. Biol Psychiatry. 2005;58 (5):382–391. doi: 10.1016/j.biopsych.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 21.Bennur S, Chattarji S. Effects of chronic stress on intrinsic and synaptic plasticity in principal neurons of the basolateral amygdala. Society for Neuroscience Abstracts. 2004:511.2. [Google Scholar]
- 22.Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci. 2002;22(15):6810–6818. doi: 10.1523/JNEUROSCI.22-15-06810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vyas A, Pillai AG, Chattarji S. Recovery after chronic stress fails to reverse amygdaloid neuronal hypertrophy and enhanced anxiety-like behavior. Neuroscience. 2004;128(4):667–673. doi: 10.1016/j.neuroscience.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 24.Vyas A, Jadhav S, Chattarji S. Prolonged behavioral stress enhances synaptic connectivity in the basolateral amygdala. Neuroscience. 2006;143(2):387–393. doi: 10.1016/j.neuroscience.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 25.McEwen BS. Protection and damage from acute and chronic stress: allostasis and allostatic overload and relevance to the pathophysiology of psychiatric disorders. Ann N Y Acad Sci. 2004;1032:1–7. doi: 10.1196/annals.1314.001. [DOI] [PubMed] [Google Scholar]
- 26.Mitra R, Jadhav S, McEwen BS, Vyas A, Chattarji S. Stress duration modulates the spatiotemporal patterns of spine formation in the basolateral amygdala. Proc Natl Acad Sci U S A. 2005;102(26):9371–9376. doi: 10.1073/pnas.0504011102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6(6):463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- 28.Mitra R, Ferguson D, Sapolsky RM. SK2 potassium channel overexpression in basolateral amygdala reduces anxiety, stress-induced corticosterone secretion and dendritic arborization. Mol Psychiatry. 2009;14(9):847–855. 827. doi: 10.1038/mp.2009.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitra R, Sapolsky RM. Acute corticosterone treatment is sufficient to induce anxiety and amygdaloid dendritic hypertrophy. Proc Natl Acad Sci U S A. 2008;105(14):5573–5578. doi: 10.1073/pnas.0705615105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Govindarajan A, Rao BS, Nair D, Trinh M, Mawjee N, Tonegawa S, et al. Transgenic brain-derived neurotrophic factor expression causes both anxiogenic and antidepressant effects. Proc Natl Acad Sci U S A. 2006;103(35):13208–13213. doi: 10.1073/pnas.0605180103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83(3):1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- 32.Ramikie TS, Patel S. Endocannabinoid signaling in the amygdala: anatomy, synaptic signaling, behavior, and adaptations to stress. Neuroscience. 2012;204:38–52. doi: 10.1016/j.neuroscience.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patel S, Roelke CT, Rademacher DJ, Hillard CJ. Inhibition of restraint stress-induced neural and behavioural activation by endogenous cannabinoid signalling. Eur J Neurosci. 2005;21(4):1057–1069. doi: 10.1111/j.1460-9568.2005.03916.x. [DOI] [PubMed] [Google Scholar]
- 34.Patel S, Kingsley PJ, Mackie K, Marnett LJ, Winder DG. Repeated homotypic stress elevates 2-arachidonoylglycerol levels and enhances short-term endocannabinoid signaling at inhibitory synapses in basolateral amygdala. Neuropsychopharmacology. 2009;34(13):2699–2709. doi: 10.1038/npp.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hill MN, McLaughlin RJ, Morrish AC, Viau V, Floresco SB, Hillard CJ, et al. Suppression of amygdalar endocannabinoid signaling by stress contributes to activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology. 2009;34(13):2733–2745. doi: 10.1038/npp.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hill MN, McLaughlin RJ, Bingham B, Shrestha L, Lee TT, Gray JM, et al. Endogenous cannabinoid signaling is essential for stress adaptation. Proc Natl Acad Sci U S A. 2010;107(20):9406–9411. doi: 10.1073/pnas.0914661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rademacher DJ, Meier SE, Shi L, Ho WS, Jarrahian A, Hillard CJ. Effects of acute and repeated restraint stress on endocannabinoid content in the amygdala, ventral striatum, and medial prefrontal cortex in mice. Neuropharmacology. 2008;54 (1):108–116. doi: 10.1016/j.neuropharm.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 38.Sumislawski JJ, Ramikie TS, Patel S. Reversible gating of endocannabinoid plasticity in the amygdala by chronic stress: a potential role for monoacylglycerol lipase inhibition in the prevention of stress-induced behavioral adaptation. Neuropsychopharmacology. 2011;36(13):2750–2761. doi: 10.1038/npp.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Azad SC, Monory K, Marsicano G, Cravatt BF, Lutz B, Zieglgansberger W, et al. Circuitry for associative plasticity in the amygdala involves endocannabinoid signaling. J Neurosci. 2004;24(44):9953–9961. doi: 10.1523/JNEUROSCI.2134-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kodirov SA, Jasiewicz J, Amirmahani P, Psyrakis D, Bonni K, Wehrmeister M, et al. Endogenous cannabinoids trigger the depolarization-induced suppression of excitation in the lateral amygdala. Learn Mem. 2009;17(1):43–49. doi: 10.1101/lm.1663410. [DOI] [PubMed] [Google Scholar]
- 41.Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418(6897):530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- 42.Shin RM, Tully K, Li Y, Cho JH, Higuchi M, Suhara T, et al. Hierarchical order of coexisting pre- and postsynaptic forms of long-term potentiation at synapses in amygdala. Proc Natl Acad Sci U S A. 107(44):19073–19078. doi: 10.1073/pnas.1009803107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chevaleyre V, Heifets BD, Kaeser PS, Sudhof TC, Castillo PE. Endocannabinoid-mediated long-term plasticity requires cAMP/PKA signaling and RIM1alpha. Neuron. 2007;54(5):801–812. doi: 10.1016/j.neuron.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang YC, Wang SJ, Chiou LC, Gean PW. Mediation of amphetamine-induced long-term depression of synaptic transmission by CB1 cannabinoid receptors in the rat amygdala. J Neurosci. 2003;23(32):10311–10320. doi: 10.1523/JNEUROSCI.23-32-10311.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, et al. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci U S A. 2001;98 (16):9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Omeir RL, Chin S, Hong Y, Ahern DG, Deutsch DG. Arachidonoyl ethanolamide-[1,2–14C] as a substrate for anandamide amidase. Life Sci. 1995;56 (23–24):1999–2005. doi: 10.1016/0024-3205(95)00181-5. [DOI] [PubMed] [Google Scholar]
- 47.Hillard CJ, Wilkison DM, Edgemond WS, Campbell WB. Characterization of the kinetics and distribution of N-arachidonylethanolamine (anandamide) hydrolysis by rat brain. Biochim Biophys Acta. 1995;1257(3):249–256. doi: 10.1016/0005-2760(95)00087-s. [DOI] [PubMed] [Google Scholar]
- 48.Patel S, Carrier EJ, Ho WS, Rademacher DJ, Cunningham S, Reddy DS, et al. The postmortal accumulation of brain N-arachidonylethanolamine (anandamide) is dependent upon fatty acid amide hydrolase activity. J Lipid Res. 2005;46(2):342–349. doi: 10.1194/jlr.M400377-JLR200. [DOI] [PubMed] [Google Scholar]
- 49.Johnson SA, Wang JF, Sun X, McEwen BS, Chattarji S, Young LT. Lithium treatment prevents stress-induced dendritic remodeling in the rodent amygdala. Neuroscience. 2009;163(1):34–39. doi: 10.1016/j.neuroscience.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 50.Hill MN, Hillard CJ, McEwen BS. Alterations in Corticolimbic Dendritic Morphology and Emotional Behavior in Cannabinoid CB1 Receptor-Deficient Mice Parallel the Effects of Chronic Stress. Cereb Cortex. 2011;21:2056–2064. doi: 10.1093/cercor/bhq280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wellman CL, Izquierdo A, Garrett JE, Martin KP, Carroll J, Millstein R, et al. Impaired stress-coping and fear extinction and abnormal corticolimbic morphology in serotonin transporter knock-out mice. J Neurosci. 2007;27(3):684–691. doi: 10.1523/JNEUROSCI.4595-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feldman ML, Peters A. A technique for estimating total spine numbers on Golgi-impregnated dendrites. J Comp Neurol. 1979;188(4):527–542. doi: 10.1002/cne.901880403. [DOI] [PubMed] [Google Scholar]
- 53.Trommald M, Hulleberg G. Dimensions and density of dendritic spines from rat dentate granule cells based on reconstructions from serial electron micrographs. J Comp Neurol. 1997;377(1):15–28. doi: 10.1002/(sici)1096-9861(19970106)377:1<15::aid-cne3>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 54.Trommald M, Jensen V, Andersen P. Analysis of dendritic spines in rat CA1 pyramidal cells intracellularly filled with a fluorescent dye. J Comp Neurol. 1995;353(2):260–274. doi: 10.1002/cne.903530208. [DOI] [PubMed] [Google Scholar]
- 55.Horner CH, Arbuthnott E. Methods of estimation of spine density--are spines evenly distributed throughout the dendritic field? J Anat. 1991;177:179–184. [PMC free article] [PubMed] [Google Scholar]
- 56.Hill MN, Carrier EJ, McLaughlin RJ, Morrish AC, Meier SE, Hillard CJ, et al. Regional alterations in the endocannabinoid system in an animal model of depression: effects of concurrent antidepressant treatment. J Neurochem. 2008;106(6):2322–2336. doi: 10.1111/j.1471-4159.2008.05567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mozhui K, Karlsson RM, Kash TL, Ihne J, Norcross M, Patel S, et al. Strain differences in stress responsivity are associated with divergent amygdala gene expression and glutamate-mediated neuronal excitability. J Neurosci. 2010;30 (15):5357–5367. doi: 10.1523/JNEUROSCI.5017-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Campolongo P, Roozendaal B, Trezza V, Cuomo V, Astarita G, Fu J, et al. Fat-induced satiety factor oleoylethanolamide enhances memory consolidation. Proc Natl Acad Sci U S A. 2009;106(19):8027–8031. doi: 10.1073/pnas.0903038106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Musella A, De Chiara V, Rossi S, Prosperetti C, Bernardi G, Maccarrone M, et al. TRPV1 channels facilitate glutamate transmission in the striatum. Mol Cell Neurosci. 2009;40(1):89–97. doi: 10.1016/j.mcn.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 60.Rossi S, De Chiara V, Musella A, Sacchetti L, Cantarella C, Castelli M, et al. Preservation of striatal cannabinoid CB1 receptor function correlates with the antianxiety effects of fatty acid amide hydrolase inhibition. Mol Pharmacol. 2010;78 (2):260–268. doi: 10.1124/mol.110.064196. [DOI] [PubMed] [Google Scholar]
- 61.Gubellini P, Picconi B, Bari M, Battista N, Calabresi P, Centonze D, et al. Experimental parkinsonism alters endocannabinoid degradation: implications for striatal glutamatergic transmission. J Neurosci. 2002;22(16):6900–6907. doi: 10.1523/JNEUROSCI.22-16-06900.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5(5):446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- 63.Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, et al. Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur J Neurosci. 2004;20(2):441–458. doi: 10.1111/j.1460-9568.2004.03428.x. [DOI] [PubMed] [Google Scholar]
- 64.Domenici MR, Azad SC, Marsicano G, Schierloh A, Wotjak CT, Dodt HU, et al. Cannabinoid receptor type 1 located on presynaptic terminals of principal neurons in the forebrain controls glutamatergic synaptic transmission. J Neurosci. 2006;26 (21):5794–5799. doi: 10.1523/JNEUROSCI.0372-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karst H, Berger S, Erdmann G, Schutz G, Joels M. Metaplasticity of amygdalar responses to the stress hormone corticosterone. Proc Natl Acad Sci U S A. 2010;107(32):14449–14454. doi: 10.1073/pnas.0914381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haller J, Barna I, Barsvari B, Gyimesi Pelczer K, Yasar S, Panlilio LV, et al. Interactions between environmental aversiveness and the anxiolytic effects of enhanced cannabinoid signaling by FAAH inhibition in rats. Psychopharmacology (Berl) 2009;204:607–616. doi: 10.1007/s00213-009-1494-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Naidu PS, Varvel SA, Ahn K, Cravatt BF, Martin BR, Lichtman AH. Evaluation of fatty acid amide hydrolase inhibition in murine models of emotionality. Psychopharmacology (Berl) 2007;192(1):61–70. doi: 10.1007/s00213-006-0689-4. [DOI] [PubMed] [Google Scholar]
- 68.Patel S, Hillard CJ. Pharmacological evaluation of cannabinoid receptor ligands in a mouse model of anxiety: further evidence for an anxiolytic role for endogenous cannabinoid signaling. J Pharmacol Exp Ther. 2006;318(1):304–311. doi: 10.1124/jpet.106.101287. [DOI] [PubMed] [Google Scholar]
- 69.Moreira FA, Kaiser N, Monory K, Lutz B. Reduced anxiety-like behaviour induced by genetic and pharmacological inhibition of the endocannabinoid-degrading enzyme fatty acid amide hydrolase (FAAH) is mediated by CB1 receptors. Neuropharmacology. 2008;54(1):141–150. doi: 10.1016/j.neuropharm.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 70.Hill MN, Karacabeyli ES, Gorzalka BB. Estrogen recruits the endocannabinoid system to modulate emotionality. Psychoneuroendocrinology. 2007;32(4):350–357. doi: 10.1016/j.psyneuen.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 71.Bambico FR, Cassano T, Dominguez-Lopez S, Katz N, Walker CD, Piomelli D, et al. Genetic deletion of fatty acid amide hydrolase alters emotional behavior and serotonergic transmission in the dorsal raphe, prefrontal cortex, and hippocampus. Neuropsychopharmacology. 2010;35(10):2083–2100. doi: 10.1038/npp.2010.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hill MN, McEwen BS. Involvement of the endocannabinoid system in the neurobehavioural effects of stress and glucocorticoids. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(5):791–797. doi: 10.1016/j.pnpbp.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Di Marzo V, De Petrocellis L. Endocannabinoids as regulators of transient receptor potential (TRP) channels: A further opportunity to develop new endocannabinoid-based therapeutic drugs. Curr Med Chem. 17(14):1430–1449. doi: 10.2174/092986710790980078. [DOI] [PubMed] [Google Scholar]
- 74.Hill MN, Miller GE, Ho WS, Gorzalka BB, Hillard CJ. Serum endocannabinoid content is altered in females with depressive disorders: a preliminary report. Pharmacopsychiatry. 2008;41(2):48–53. doi: 10.1055/s-2007-993211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sipe JC, Scott TM, Murray S, Harismendy O, Simon GM, Cravatt BF, et al. Biomarkers of endocannabinoid system activation in severe obesity. PLoS One. 5(1):e8792. doi: 10.1371/journal.pone.0008792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hariri AR, Gorka A, Hyde LW, Kimak M, Halder I, Ducci F, et al. Divergent effects of genetic variation in endocannabinoid signaling on human threat- and reward-related brain function. Biol Psychiatry. 2009;66(1):9–16. doi: 10.1016/j.biopsych.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hill MN, Hillard CJ, Bambico FR, Patel S, Gorzalka BB, Gobbi G. The therapeutic potential of the endocannabinoid system for the development of a novel class of antidepressants. Trends Pharmacol Sci. 2009;30(9):484–493. doi: 10.1016/j.tips.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 78.Gaetani S, Dipasquale P, Romano A, Righetti L, Cassano T, Piomelli D, et al. The endocannabinoid system as a target for novel anxiolytic and antidepressant drugs. Int Rev Neurobiol. 2009;85:57–72. doi: 10.1016/S0074-7742(09)85005-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.