

Abstract
Objective
Peptidylarginine deiminase 4 (PAD4) is a citrullinating enzyme that has multiple associations with inflammation. In rheumatoid arthritis, PAD4 and protein citrullination are increased in inflamed joints, and anti–citrullinated protein antibodies (ACPAs) form against citrullinated antigens are formed. ACPA immune complexes can deposit in the joint and induce the production of tumor necrosis factor α (TNFα), a critical inflammatory cytokine in the pathogenesis of rheumatoid arthritis. Further, in other settings, TNFα has been shown to induce PAD4 activity and modulate antibody formation. We undertook this study to investigate whether TNFα and PAD4 may synergistically exacerbate autoantibody production and inflammatory arthritis.
Methods
To determine whether TNFα and PAD4 augment autoantibody production and inflammatory arthritis, we first used a multiplex assay to determine whether mice with chronic inflammatory arthritis due to overexpression of TNFα develop autoantibodies against native and citrullinated antigens. With TNF+ PAD4+/+ and TNF+PAD4−/− mice, we then compared serum autoantibody levels by multiplex array, lymphocyte activation by flow cytometry, total serum IgG levels by enzyme-linked immunosorbent assay, arthritis by clinical and histologic scoring, and systemic inflammation using microfluidic devices.
Results
TNFα-overexpressing mice had increased levels of autoantibodies reactive against native and citrullinated antigens. PAD4−/− mice with TNFα-induced arthritis had lower levels of autoantibodies reactive against native and citrullinated antigens, decreased T cell activation and total IgG levels, and reduced inflammation and arthritis compared to PAD4+/+ TNFα-overexpressing mice.
Conclusion
PAD4 mediates autoantibody production and inflammatory arthritis downstream of TNFα.
Rheumatoid arthritis has long been known to be an inflammatory arthritis, with tumor necrosis factor α (TNFα) playing a leading role. However, more recent evidence demonstrates that rheumatoid arthritis is also an autoimmune disease characterized by autoantibodies such as anti–citrullinated protein antibodies (ACPAs). ACPAs are specific for rheumatoid arthritis, predictive of more severe disease, and implicated in disease pathogenesis (1). Although the pathophysiology of rheumatoid arthritis is incompletely understood, it is hypothesized (2,3) that rheumatoid arthritis can be triggered by protein citrullination, potentially from environmental exposures such as tobacco smoke (4) or Porphyromonas gingivalis infection (5), followed by the development of ACPAs in genetically predisposed individuals. The incorporation of citrullinated antigens into ACPA immune complexes can result in immune complex deposition in the joint stimulating macrophage activation, TNFα production, inflammation, and ultimately clinical rheumatoid arthritis. However, many of the factors that lead to protein citrullination, ACPAs, and arthritis are not clearly defined.
Citrullination is the conversion of a protein’s arginine residues to citrulline, and it is catalyzed by peptidylarginine deiminases (PADs). Citrullination is increased in the rheumatoid joint (6), and inhibition of PADs with Cl-amidine decreases murine collagen-induced arthritis (7), supporting a role for the PADs in rheumatoid arthritis. Since PAD2 and PAD4 are expressed in inflammatory cells and up-regulated in inflamed joints (8), they may be the main PADs responsible for citrullination in arthritis. PAD4 is particularly interesting since its gene contains single-nucleotide polymorphisms associated with rheumatoid arthritis (9). Further, PAD4 is critical for the formation of neutrophil extracellular traps (NETs) (10), which are inflammatory and present some of the same citrullinated antigens that can be targeted by ACPAs (11). Thus, PAD4 could contribute to rheumatoid arthritis pathogenesis due to a role in inflammation and/or antigen citrullination. However, PAD4 is not essential for acute murine K/BxN arthritis (12), a model of the effector component of rheumatoid arthritis that is dependent upon neutrophils (13). Thus, the role of PAD4 in rheumatoid arthritis remains unclear. Fully understanding the contributions of PAD4 to rheumatoid arthritis is particularly important, since drugs targeting PAD4 are under development (14).
As mentioned above, protein citrullination is sometimes considered a starting point for the development of rheumatoid arthritis (2), but citrullination is associated with inflammation of many types (15) and may be a consequence of rheumatoid inflammation (16) as well as a trigger. Interestingly, TNFα, which is present at high levels in rheumatoid arthritis, can induce PAD4 nuclear translocation, histone citrullination (17,18), and NET formation (11,19). Therefore, TNFα may propagate inflammation in rheumatoid arthritis in part through PAD4. Further, TNFα is known to positively regulate B cell proliferation and antibody production (20,21) and could thus augment ACPA production. Indeed, the ACPA repertoire expands and TNFα levels increase prior to the development of clinical rheumatoid arthritis (22), but it has been hypothesized that TNFα up-regulation is downstream of antigen citrullination and ACPA production (2). This idea is consistent with the ability of citrullinated proteins and ACPAs to induce TNFα production by macrophages (23). However, the ability of ACPA immune complexes to induce TNFα does not exclude the possibility that TNFα could also augment ACPA production. There could be a complex positive feedback network involving TNFα, PAD4, citrullination, and ACPAs driving rheumatoid arthritis, but most work has focused on citrullination and autoantibodies upstream of TNFα, not downstream. Overexpression of TNFα in mice causes a chronic, erosive inflammatory arthritis similar to rheumatoid arthritis (24), but little is known about the production of autoantibodies or the role of PAD4 in this model. Here we show that overexpression of TNFα amplifies autoantibody production, and PAD4 mediates TNFα-induced autoantibodies, inflammation, and chronic inflammatory arthritis.
MATERIALS AND METHODS
Animals
Mice that overexpress 1 copy of the TNFα transgene (Tg3647-transgenic mice) (24) on a C57BL/6 background were provided by Dr. Edward Schwarz (University of Rochester Medical Center, Rochester, NY), and permission for their use was granted by Dr. George Kollias and the Alexander Fleming Biomedical Sciences Research Center (Athens, Greece). TNFα-overexpressing mice were crossed with PAD4-deficient mice on a 129 background (10) to ultimately generate TNF+PAD4+/+ and TNF+PAD4−/− mice. Mice were cared for and euthanized in a manner approved by the University of Wisconsin Animal Care and Use Committee.
Multiplex autoantibody immunoassay
Antibodies targeting 37 putative rheumatoid arthritis–associated autoantigens were measured using a custom bead-based immunoassay on the Bio-Plex platform (Bio-Rad) as previously described (22,25). Of the 37 antigens, 30 are citrullinated and 7 are native (native histone H2A, histone H2B, apolipoprotein A–I [Apo A–I], filaggrin 48–65 peptide, vimentin, fibrinogen, and Apo A–I 231–248 peptide). Briefly, serum was diluted and mixed with spectrally distinct fluorescent beads conjugated with putative rheumatoid arthritis–associated autoantigens, followed by incubation with phycoerythrin-conjugated anti-mouse antibody and analysis on a Luminex 200 instrument. Values are reported as relative median fluorescence intensity above background as a semiquantitative measure of serum autoantibody level.
Western blot analysis
Native or in vitro–citrullinated vimentin, histone H2A, and histone H2B were denatured, subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), blotted to nitrocellulose, and exposed overnight at 4°C to serum from TNFα-overexpressing or control mice at a dilution of 1:250 for histone blots and 1:20 for vimentin blots. Blots were washed, incubated with goat anti-mouse IgG conjugated to IRDYE800 (Rockland Immuno-chemicals), washed, and imaged using an Odyssey Imager (Li-Cor). Band density was determined with Odyssey software.
Color development reagent assay
Color development reagent assay was performed in 96-well dishes as previously described (26). Briefly, 6 μl of serum (previously desalted using Zeba spin columns according to the instructions of the manufacturer [Thermo Scientific]) was diluted with 54 μl of color development reagent buffer (50 mM NaCl, 10 mM CaCl2, 2 mM dithiothreitol, 100 mM Tris, pH 7.4), added to 200 μl of color development reagent (1 part 80 mM diacetyl monoxime and 2.0 mM thiosemicarbazide; 3 parts 3M H3PO4, 6M H2SO4, and 2 mM NH4Fe[SO4]2), mixed, and incubated at 95°C for 30 minutes, and the absorbance was read at 540 nm using a Victor multilabel plate reader.
Enzyme-linked immunosorbent assay (ELISA)
To detect antibodies against native and citrullinated antigens, peptides (10 μg/ml) or proteins (20 μg/ml) were coated onto 96-well flat-bottomed polystyrene plates overnight at 4°C. After washing, plates were blocked with 1% bovine serum albumin (BSA) in phosphate buffered saline (PBS) for 1 hour at room temperature followed by incubation with serum at a dilution of 1:50 (protein ELISA) or 1:100 (peptide ELISA) in PBS with 0.05% Tween 20 for 2 hours at room temperature. After washing, wells were incubated for 1 hour at room temperature with horseradish peroxidase–conjugated anti-mouse IgG antibodies (Peroxidase-AffiniPure Goat Anti-Mouse IgG [H+L]; Jackson ImmunoResearch) at 1:10,000 dilution in PBS with 0.05% Tween 20. Bound secondary antibodies were detected by chemiluminescence at 450 nm (1-step Ultra TMB-ELISA; Pierce). For total IgG ELISA, sera were diluted 1:20,000 or 1:100,000 and used with a mouse IgG ELISA kit (Bethyl Laboratories) according to the manufacturer’s instructions. Absorbance was read at 450 nm on a Victor multilabel plate reader.
Flow cytometry
Bone marrow was flushed, spleens were dissociated, and red blood cells were lysed using standard methods, followed by resuspending cells in a buffer of 1% BSA, 2% bovine calf serum, 0.03% NaN3, and 2 mM EDTA in PBS. Two million cells were stained at a 1:100 dilution of the following antibodies: allophycocyanin-conjugated anti-B220 (clone RA3-6B2; eBioscience), phycoerythrin-conjugated anti-CD138 (clone 281-2; BD Biosciences), phycoerythrin-conjugated anti-CD4 (clone RM4.5; eBioscience), fluorescein isothiocyanate–conjugated anti-CD8b (clone eBioH35.17.2; eBioscience), and allophycocyanin-conjugated anti-CD69 (H1.2F3; eBioscience). Samples were washed, fixed in 1% paraformaldehyde, and run on a FACSCalibur flow cytometer followed by analysis with FlowJo software (Tree Star). Debris was excluded using forward and side scatter gating.
Clinical arthritis scores
Arthritis was scored by the same investigator (MAS) in a blinded manner on a scale of 0–3, as follows: 0 = no arthritis; 0.5 = mild joint deformity, mild swelling; 1.0 = moderate joint deformity, moderate swelling; 1.5 = moderate/severe joint deformity, moderate swelling, decreased grip strength on a metal wire; 2.0 = severe joint deformity, moderate swelling, no grip strength; 2.5 = severe joint deformity, moderate/severe swelling, no grip strength; and 3.0 = severe joint deformity and swelling, no grip strength.
Pathologic analysis
Hind legs were fixed in 10% neutral buffered formalin and decalcified for 30 hours with Surgipath Decalcifier 1 (Leica Biosystems). Tissue was embedded, sectioned, and stained with hematoxylin and eosin using standard methods. The tibiotalar joint was scored on a scale of 0–4 for severity of synovitis and cartilage/bone erosion by a single pathologist (TFW) who was blinded to the genotype of the mice.
Microfluidic analysis
As described previously (27), a Kit-On-A-Lid-Assay (KOALA) microfluidic chemotaxis device (made by EKS at the laboratory of DJB at the University of Wisconsin–Madison) was coated with mouse recombinant P-selectin (R&D Systems) at 4°C for at least 30 minutes, followed by washing with PBS. Three microliters of blood was collected, diluted in 18 μl of PBS, and pipetted into the KOALA device. Neutrophils were allowed to adhere for 4 minutes followed by 3 washes with 3 μl of PBS to purify the neutrophils from the other components of whole blood. The microchannels were imaged using an Olympus IX-81 microscope, and cells were counted manually with ImageJ software (National Institutes of Health).
Statistical analysis
Paired and unpaired t-tests as well as Wilcoxon’s matched pairs signed rank test were used as appropriate with GraphPad Prism software. Chauvenet’s criterion was used to exclude a single pair of samples for the microfluidic analysis and ELISA. Significance Analysis of Microarrays was used to analyze all array data, with statistically significant autoantigens displayed on heatmaps.
RESULTS
Association of TNFα overexpression with increased autoantibody production and citrullination
Mice that overexpress TNFα start to develop arthritis between the ages of 4 weeks and 8 weeks, depending on the number of copies of the TNFα transgene (24). The arthritis is thought to be primarily due to activation of the innate immune system (28), but the adaptive immune system is also activated. Autoantibodies have been detected in TNFα-overexpressing mice, but antibodies against cyclic citrullinated peptide, an antigen often used to detect ACPAs, were not detected by age 14 weeks in mice with TNFα-induced arthritis (29). To better understand the role of TNFα in inflammatory arthritis and to better characterize this model for studying PAD4, we determined whether ACPAs formed later in TNFα-induced arthritis. Sera from TNFα-overexpressing mice and littermate controls at ages 2, 3.5, and 5 months were subjected to a bead-based multiplex assay to detect multiple different autoantibodies against citrullinated and native antigens. As shown in Figure 1A, there was an increase in the levels of autoantibodies against citrullinated and native antigens in mice that overexpress TNFα compared to littermate controls at age 5 months. There was no significant increase in the levels of auto-antibodies in TNFα-overexpressing mice at ages 2 and 3.5 months (data not shown), suggesting that autoanti-bodies develop late in TNFα-induced arthritis.
Figure 1.
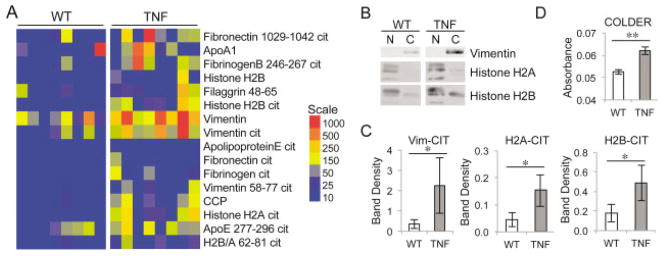
Overexpression of tumor necrosis factor α (TNFα) amplifies autoantibody production and serum citrulline. A, Sera from 5-month-old TNFα-overexpressing (TNF) mice and wild-type (WT) littermates were subjected to a multiplex assay to detect autoantibodies against citrullinated (Cit) and native antigens. The median fluorescence intensity of individual serum samples is displayed in a heatmap of antigens against which antibodies are present at higher levels in sera from TNFα-overexpressing mice compared to sera from WT mice (false discovery rate <0.1% by Significance Analysis of Microarrays). B and C, Native (N) and citrullinated (C) vimentin (Vim), histone H2A, and histone H2B were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to nitrocellulose, and probed with sera from WT and TNFα-overexpressing mice to detect autoantibodies against native and citrullinated antigens. Representative blots are shown in B, with the mean ± SEM of the band density shown in C (n = 8 for citrullinated histones and n = 6 for citrullinated vimentin). * = P ≤ 0.05 by Wilcoxon matched pairs signed rank test. D, Desalted sera from 5-month-old TNFα-overexpressing and WT mice were subjected to the color development reagent (COLDER) assay to quantify citrulline. Graph depicts the mean ± SEM relative absorbance. Samples were tested in duplicate (n = 5). ** = P < 0.01 by paired t-test.
To complement the array data, we performed Western blot analysis for 3 of the proteins against which autoantibody levels were increased in TNFα-overexpressing mice. Native and in vitro citrullinated vimentin, histone H2A, and histone H2B were subjected to SDS-PAGE, blotted, and probed with sera from TNFα-overexpressing or control mice. As shown in Figures 1B and C, antibodies against citrullinated vimentin, histone H2A, and histone H2B were present at higher levels in TNFα-overexpressing mice than in controls. Antibodies against native vimentin could not be detected even at higher concentrations of serum, but antibodies against native histone H2A and H2B were present in controls and increased in TNFα-overexpressing mice (Figure 1B; densitometry results not shown). Thus, chronic overexpression of TNFα can drive autoantibody production, including autoantibodies reactive against citrullinated antigens, but the autoantibody repertoire generated does not specifically target citrullinated epitopes.
Given the presence of autoantibodies against citrullinated antigens and the fact that protein citrullination is associated with inflammation, we evaluated whether TNFα-induced inflammation is associated with increased citrullination. We desalted sera from 5-month-old TNFα-overexpressing mice and control mice to remove free citrulline, and we subjected the sera to the color development reagent assay to obtain an approximate measure of overall citrullination. As shown in Figure 1D, serum citrulline was elevated in TNFα-overexpressing mice compared to controls. These data suggest that chronic overexpression of TNFα in mice amplifies autoantibody production and protein citrullination, but does not lead to a classic ACPA response which exclusively targets citrullinated antigens.
Requirement of PAD4 for maximal autoantibody production downstream of TNFα
We next determined whether PAD4 might be important downstream of TNFα for overall levels of citrullination and development of autoantibodies against native or citrullinated antigens. We crossed mice deficient in PAD4 with mice that overexpress TNFα to ultimately generate TNF+PAD4+/+ and TNF+PAD4−/− mice. We confirmed the absence of PAD4 activity in TNF+PAD4−/− mice by lack of citrullinated histone H4 in peripheral blood leukocytes (data not shown).
To determine the presence of altered autoantibody production in PAD4-deficient mice that overexpress TNFα, we subjected sera from TNF+PAD4+/+ and TNF+PAD4−/− littermates at ages 2, 3.5, and 5 months to the same array described above. We detected elevated levels of autoantibodies in both TNF+PAD4+/+ and TNF+PAD4−/− mice at age 5 months (data not shown); however, the TNF+PAD4−/− mice had reduced levels of several autoantibodies against citrullinated and native antigens (Figure 2A). There was no difference in autoantibody production between TNF+PAD4+/+ and TNF+PAD4−/− littermates at age 2 months, and only 3 autoantibodies showed reduced levels at age 3.5 months (data not shown). To confirm the decreased levels of autoantibodies against 2 key antigens seen in the multiplex array, serum samples from TNF+PAD4+/+ and TNF+ PAD4−/− littermates at age 5 months were subjected to ELISA using plates coated with native or citrullinated vimentin or histone H2B antigens. As shown in Figure 2B, TNF+PAD4−/− mice had decreased levels of antibodies against both native and citrullinated histone H2B and vimentin antigens.
Figure 2.

Reduction of autoantibody levels, but not serum citrulline levels, in peptidylarginine deiminase 4 (PAD4)–deficient mice with tumor necrosis factor α (TNFα)–induced arthritis. Sera from TNF+ PAD4+/+ and TNF+PAD4−/− littermates at age 5 months were subjected to multiplex assay to detect autoantibodies against individual citrullinated (Cit) and native antigens. A, Median fluorescence intensity of individual serum samples is displayed in a heatmap of antigens against which antibodies are present at higher levels in sera from TNF+PAD4+/+ mice compared to sera from TNF+PAD4−/− mice (false discovery rate <0.1% by Significance Analysis of Microarrays). B, Sera from TNF+PAD4+/+ mice (shaded bars) and TNF+PAD4−/− littermates (open bars) at age 5 months were analyzed by enzyme-linked immunosorbent assay to detect autoantibodies against citrullinated and native histone H2B and vimentin (Vim) antigens (n = 6 littermate pairs). C, Sera from TNF+PAD4+/+ and TNF+PAD4−/− littermates at age 5 months were desalted and subjected to the color development reagent (COLDER) assay to quantify citrulline. Values are the mean ± SEM. * = P < 0.05; ** = P < 0.01 by paired t-test.
Since decreased citrullination with its associated protein unfolding could lead to reduced exposure of both native and citrullinated epitopes, we investigated whether a general reduction in antigen citrullination occurs when PAD4 is absent in TNFα-induced arthritis. The color development reagent assay was performed as described above on desalted serum from TNF+ PAD4+/+and TNF+PAD4−/− littermates at age 5 months. As shown in Figure 2C, there was no significant reduction in the serum citrulline level in TNF+PAD4−/− mice. Taken together, these data suggest that in TNFα-induced arthritis, PAD4 contributes to autoantibody production in general, but not specifically to the generation of antibodies against citrullinated antigens. Further, although the color development reagent assay does not detect minor differences in citrullination of individual residues, PAD4 does not appear to be required for the general increase in citrullination seen with overexpression of TNFα.
Contribution of PAD4 to B and T cell activation downstream of TNFα
Given the general reduction in the levels of autoantibodies in TNF+PAD4−/− mice, we evaluated whether plasma cell and total antibody levels were altered. To determine whether there was a decrease in the terminally differentiated, antibody-secreting cells of the B cell lineage, we harvested bone marrow from TNF+PAD4+/+ and TNF+PAD4−/− littermates at age 5 months and quantified B220low CD138high plasma cells by flow cytometry. As shown in Figure 3A, there was no difference in the percentage of plasma cells in the bone marrow of TNF+PAD4−/− mice compared to TNF+ PAD4+/+ littermates. We then examined total serum IgG levels in TNF+PAD4−/− and TNF+PAD4+/+ littermates at age 5 months by ELISA. As shown in Figure 3B, TNF+PAD4−/− mice had decreased IgG levels compared to TNF+PAD4+/+ littermates. There was no difference in total serum IgG levels in PAD4−/− and PAD4+/+ littermates that did not overexpress TNFα at age 3 months (Figure 3C), suggesting that baseline IgG production is unaltered in the absence of PAD4.
Figure 3.

Normal plasma cell numbers but reduced total serum IgG in PAD4−/− mice with TNFα-induced arthritis. Bone marrow from TNF+PAD4+/+ and TNF+PAD4−/− littermates at age 5 months was stained with B220 and CD138 for flow cytometry. A, Shown are a representative dot plot with plasma cells in the boxed area (representative of all mice from 7 experiments) (left) and plasma cell percentages (n = 7 littermate pairs) (right). B and C, Enzyme-linked immunosorbent assay was used to measure IgG levels in sera from TNF+PAD4+/+ and TNF+PAD4−/− littermates at age 5 months (n = 8 littermate pairs) (B) and PAD4+/+ and PAD4−/− littermates at age 3 months (n = 4 littermate pairs) (C). Values are the mean ± SEM. * = P < 0.05 by unpaired t-test. See Figure 2 for definitions.
Since IgG levels were reduced in PAD4-deficient mice that overexpress TNFα, we investigated whether T cell activation was also affected by PAD4. Splenocytes from TNF+PAD4+/+ and TNF+PAD4−/− littermates at age 5 months were stained for CD4, CD8, and CD69 (a marker of activated cells) and analyzed by flow cytometry. We found no difference in the numbers of CD4+ or CD8+ T cells in TNF+PAD4−/− mice compared to TNF+PAD4+/+ littermates (data not shown). However, there were fewer CD69+CD4+ T cells in TNF+ PAD4−/− mice compared to TNF+PAD4+/+ littermates (Figure 4A). Further, the mean fluorescence intensity of CD69 staining was lower in CD8+ T cells from TNF+PAD4−/− mice compared to littermate controls (Figure 4B). Splenic T cells were also evaluated for CD69 levels in PAD4+/+ and PAD4−/− littermates at age 3 months. As shown in Figures 4C and D, there were no differences in CD69 levels in either CD4+ or CD8+ cells in PAD4+/+ versus PAD4−/− mice. Therefore, PAD4 appears to contribute preferentially to T cell activation in TNFα-induced arthritis.
Figure 4.

Reduced T cell activation in PAD4−/− mice with TNFα-induced arthritis. A and B, Spleens of TNF+PAD4+/+ and TNF+PAD4−/− littermates at age 5 months were stained with CD4, CD8, and CD69, analyzed by flow cytometry, and gated on CD4+ and CD8+ cells. CD69 levels on CD4+ cells (A) and CD8+ cells (B) from TNF+PAD4+/+ mice (solid line) and TNF+PAD4−/− mice (dashed line) are displayed on histograms (representative of 6 experiments). Graphs depict the percentage of CD4+ cells that are CD69+ (A) and the mean fluorescence intensity (MFI) of CD69 levels on CD8+ cells (B) (n = 6 littermate pairs). C and D, Similar experiments were performed on PAD4+/+ and PAD4−/− littermates at age 3 months. CD69 levels on CD4+ cells (C) and CD8+ cells (D) from the spleens of PAD4+/+ (solid line) and PAD4−/− littermates (dashed line) are displayed on histograms (representative of 3 experiments). Graphs depict the percentage of CD4+ cells that are CD69+ (C) and the MFI of CD69 levels on CD8+ cells (D) (n = 3 littermate pairs). Values are the mean ± SEM. * = P < 0.05 by paired t-test. See Figure 2 for other definitions.
PAD4 exacerbation of arthritis and inflammation downstream of TNFα
PAD4 is not required for acute K/BxN arthritis, but given the reduction in autoantibody levels and decreased activation of the adaptive immune system in PAD4-deficient mice with TNFα-induced arthritis, we hypothesized that PAD4 might contribute to rheumatoid arthritis and murine models of chronic inflammatory arthritis. To test whether arthritis is affected in the absence of PAD4 in TNFα-induced inflammatory arthritis, we clinically scored arthritis in TNF+PAD4+/+ and TNF+PAD4−/− littermates until the age of 5 months. Arthritis could not be scored after this time due to the poor health and early death of mice overexpressing TNFα, regardless of PAD4 status. As shown in Figure 5A, we found that arthritis was initially equivalent in TNF+PAD4+/+ and TNF+PAD4−/− littermates, similar to acute arthritis in the K/BxN model. However, over time, the arthritis in mice without PAD4 did not worsen as much as that in their TNF+PAD4+/+ littermates. By age 5 months, arthritis was significantly reduced in TNF+PAD4−/− mice compared to their TNF+PAD4+/+littermates (Figure 5A). We also assessed arthritis histologically. The tibiotalar joints of 5-month-old TNF+PAD4+/+ and TNF+PAD4−/− littermates were fixed, decalcified, sectioned, stained, and arthritis severity was scored on a scale of 0–4. As shown in Figures 5B and C, there was a reduction in synovitis and erosions of cartilage and bone in TNF+PAD4−/− mice compared to their TNF+PAD4+/+ littermates.
Figure 5.

Reduced arthritis and inflammation in PAD4−/− mice with TNFα-induced arthritis. A, Severity of clinical arthritis was scored in TNF+PAD4+/+ mice (solid circles) and TNF+PAD4−/− mice (open circles). Graph depicts the mean ± SEM of 12 pairs of mice. *** = P = 0.0001 versus TNF+PAD4+/+ mice, by paired t-test. B, Hind legs from TNF+PAD4+/+ and TNF+PAD4−/− littermates at age 5 months were assessed histologically. Images are representative of 12 pairs of mice and show inflamed synovium/pannus invading cartilage and bone (arrowheads) at the tibiotalar joint. Original magnification × 100. Bar = 200 μm. C, The extent of synovitis and erosion at the tibiotalar joint was scored in a blinded manner on a scale of 0–4, and the graph depicts the mean ± SEM (n = 12 pairs of mice). * = P < 0.05 by paired t-test. D, Blood from TNF+PAD4+/+ and TNF+PAD4−/− mice at age 5 months was pipetted into Kit-On-A-Lid-Assay microfluidic devices. Neutrophils were captured by the P-selectin–coated surface and counted. All experiments were performed at least in triplicate. Graph depicts the mean ± SEM number of captured neutrophils from the TNF+PAD4−/− mouse normalized to the number from the TNF+PAD4+/+ littermate in 7 experiments. *** = P < 0.0001 by unpaired t-test. See Figure 2 for definitions.
Although clinical arthritis and histologic severity were scored in a blinded manner, they are subjective measures. Previously, we used a novel microfluidic device that captures a pure population of neutrophils to demonstrate increased neutrophil capture from TNFα-overexpressing mice compared to wild-type mice (27), consistent with increased inflammation. Therefore, these microfluidic devices can be used as a tool to objectively quantify systemic inflammation. We subjected blood from TNF+PAD4+/+ and TNF+PAD4−/− mice at age 5 months to KOALA microfluidic devices, and, as shown in Figure 5D, fewer neutrophils were captured in samples from TNF+PAD4−/− mice than in those from TNF+PAD4+/+ controls. Taken together, these findings suggest that PAD4 contributes to inflammation and arthritis downstream of TNFα.
DISCUSSION
Here we provide the first evidence that PAD4 contributes to inflammatory arthritis. Several lines of evidence suggested that PAD4 would be important for inflammatory arthritis, as discussed above, but PAD4 was not found to be essential in acute K/BxN arthritis (12), making the role of PAD4 unclear. We found that the greatest reduction in TNFα-induced arthritis in PAD4-deficient mice is late in disease (Figure 5). Further, we detected reduced autoantibody levels (Figure 2), overall IgG levels (Figure 3), and T cell activation (Figure 4), which would not be expected to be important in K/BxN arthritis, an acute arthritis model dependent on the innate immune system. Thus, PAD4 may uniquely exacerbate chronic inflammatory arthritides such as rheumatoid arthritis.
PAD4 could contribute to inflammatory arthritis downstream of TNFα in several ways. One mechanism to consider is a role of PAD4 in antigen citrullination and ACPA production. However, although we saw an increase in the levels of autoantibodies reactive to citrullinated antigens in TNFα-induced arthritis (Figure 1), the autoantibody repertoire did not exclusively target citrullinated antigens as in a human rheumatoid arthritis ACPA response (30). This finding is consistent with a recent report that some murine models of rheumatoid arthritis do not have true ACPAs (31). Further, in TNFα-induced arthritis in the absence of PAD4, we saw a decrease in the levels of autoantibodies against citrullinated and native antigens (Figure 2) as well as a reduction in the levels of total IgG (Figure 3), suggesting that the decrease in autoantibody levels may be related to reduced total antibody levels. In addition, loss of PAD4 did not alter overall citrullination downstream of TNFα (Figure 2), suggesting redundancy with other PADs. Citrullinated proteins and PAD2 are released by mast cells (32), and PAD2 may contribute to macrophage extracellular trap formation (33). Both mast cells and macrophage extracellular traps are possible sources of citrullinated antigens against which antibodies could form. Taken together, although PAD4 could contribute to ACPAs in human rheumatoid arthritis, our studies suggest that PAD4 may have important functions in autoantibody production and arthritis independently of antigen citrullination and ACPAs. Consistent with this, the genetic risk of radiographic progression in human rheumatoid arthritis related to polymorphisms of the PAD4 gene is independent of ACPA status (34).
In considering an ACPA-independent role for PAD4 in arthritis, there are several possibilities. PAD4 is expressed in neutrophils and is critical for NET formation (10). Since NETs contain native and citrullinated antigens and stimulate inflammatory cytokines (11), a loss of NETs in PAD4-deficient mice could reduce levels of inflammation as well as the presentation of citrullinated and native antigens to lymphocytes and thus reduce autoantibody production and inflammation. Indeed, we did detect reduced autoantibody production in PAD4-deficient mice with TNFα-induced arthritis (Figure 2), although this may have been related to decreased total IgG levels (Figure 3). Also, we detected reduced neutrophil capture in TNF+PAD4−/− mice (Figure 5), which could be a sign of either reduced inflammation or defective neutrophil function. Since there was no effect of PAD4 deficiency in acute K/BxN arthritis (12), any abnormalities in neutrophils would need to affect chronic arthritis preferentially, an interesting possibility considering our evolving understanding of the role of neutrophils in chronic inflammation (35).
We also observed decreased T cell activation and IgG levels in TNF+PAD4−/− mice (Figures 3 and 4). Others have shown that Cl-amidine induces lymphocyte apoptosis in a model of ulcerative colitis (36). Together, these data suggest that lymphocytes are affected by loss of PAD activity. There is some evidence that PAD4 is expressed in lymphocytes (6), and PAD4 regulates gene expression (37), so PAD4 could directly affect lymphocyte function. However, the decreased lymphocyte activation in TNF+PAD4−/− mice could also be indirect, possibly related to the overall reduced inflammation. Finally, PAD4 is present in monocytes and macrophages (38), which are key players in rheumatoid arthritis pathogenesis (39). Further studies are needed to clarify the role of PAD4 in different immune cells and arthritis.
In addition to our findings related to PAD4, we found increased autoantibody production and serum protein citrullination in TNFα-induced arthritis. In contrast to previous work (29), we detected autoantibodies against citrullinated antigens in TNFα-induced arthritis, including antibodies against citrullinated Apo E, fibrinogen, histone H2A, histone H2B, and vimentin, which have been shown to be elevated in preclinical human rheumatoid arthritis using a similar multiplex array (22). Although we do not see exclusive reactivity to citrullinated antigens as in human rheumatoid arthritis, over-expression of TNFα does amplify the production of autoantibodies with reactivity against citrullinated proteins. Thus, in the setting of rheumatoid arthritis in genetically susceptible individuals, TNFα might augment true ACPA production.
In conclusion, we have shown that TNFα amplifies autoantibody production and PAD4 mediates TNFα-induced autoantibodies, inflammation, and arthritis. These findings, combined with the work of others, raise the question of a complex positive feedback network involving PAD4, citrullinated antigens, ACPAs, and TNFα in the exacerbation of rheumatoid arthritis. Further work is needed to better understand the mechanisms by which PAD4 contributes to rheumatoid arthritis pathogenesis.
Acknowledgments
We thank Beth Gray for excellent preparation of pathology samples.
Dr. Shelef’s work was supported by a Rheumatology Research Foundation Scientist Development award. Dr. Sokolove’s work was supported by an Arthritis Foundation Innovative Research award and a VA Career Development award. Dr. Wang’s work was supported by NIH grant R01-CA-136856. Dr. Beebe’s work was supported by NIH grant R01-EB-010039. Dr. Robinson’s work was supported by National Heart, Lung, and Blood Institute, NIH, Proteomics Center grant N01-HV-00242 and by the Department of Veterans Affairs. Dr. Huttenlocher’s work was supported by a Burroughs Wellcome Fund grant and NIH grant R56-AI-094923.
Footnotes
Drs. Sackmann and Beebe have ownership interest in Salus Discovery LLC, which has licensed technology (Kit-On-A-Lid-Assay [KOALA] devices) and patents pending for microfluidics technology reported in this work.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Shelef had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Shelef, Sokolove, Beebe, Robinson, Huttenlocher.
Acquisition of data. Shelef, Lahey, Wagner, Sackmann, Warner, Wang.
Analysis and interpretation of data. Shelef, Sokolove, Lahey, Sackmann, Wang, Robinson, Huttenlocher.
References
- 1.Courvoisier N, Dougados M, Cantagrel A, Goupille P, Meyer O, Sibilia J, et al. Prognostic factors of 10-year radiographic outcome in early rheumatoid arthritis: a prospective study. Arthritis Res Ther. 2008;10:R106. doi: 10.1186/ar2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quirke AM, Fisher BA, Kinloch AJ, Venables PJ. Citrullination of autoantigens: upstream of TNFα in the pathogenesis of rheumatoid arthritis. FEBS Lett. 2011;585:3681–8. doi: 10.1016/j.febslet.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Klareskog L, Malmstrom V, Lundberg K, Padyukov L, Alfredsson L. Smoking, citrullination and genetic variability in the immunopathogenesis of rheumatoid arthritis. Semin Immunol. 2011;23:92–8. doi: 10.1016/j.smim.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 4.Klareskog L, Stolt P, Lundberg K, Kallberg H, Bengtsson C, Grunewald J, et al. the Epidemiological Investigation of Rheumatoid Arthritis Study Group. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA–DR (shared epitope)–restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2006;54:38–46. doi: 10.1002/art.21575. [DOI] [PubMed] [Google Scholar]
- 5.Liao F, Li Z, Wang Y, Shi B, Gong Z, Cheng X. Porphyromonas gingivalis may play an important role in the pathogenesis of periodontitis-associated rheumatoid arthritis. Med Hypotheses. 2009;72:732–5. doi: 10.1016/j.mehy.2008.12.040. [DOI] [PubMed] [Google Scholar]
- 6.Chang X, Yamada R, Suzuki A, Sawada T, Yoshino S, Tokuhiro S, et al. Localization of peptidylarginine deiminase 4 (PADI4) and citrullinated protein in synovial tissue of rheumatoid arthritis. Rheumatology (Oxford) 2005;44:40–50. doi: 10.1093/rheumatology/keh414. [DOI] [PubMed] [Google Scholar]
- 7.Willis VC, Gizinski AM, Banda NK, Causey CP, Knuckley B, Cordova KN, et al. N-α-benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. J Immunol. 2011;186:4396–404. doi: 10.4049/jimmunol.1001620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Foulquier C, Sebbag M, Clavel C, Chapuy-Regaud S, Al Badine R, Mechin MC, et al. Peptidyl arginine deiminase type 2 (PAD-2) and PAD4 but not PAD-1, PAD-3, and PAD-6 are expressed in rheumatoid arthritis synovium in close association with tissue inflammation. Arthritis Rheum. 2007;56:3541–53. doi: 10.1002/art.22983. [DOI] [PubMed] [Google Scholar]
- 9.Kurko J, Besenyei T, Laki J, Glant TT, Mikecz K, Szekanecz Z. Genetics of rheumatoid arthritis—a comprehensive review. Clin Rev Allergy Immunol. 2013;45:170–9. doi: 10.1007/s12016-012-8346-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207:1853–62. doi: 10.1084/jem.20100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. 2013;5:178ra40. doi: 10.1126/scitranslmed.3005580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rohrbach AS, Hemmers S, Arandjelovic S, Corr M, Mowen KA. PAD4 is not essential for disease in the K/BxN murine auto-antibody-mediated model of arthritis. Arthritis Res Ther. 2012;14:R104. doi: 10.1186/ar3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol. 2001;167:1601–8. doi: 10.4049/jimmunol.167.3.1601. [DOI] [PubMed] [Google Scholar]
- 14.Jones JE, Slack JL, Fang P, Zhang X, Subramanian V, Causey CP, et al. Synthesis and screening of a haloacetamidine containing library to identify PAD4 selective inhibitors. ACS Chem Biol. 2012;7:160–5. doi: 10.1021/cb200258q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Makrygiannakis D, af Klint E, Lundberg IE, Lofberg R, Ulfgren AK, Klareskog L, et al. Citrullination is an inflammation-dependent process. Ann Rheum Dis. 2006;65:1219–22. doi: 10.1136/ard.2005.049403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makrygiannakis D, Revu S, Engstrom M, af Klint E, Nicholas AP, Pruijn GJ, et al. Local administration of glucocorticoids decreases synovial citrullination in rheumatoid arthritis. Arthritis Res Ther. 2012;14:R20. doi: 10.1186/ar3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mastronardi FG, Wood DD, Mei J, Raijmakers R, Tseveleki V, Dosch HM, et al. Increased citrullination of histone H3 in multiple sclerosis brain and animal models of demyelination: a role for tumor necrosis factor-induced peptidylarginine deiminase 4 translocation. J Neurosci. 2006;26:11387–96. doi: 10.1523/JNEUROSCI.3349-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neeli I, Khan SN, Radic M. Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol. 2008;180:1895–902. doi: 10.4049/jimmunol.180.3.1895. [DOI] [PubMed] [Google Scholar]
- 19.Keshari RS, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, et al. Cytokines induced neutrophil extracellular traps formation: implication for the inflammatory disease condition. PLoS One. 2012;7:e48111. doi: 10.1371/journal.pone.0048111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boussiotis VA, Nadler LM, Strominger JL, Goldfeld AE. Tumor necrosis factor α is an autocrine growth factor for normal human B cells. Proc Natl Acad Sci U S A. 1994;91:7007–11. doi: 10.1073/pnas.91.15.7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobie JJ, Zheng B, Bryk P, Barnes M, Ritchlin CT, Tabechian DA, et al. Decreased influenza-specific B cell responses in rheumatoid arthritis patients treated with anti-tumor necrosis factor. Arthritis Res Ther. 2011;13:R209. doi: 10.1186/ar3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sokolove J, Bromberg R, Deane KD, Lahey LJ, Derber LA, Chandra PE, et al. Autoantibody epitope spreading in the pre-clinical phase predicts progression to rheumatoid arthritis. PLoS One. 2012;7:e35296. doi: 10.1371/journal.pone.0035296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sokolove J, Zhao X, Chandra PE, Robinson WH. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcγ receptor. Arthritis Rheum. 2011;63:53–62. doi: 10.1002/art.30081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Douni E, Akassoglou K, Alexopoulou L, Georgopoulos S, Haralambous S, Hill S, et al. Transgenic and knockout analyses of the role of TNF in immune regulation and disease pathogenesis. J Inflamm. 1995;47:27–38. [PubMed] [Google Scholar]
- 25.Sokolove J, Lindstrom TM, Robinson WH. Development and deployment of antigen arrays for investigation of B-cell fine specificity in autoimmune disease. Front Biosci (Elite Ed) 2012;4:320–30. doi: 10.2741/379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knipp M, Vasak M. A colorimetric 96-well microtiter plate assay for the determination of enzymatically formed citrulline. Anal Biochem. 2000;286:257–64. doi: 10.1006/abio.2000.4805. [DOI] [PubMed] [Google Scholar]
- 27.Sackmann EK, Berthier E, Young EW, Shelef MA, Wernimont SA, Huttenlocher A, et al. Microfluidic kit-on-a-lid: a versatile platform for neutrophil chemotaxis assays. Blood. 2012;120:e45–53. doi: 10.1182/blood-2012-03-416453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li P, Schwarz EM. The TNF-α transgenic mouse model of inflammatory arthritis. Springer Semin Immunopathol. 2003;25:19–33. doi: 10.1007/s00281-003-0125-3. [DOI] [PubMed] [Google Scholar]
- 29.Hoffmann M, Hayer S, Steiner G. Immmunopathogenesis of rheumatoid arthritis: induction of arthritogenic autoimmune responses by proinflammatory stimuli. Ann N Y Acad Sci. 2009;1173:391–400. doi: 10.1111/j.1749-6632.2009.04753.x. [DOI] [PubMed] [Google Scholar]
- 30.Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest. 1998;101:273–81. doi: 10.1172/JCI1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cantaert T, Teitsma C, Tak PP, Baeten D. Presence and role of anti–citrullinated protein antibodies in experimental arthritis models. Arthritis Rheum. 2013;65:939–48. doi: 10.1002/art.37839. [DOI] [PubMed] [Google Scholar]
- 32.Arandjelovic S, McKenney KR, Leming SS, Mowen KA. ATP induces protein arginine deiminase 2-dependent citrullination in mast cells through the P2X7 purinergic receptor. J Immunol. 2012;189:4112–22. doi: 10.4049/jimmunol.1201098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mohanan S, Horibata S, McElwee JL, Dannenberg AJ, Coonrod SA. Identification of macrophage extracellular trap-like structures in mammary gland adipose tissue: a preliminary study. Front Immunol. 2013;4:67. doi: 10.3389/fimmu.2013.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki T, Ikari K, Yano K, Inoue E, Toyama Y, Taniguchi A, et al. PADI4 and HLA-DRB1 are genetic risks for radiographic progression in RA patients, independent of ACPA status: results from the IORRA Cohort Study. PloS One. 2013;8:e61045. doi: 10.1371/journal.pone.0061045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shelef MA, Tauzin S, Huttenlocher A. Neutrophil migration: moving from zebrafish models to human autoimmunity. Immunol Rev. 2013;256:269–81. doi: 10.1111/imr.12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chumanevich AA, Causey CP, Knuckley BA, Jones JE, Poudyal D, Chumanevich AP, et al. Suppression of colitis in mice by Cl-amidine: a novel peptidylarginine deiminase inhibitor. Am J Physiol Gastrointest Liver Physiol. 2011;300:G929–38. doi: 10.1152/ajpgi.00435.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li P, Wang D, Yao H, Doret P, Hao G, Shen Q, et al. Coordination of PAD4 and HDAC2 in the regulation of p53-target gene expression. Oncogene. 2010;29:3153–62. doi: 10.1038/onc.2010.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vossenaar ER, Radstake TR, van der Heijden A, van Mansum MA, Dieteren C, de Rooij DJ, et al. Expression and activity of citrullinating peptidylarginine deiminase enzymes in monocytes and macrophages. Ann Rheum Dis. 2004;63:373–81. doi: 10.1136/ard.2003.012211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davignon JL, Hayder M, Baron M, Boyer JF, Constantin A, Apparailly F, et al. Targeting monocytes/macrophages in the treatment of rheumatoid arthritis. Rheumatology (Oxford) 2013;52:590–8. doi: 10.1093/rheumatology/kes304. [DOI] [PubMed] [Google Scholar]