

Key Points
The JAK2/STAT5 pathway is a relevant therapeutic target in CML SPCs.
Targeting the JAK2/STAT5 pathway by nilotinib and RUX in combination leads to enhanced eradication of primitive CML stem cells.
Abstract
Chronic myeloid leukemia (CML) stem cell survival is not dependent on BCR-ABL protein kinase and treatment with ABL tyrosine kinase inhibitors cures only a minority of CML patients, thus highlighting the need for novel therapeutic targets. The Janus kinase (JAK)2/signal transducer and activator of transcription (STAT)5 pathway has recently been explored for providing putative survival signals to CML stem/progenitor cells (SPCs) with contradictory results. We investigated the role of this pathway using the JAK2 inhibitor, ruxolitinib (RUX). We demonstrated that the combination of RUX, at clinically achievable concentrations, with the specific and potent tyrosine kinase inhibitor nilotinib, reduced the activity of the JAK2/STAT5 pathway in vitro relative to either single agent alone. These effects correlated with increased apoptosis of CML SPCs in vitro and a reduction in primitive quiescent CML stem cells, including NOD.Cg-Prkdcscid IL2rgtm1Wjl /SzJ mice repopulating cells, induced by combination treatment. A degree of toxicity toward normal SPCs was observed with the combination treatment, although this related to mature B-cell engraftment in NOD.Cg-Prkdcscid IL2rgtm1Wjl /SzJ mice with minimal effects on primitive CD34+ cells. These results support the JAK2/STAT5 pathway as a relevant therapeutic target in CML SPCs and endorse the current use of nilotinib in combination with RUX in clinical trials to eradicate persistent disease in CML patients.
Introduction
Chronic myeloid leukemia (CML) arises in a hemopoietic stem cell (HSC) as a result of the reciprocal translocation between chromosomes 9 and 22 (t9;22), leading to the formation of the fusion oncogene BCR-ABL. The BCR-ABL protein has constitutive tyrosine kinase (TK) activity which drives myeloid progenitor cell expansion and is both necessary and sufficient for the transformed phenotype.1 The introduction of ABL TK inhibitors (TKI) has dramatically changed the management of newly diagnosed chronic phase (CP) CML patients, with the vast majority now achieving deep molecular responses, while enjoying a good quality of life.2 However, 15% to 20% of patients show variable degrees of resistance to currently available TKI,3 and even in patients achieving deep responses, including those with undetectable BCR-ABL transcript levels, there is evidence of persistence of BCR-ABL+ cells at the stem-cell level4,5 and of positivity for BCR-ABL genomic DNA by polymerase chain reaction (PCR).6,7 Furthermore, over 50% of patients achieving sustained undetectable BCR-ABL transcript levels showed evidence of molecular relapse upon TKI discontinuation.8 Leukemic stem cell (LSC) persistence determines the need for lifelong TKI treatment in the ever growing CML patient population, with associated implications in terms of compliance, adverse events, and costs. Recent evidence has demonstrated that CML-LSC persistence is secondary to their insensitivity to TKI despite effective BCR-ABL kinase inhibition, suggesting that other pathways contribute to their survival.9,10 Identifying such pathways and trying to exploit them therapeutically is paramount to achieving CML-LSC eradication and disease cure.
During normal hemopoiesis, the intracellular TK Janus kinase (JAK)2 is activated following binding of hemopoietic growth factors (GF) to their receptors. JAK2 subsequently phosphorylates the signal transducer and activator of transcription (STAT)5 factor, leading to its nuclear relocation. Nuclear STAT5 exerts its activity by regulating the transcription of genes involved in normal hemopoiesis.11 The central role of the JAK2/STAT5 axis is clearly demonstrated by the profound effects on hemopoiesis, resulting in embryonic lethality of JAK2 knockout (KO) mice.12-14 Both JAK2 and STAT5 are constitutively active in BCR-ABL+ cells15,16 with evidence supporting a role for each in CML leukemogenesis. BCR-ABL+ cell clones transfected with kinase inactive JAK2 mutant displayed reduced clonogenic potential and tumorogenic activity.17 Recently, the existence of a JAK2/BCR-ABL protein complex, which helps to stabilize BCR-ABL kinase activity, has been demonstrated.18,19 Disrupting this complex using either JAK2 chemical inhibitors or RNA interference was shown to increase eradication of BCR-ABL+ cells, including primary CML CD34+ cells.18,20 Similarly, STAT5-deleted murine bone marrow (BM) cells transduced with BCR-ABL retrovirus were unable to generate and maintain BCR-ABL+ leukemias in vivo.21-23 Furthermore, high levels of STAT5 were protective for BCR-ABL+ cells treated with TKI24 and specific targeting of STAT5 activity increased eradication of BCR-ABL+ cells, including primary CML CD34+ cells and CML cells resistant to TKI.25 Pharmacologic interference with the JAK2/STAT5 pathway, therefore, represents an attractive therapeutic strategy in CML; however, STAT5 is a difficult drug target as it lacks an enzymatic domain and a simpler approach to interfere with STAT5 function is to inhibit its predominant activating kinase, JAK2.
In contrast to normal hemopoiesis, the role of JAK2 in activating STAT5 in BCR-ABL+ leukemias remains controversial. Investigations using both a dominant negative JAK2 mutant26 and BCR-ABL transduced JAK2 KO murine BM cells27 suggested that BCR-ABL is able to directly phosphorylate STAT5, rendering the role of JAK2 dispensable. It has also been suggested that the reported effects of most JAK2 inhibitors on BCR-ABL+ cells were secondary to their off-target inhibition of BCR-ABL kinase.27,28 These data have questioned the role of JAK2 as a bona fide therapeutic target in CML.
The relevance of understanding the role of the JAK/STAT pathway in CML has increased with the clinical development of numerous JAK2 inhibitors. Among these, ruxolitinib (RUX) has emerged as a potent and orally bioavailable JAK1/2 inhibitor29 which is now licensed for the treatment of primary myelofibrosis following results from phase 3 clinical trials.30,31 As a result, a therapeutic strategy employing RUX in CML could now easily be pursued, and early phase clinical studies aiming to assess the ability of RUX and TKI in combination, to eradicate CML stem/progenitor cells (SPCs) are already underway (ClinicalTrials.gov identifiers: #NCT01702064 and #NCT01751425). In this study, we aimed to further characterize the role of JAK2 in human primary CML CD34+ cells and complementary mouse models. The effects of RUX alone and in combination with nilotinib (NL) on JAK2 and STAT5 activity were assessed, aiming to clarify whether JAK2 modulated STAT5 activity in CML cells, especially in the context of a fully inhibited BCR-ABL kinase. Moreover, the effects of RUX, with or without NL, on the survival and proliferation of primary human CD34+ CML and normal cells in vitro and on leukemia engraftment in vivo, were tested to assess their efficacy in CML and potential toxicity to normal cells.
Methods
Reagents
RUX and NL were supplied by Novartis Pharmaceuticals and stored at 10 mM in dimethylsulfoxide at −20°C.
Primary cell samples and in vitro culture
Primary cells were obtained following consent, according to the Declaration of Helsinki, from peripheral blood leukapheresis samples of newly diagnosed CP CML patients (see supplemental Table on the Blood Web site) and lymphoma patients without BM involvement or cord blood (CB) as normal controls. Enrichment for CD34+ expression, fluorescence-activated cell sorting (FACS) of CML CD34+ CD38−, detection of BCR-ABL fusion in CML SPC/LSC cells by fluorescence in situ hybridization (FISH), and in vitro culture in physiological (CML/CB) or high (adult normal CD34+) GF-supplemented serum-free medium were conducted, as previously reported.9,32 Total cell counts were performed using the trypan blue dye exclusion method or CountBright Absolute Counting Beads for flow cytometry (Life Technologies). Storage and use of primary cell samples for research purposes was approved by West of Scotland Research Ethics Committee 4 (ref #10/S0704/2).
Colony-forming cell assays, apoptosis assessment, and cell division tracking
Colony-forming cell (CFC) assays, apoptosis measurement by Annexin-V staining, and cell division tracking using carboxyfluorescein diacetate succinimidyl ester (CFSE; Life Technologies) of CML and normal CD34+ cells were performed according to standard protocols, as previously described.9
Engraftment of human cells in immunodeficient mice
CML CD34+ cells (1 × 106 cells/mouse) or CB CD34+ cells (1 × 105 cells/mouse) were cultured for 72 hours in the absence of drug (control), or with addition of NL (1 μM) alone, RUX (500 nM) alone, or their combination. Cells were then harvested, washed, and transplanted via tail vein injection into sublethally irradiated (300 cGy) 8-week-old NOD. Cg-Prkdcscid IL2rgtm1Wjl /SzJ (NSG) mice (The Jackson Laboratory). Mice were euthanized after 16 (CB) and 18 (CML) weeks and marrow contents of femurs, spleen cells, and blood cells were obtained at necropsy. To assess human cell engraftment, cells were labeled with antihuman CD45-allophycocyanin antibody (#17-9459-42; eBioscience) and analyzed by flow cytometry. Specific human cell subsets were detected by staining with antibodies to human CD34-PE-Cy7 (#25-0349-42; eBioscience), CD33-PE (#347787; BD Biosciences), and CD19-efluoro450 (#48-0199-42; eBioscience). Human CD45+ cells were selected by immunomagnetic column selection. To assess engraftment of malignant BCR-ABL expressing cells, CD45+ selected cells were evaluated for BCR-ABL messenger RNA (mRNA) levels by quantitative reverse-transcription PCR (qRT-PCR) using custom made primers and probes for both BCR-ABL (forward primer GGGCTCTATGGGTTTCTGAATG; reverse primer CGCTGAAGGGCTTTTGAACT; probe CATCGTCCACTCAGCCACTGGATTTAAGC), and BCR (forward primer CCTTCGACGTCAATAACAAGGAT; reverse primer CCTGCGATGGCGTTCAC; and probe TCCATCTCGCTCATCATCACCGACA) as the reference gene.
FACS and western blotting
FACS for intracellular protein staining and western blot analysis was performed as previously reported32 using the following antibodies: p-JAK2Tyr1007/1008 (#1477-1) (Epitomics), p-STAT5Tyr694 (#9351), p-CrkLTyr207 (#3181), p-c-AblTyr245 (#2868), and β-tubulin (#2128) (New England BioLabs), SH-PTP2 (#sc-280) (Santa Cruz Biotechnology).
RNA extraction, complementary DNA synthesis, and qRT-PCR
Total RNA was isolated from pellets using the RNeasy Mini Kit (Qiagen, United Kingdom) according to the manufacturer’s instructions. complementary DNA (cDNA) was generated using the High Capacity cDNA Reverse Transcription Kit and specific target amplification was performed using the TaqMan PreAmp Master Mix Kit (Life Technologies) according to manufacturer’s protocol. qRT-PCR was performed using the Fluidigm BioMark HD System with the following TaqMan validated gene expression assays: BCL-XL (Hs01067345_g1); BCL6 (Hs00277037_m1); Cyclin D1 (Hs00765553_m1); Cyclin D2 (Hs00277041_m1); Cyclin D3 (Hs00426901_m1); CDKN1B/p27 (Hs00153277_m1); GAPDH (Hs99999905_m1); ID1 (Hs03676575_s1); MYC (Hs00905030_m1); PTEN (Hs03673482_s1); and TBP (Hs99999910_m1).
Statistical analysis
All data from independent experiments are presented as mean ± standard error of the mean (SEM). Statistical analyses were performed using the paired Student t test for matched samples, the Mann-Whitney test for unpaired samples, and the one-way ANOVA with post hoc testing for multiple comparisons. P ≤ .05 was considered statistically significant.
Results
The combination of NL and RUX deeply inhibits JAK2 and STAT5 activities, with no off-target effects on BCR-ABL kinase
To assess the respective activities of JAK2 and STAT5 in CML CD34+ cells following treatment with NL or RUX or both, levels of p-JAK2 and p-STAT5 were measured by flow cytometry over 72 hours of culture in medium supplemented with physiological GFs to mimic microenvironmental signaling.33 Interestingly, while both p-JAK2 and p-STAT5 levels were reduced by NL treatment, which infers that both are partially regulated by BCR-ABL kinase, the addition of RUX caused a faster and deeper reduction in the level of both phosphoproteins (Figure 1A-B and supplemental Figure 1A). Further evidence of this more pronounced inhibition of STAT5 activity by the combination treatment was derived by gene expression analysis of several STAT5 target genes. Treatment with the combined agents, as compared with NL or RUX alone, caused further downregulation of genes known to be positively regulated by STAT5, such as the antiapoptotic gene BCL-XL, the positive cell cycle regulators Cyclin D1, D2, D3, MYC, and the transcription factor ID1,34 which plays a key role in HSC self-renewal and differentiation.35,36 Conversely, genes normally negatively regulated by STAT5 were upregulated, including the negative regulator of the AKT pathway, PTEN, the negative regulator of cell cycle, CDKN1B/p27, and the transcription factor, BCL6, which has been reported to be important for the maintenance of CML LSC37 (Figure 1C). When these cultures were repeated in the absence of paracrine GFs, the enhanced STAT5 inhibition with the combination was no longer observed (supplemental Figure 1B).
Figure 1.

Effects of the NL and RUX combination on JAK2/STAT5 signaling. CML CD34+ samples (n = 3) were either left untreated (UT), or treated with NL (5 µM) or RUX (200 nM) or their combination. After 24 hours of incubation in suspension cultures, p-STAT5Tyr694 (A) and p-JAK2Tyr1007/1008 (B) levels were measured by intracellular flow cytometry. Levels of phosphorylation of both proteins were expressed as a ratio of the mean fluorescence intensity of p-STAT5Tyr694 (A) and p-JAK2Tyr1007/1008 (B) antibody stained cells over the mean fluorescence intensity of cells stained with a matched isotype control. The average of UT values was normalized to 100% and changes following treatment expressed as % change from UT. (C) Candidate STAT5 target genes mRNA expression changes were measured in CML CD34+ samples (n = 5) following 8 hours in suspension culture with NL (5 µM) or RUX (1000 nM), or their combination. Differences in gene expression levels following treatment were calculated using the 2−ΔΔCt method after normalization within each sample of candidate gene expression levels against the expression levels of the reference genes (GAPDH and TBP). Relative quantitation of candidate genes mRNA expression following NL, and NL and RUX treatment, was then plotted as log2 of the 2−ΔΔCt values (with the RUX-treated cells having a value of 0 in the graph and being the calibrator). All data from independent experiments are presented as mean ± SEM. Significance values: *P < .05; †P < .01; ‡P < .001. ns, not significant.
Analysis of the phosphorylation levels of BCR-ABL and its kinase substrate CrkL demonstrated that full inhibition of BCR-ABL kinase activity had been achieved upon treatment with NL. No significant changes in neither p-BCR-ABL nor p-CrkL levels were observed when cells were treated with RUX as a single agent (Figure 2A-B). These data eliminate any concerns that the additive effects on STAT5 signaling of the combined inhibitor treatment were secondary to RUX-induced off-target effects on BCR-ABL kinase activity, as had been suggested for other JAK2 inhibitors and for RUX at higher concentrations (>20 µM).27
Figure 2.

Effects of the NL and RUX combination on BCR-ABL kinase activity. CML CD34+ cells were either left UT, or treated for 8 hours with NL (5 µM) or RUX (1000 nM) or their combination, prior to protein extraction and measurement of (A) p-BCR-ABL (p-c-AblTyr245) (n = 2), and (B) p-CrkLTyr207 (n = 4, 2 representative blots shown) levels by western blot. SH-PTP2 (A) and β-tubulin (B) were used as loading controls.
Overall, these data showed that the combination of NL and RUX caused a more rapid and profound inhibition of p-STAT5 activity as compared with single agents and that this was not secondary to off-target effects of the JAK2 inhibitor on BCR-ABL kinase activity.
The combination of NL and RUX increases kill and reduces CFC output of human CML CD34+ cells, while sparing normal CD34+ cells in vitro
Given the additive effects of NL in combination with RUX on STAT5 signaling, the biological effects of these agents against primary CML CD34+ cells were investigated in vitro. Following preliminary experiments to establish the IC50 of RUX in this cell population (supplemental Figure 2), a concentration of 200 nM was selected. This concentration was commensurate with that used in published preclinical studies of cell lines harboring the JAK2V617F mutation29 and below the maximal concentration of 1000 nM achieved in the plasma of patients treated with currently licensed doses of RUX.38 Moreover, this concentration was 100-fold less than the RUX concentration reported to produce off-target effects on BCR-ABL kinase.27 While neither NL or RUX showed limited effects on CML CD34+ cell viability, their combination induced higher levels of apoptosis (Figure 3A). Similarly, while RUX had no effect and NL reduced CML CD34+ CFC output by 47% relative to starting CFC output (baseline), their combination produced a further 49% reduction in the total number of CFC produced relative to NL as a single agent (Figure 3B). Similar effects were seen for both erythroid and granulocyte-macrophage colonies (Figure 3C-D), and NL and RUX combined were also noted to reduce the size of the colonies (Figure 3E). Interestingly, these effects were not observed when CML CD34+ cells were grown in the absence of any GFs (supplemental Figure 3). A similar series of experiments was then performed using normal CD34+ cells to determine if there was a selectivity toward CML CD34+ cells over normal SPC for the drug combination. Following combination treatment, apoptosis levels were only mildly, and not significantly, increased for the combination vs each single agent and this did not translate to a significant reduction in normal CFC output (Figure 3F-G). Overall, these data showed that the combination of NL and RUX was more effective than NL treatment alone in eradicating CML CD34+ cells, including CFC, in short term culture assays. These effects were observed only in the presence of GFs. Although a degree of toxicity toward mature normal progenitors was observed, this was less marked than for CML CD34+.
Figure 3.

Effects of the NL and RUX combination on CML and normal CD34+ cell viability and CFC output. (A) CML CD34+ cells (n = 3) were either left UT, or treated with NL (5 µM) or RUX (200 nM) or their combination, and cultured. At 48 hours, apoptosis levels were measured by Annexin-V/7AAD staining. CML CD34+ cells (n = 3) were cultured as in (A) for 72 hours before drug washout and plating in methylcellulose progenitor assays. (B) Total CFC output was recorded after 12 days culture and compared with the CFC output for each sample prior to the start of the culture (baseline) (note that the number of colonies following 72 hours culture was adjusted for the expansion of CD34+ cells in vitro in each arm relative to baseline). (C) CFC frequency based on their morphology (erythroid-burst forming unit and erythroid-colony forming unit), and (D) granulocyte/macrophage-colony forming unit was also recorded and again compared with baseline. (E) Representative pictures of the size and morphology of recovered CFC in each treatment arm. Normal CD34+ cells (n = 3) were either left UT, or treated with NL (5 µM) or RUX (200 nM) or their combination. (F) At 48 hours, apoptosis levels were measured by Annexin-V/7AAD staining. Normal CD34+ cells (n = 3) were cultured as in (A) for 72 hours before drug washout and plating in methylcellulose progenitor assays. (G) Total CFC output was recorded after 12 days culture and compared with the CFC output at baseline as explained in (B). All data from independent experiments are presented as mean ± SEM. Significance values: *P < .05; †P < .01; ‡P < .001.
The combination of NL and RUX increases the kill of human CML CD34+ CD38− and CFSEmax cells while sparing primitive normal CD34+ cells in vitro
In order to assess the effects of the NL and RUX combination on a more primitive population of CML and normal SPC, cell division tracking experiments using CFSE staining were performed. These confirmed a marked effect on CML CD34+ cell proliferation with the combination treatment, with a reduction in undivided CFSEmax, slowly proliferating (division 1), and the total number of CD34+ cells recovered (Figure 4A-B). These effects were paralleled by an increase in apoptosis levels within the CFSEmax population with the combination treatment compared with either single agent (Figure 4C). Conversely, similar experiments on normal CD34+ cells confirmed a slight antiproliferative effect for the combination treatment; however, in contrast to results for CML, there was no significant reduction in the total number of normal CD34+ cells recovered over 72 hours, and in particular, there was no significant reduction in the CFSEmax population (Figure 4D-E). Nor was there a significant increase in apoptosis levels within the CFSEmax population (Figure 4F). Finally, similar additive effects were also observed following NL and RUX combination treatment on sorted populations of CML (BCR-ABL+ by FISH) CD34+CD38− cells, which are known to be enriched for the most primitive, quiescent population39 (Figure 4G). Taken together, these data showed that NL and RUX in combination, caused a more profound effect on the quiescent stem-cell compartment compared with either agent alone while appearing to spare the normal quiescent fraction.
Figure 4.

Effects of the NL and RUX combination on primitive and quiescent (CFSEmax) CML and normal CD34+ cells. (A) CML and (B) normal CD34+ cells (n = 3) were stained with CFSE and either left UT, or treated with NL (5 µM) or RUX (200 nM) or their combination, and cultured. At 72 hours, the percentage of starting CD34+ cells recovered within each division in each treatment arm was calculated by recording the number of viable cells seeded initially in each culture and their number following different treatment conditions. Levels of CFSE fluorescence was used to measure the percentage of cells within each division as explained previously.32 (C) Percentage of total number of starting CML, and (D) normal CD34+ cells recovered in each arm following treatment was also recorded at 72 hours. Percentage of apoptotic cells within the undivided (CFSEmax) population was measured by gating on the population double-positive for maximal CFSE expression, and Annexin-V staining at 72 hours for both (E) CML and (F) normal CD34+ cells. (G) Sorted CML, BCR-ABL+ (by FISH) CD34+ CD38− cells (n = 3) were cultured as in (A) for 72 hours. Percentage of viable cells was measured by gating on the double-negative population following Annexin-V/DAPI staining. The results were then normalized against UT within each sample. All data from independent experiments are presented as mean ± SEM. Significance values: *P < .05; †P < .01; ‡P < .001.
The combination of NL and RUX selectively eliminates NSG repopulating CML cells
The effects of NL and RUX, alone and combined, were compared against primary CML and normal SPC in NSG transplantation assays (Figure 5A). CD34+ cells treated with NL, RUX, or the combination were injected into NSG mice and engraftment was evaluated. We chose this ex vivo treatment approach since CML CP cells demonstrate relatively poor engraftment and do not develop leukemia in NSG mice which limits the feasibility of in vivo drug treatment studies to evaluate the effects of treatment on leukemia development and survival. Following 72 hours drug exposure, reconstitution of human CD45+ hemopoiesis by CML CD34+ cells was significantly reduced by either RUX or NL alone; however, the combination of NL with RUX induced a more profound reduction of short-term (6 weeks) and long-term (18 weeks) engraftment of CML cells in NSG mice (supplemental Figure 4 and Figure 5B). In particular, both CD34+ cell engraftment and myeloid engraftment were significantly reduced by the combination treatment (Figure 5C). Moreover, qRT-PCR analysis showed that the reduction in human CD45+ cell engraftment correlated with a reduction of BCR-ABL levels in residual cells (Figure 5D). Similar experiments performed using CD34+ cells derived from normal human CB showed that both NL alone, and the NL with RUX combination, reduced the reconstitution of total human CD45+ hemopoietic cells (supplemental Figure 5). However, subset analysis confirmed that the reduction in engraftment was restricted to CD19+ B cells with no such effect on either CD34+ or CD33+ myeloid cell engraftment. The reduction in B-cell engraftment was driven by NL and not further affected by the addition of RUX (Figure 5E).
Figure 5.
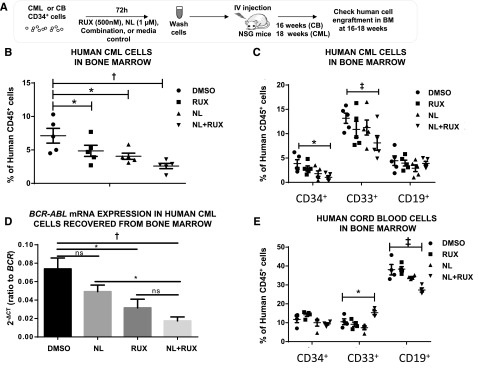
Effects of NL and RUX combination on the transplantable leukemogenic activity of CML CD34+ cells and on normal CB CD34+ cell engraftment. (A) Experimental design for the in vivo experiments. CML CD34+ cells (from 2 patients) or human CB CD34+ cells (from 2 subjects) were cultured for 72 hours with either NL (1 µM) or RUX (500 nM) or their combination, or media control. Following treatment, recovered cells were washed and injected intravenously into 8-week-old, sublethally irradiated (300 cGy) NSG mice (CML, 5 mice/per group; CB, 4 mice for controls; RUX, 4; NL, 5; and NL+RUX, 5). (B) The levels of human CML CD45+ cells, and (C) CD34+, CD33+, and CD19+ cells regenerated in the BM of mice transplanted with cells treated in different conditions were measured at 18 weeks. (D) The levels of BCR-ABL mRNA within the human CML CD45+ selected cells obtained from mouse BM at 18 weeks were evaluated by qRT-PCR and normalized to BCR mRNA expression levels. (E) The levels of human CB CD34+, CD33+, and CD19+ cells regenerated in the BM of mice transplanted with cells treated in different conditions were measured at 16 weeks. All data from independent experiments are presented as mean ± SEM. Significance values: *P < .05; †P < .01; ‡P < .001.
Overall, these results showed that the combination of NL and RUX reduced the transplantable leukemogenic activity of CML CD34+ cells more effectively than either agent alone, while having no significant effects on normal human CD34+ and myeloid cell engraftment.
Discussion
The development of JAK2 inhibitors available for clinical investigation has opened up an exciting potential avenue to explore in CML, however, further clarification of the role of JAK2 in this disease is required before pursuing it as a legitimate therapeutic target. In this study, we have provided preclinical evidence for effective targeting of JAK2 in CML CD34+ cells using the inhibitor RUX, by showing a consistent reduction in viability, colony output, and proliferation of CML CD34+ cells, including the quiescent fraction, in vitro and reduced engraftment of CML CD34+ cells in vivo, when treated with NL and RUX compared with NL treatment alone. Interestingly, the effects of RUX as a single agent were modest, but became more dramatic when combined with NL, suggesting that the role of JAK2 is particularly prominent during TKI therapy and that CML CD34+ cells become more reliant on JAK2 kinase when BCR-ABL is fully inhibited. These findings are consistent with those published recently by other research groups, mainly using nonclinically developed JAK2 inhibitors,40-43 but are contrary to those reported by Hantschel et al, showing that BCR-ABL is still able to transform murine BM cells in which JAK2 has been deleted, both in vitro and in vivo.27 Moreover, a recent report suggested that JAK2 deletion might accelerate CML development in mouse models by preferentially causing elimination of normal HSC which are more dependent on JAK2 signaling compared with CML LSC where BCR-ABL is active.13 This discrepancy might be explained by the fact that the role of JAK2 may indeed be dispensable when BCR-ABL is fully active, but becomes critical when BCR-ABL kinase is inhibited as has also been shown for other potential targets.37 Only upon inhibition of BCR-ABL do these targets become relevant, thus explaining the need to continue TKI together with any novel therapeutic agent to eradicate CML LSC, despite the evidence that CML LSC are not BCR-ABL kinase dependent for their survival.9,10 Moreover, in the report by Hantschel et al, a different model was employed (BCR-ABL transduced murine BM cells vs primary CP CML cells, respectively, with the former likely expressing higher levels of BCR-ABL compared with the latter), and most of their in vitro work was performed in the absence of exogenous GFs. We also noted that when RUX and NL were combined in CML cells grown in the absence of GFs, the additional effects of the JAK2 inhibitor were abolished, suggesting that one of the main roles of JAK2, independent of BCR-ABL kinase and in the presence of NL, is to relay survival signals from exogenous GFs which can be effectively inhibited by RUX, as also previously reported.40-43 This further helps to resolve the discrepancy between our findings and those of Hantschel et al. Our data therefore, suggested that in the absence of GFs, JAK2 signals were either absent or under the direct control of BCR-ABL, hence completely abrogated by the high doses of NL used in our experiments. Based on our data, one of the putative mechanisms of action of the NL and RUX combination in the presence of exogenous GFs was a more profound inhibition of JAK2/STAT5 activity, as shown by the correlative changes in both p-JAK2 and p-STAT5 levels, with combined treatment, associated with correlative changes in STAT5 target genes. This was not due to off-target inhibition of BCR-ABL kinase by RUX, as has been shown for other JAK2 inhibitors,27 and we were careful to use RUX at concentrations well below those previously shown to potentially cause off-target inhibition of BCR-ABL kinase (ie, more than 20 μM).27 Moreover, achieving further inhibition of BCR-ABL kinase activity by adding RUX was highly unlikely in our experimental setting as the NL concentration used was shown to achieve maximal BCR-ABL kinase inhibition. In summary, we conclude that enhanced inhibition of STAT5 activity by the NL and RUX combination was one of the major mechanisms underlying the effects seen.
However, alternative modes of action for the NL and RUX combination, independent of STAT5 activation, are possible. Although we deliberately chose NL for this study as the most specific BCR-ABL kinase inhibitor available, we cannot exclude that additional NL targets are important and required for the observed additive effects. Similarly, JAK2 has also been shown to have other targets than STAT5. It directly phosphorylates BCR-ABL on its key residue tyrosine 17720 (which is central for its transforming capacity through its ability to activate both the RAS/MAP kinase and the PI3 kinase pathways44-46), and functions as a histone kinase and chromatin modifier47 while also being able to activate MYC48 and β-catenin.49 This last effect is particularly relevant given the central role of β-catenin in CML LSC self-renewal50-52 and might explain the effects of RUX on the most primitive population, including CFSEmax, CD34+ CD38−, and NSG repopulating cells, which support those already reported by others using different, not clinically developed, JAK2 inhibitors.49 Finally, it should also be considered that RUX is a potent inhibitor of JAK1 and other oncogenic kinases at the concentrations used,28 and therefore, some of the effects seen could be mediated independently of JAK2.28 In this respect, other reports on the efficacy of RUX (and other JAK inhibitors) in BCR-ABL positive cells have shown inhibition of STAT3 activity (a downstream target of JAK1/2) as a possible mechanism-of-action for these agents.41,42 All the above might explain why the effects seen in functional assays are greater than the ones seen on the JAK2/STAT5 pathway when using RUX alone. Future work will clarify if these or other unknown mechanisms-of-action of JAK2 are present in CML SPC, which might account for the effects seen here in functional assays.
One of the main concerns for the use of JAK2 inhibitors in CML is their potential toxicity to normal BM, following a report showing toxicity of several JAK2 inhibitors toward normal hemopoiesis.41 Despite its apparently narrow therapeutic window, our in vitro and in vivo data reassuringly showed that a strategy combining RUX and NL preferentially eradicated CML compared with normal CD34+ cells, when a carefully selected concentration of RUX was chosen. The reason behind the increased sensitivity of TKI-treated CML vs normal SPCs to JAK2 inhibition is currently unclear. We speculate that in CML SPCs, the prior expression of BCR-ABL may alter signaling networks such that cells are dependent on higher levels of GFs signaling activity, possibly due to upregulation or enhanced activity of cytokine receptors as shown already in both CML53,54 and acute myeloid leukemia models.55 Upon BCR-ABL inhibition, these cells may therefore be dependent on higher levels of GF-mediated JAK2/STAT5 activation for continued viability as compared with normal cells. Moreover, it is reassuring that RUX has been used as a single agent for the treatment of patients with myelofibrosis,30,31 with no severe BM toxicity experienced, especially for patients with good BM reserve, which is the prevailing situation in CP CML patients with minimal residual disease on TKI. This suggests that the BM toxicity secondary to JAK2 inhibition in adults might be less than that observed during embryonic development in JAK2 KO mice.12-14 This is to be expected given that chemical inhibition is less complete than gene deletion and evidence that JAK2 scaffolding functions, which would not be targeted by a JAK2 kinase inhibitor, might be important for its activity.56 In conclusion, our work supports a role for JAK2 in primary CML SPC survival, and provides further preclinical evidence for studies combining TKI with RUX in CML patients. We also provide evidence of a role for JAK2 in activating STAT5-dependent survival signals in CML CD34+ cells, upon BCR-ABL kinase inhibition and in the presence of GFs, thus explaining the observed efficacy of JAK2 inhibitors. We, therefore, believe that in CML patients with minimal residual disease who have optimally responded to TKI therapy, additional therapeutic strategies targeting JAK2, in combination with TKI, warrant investigation in an attempt to achieve disease eradication.
Acknowledgments
The authors thank all CML patients and normal BM donors and United Kingdom (UK) hematology departments who contributed samples, Karen Stewart for collecting clinical information on patients, Dr Alan Hair for sample processing, Dr Laura Park for performing the FISH, and Jennifer Cassels for cell sorting.
This study was supported by the Glasgow Experimental Cancer Medicine Centre, which is funded by Cancer Research UK and the Chief Scientist’s Office, Scotland. Cell sorting facilities were funded by the Kay Kendall Leukaemia Fund (KKL501) and the Howat Foundation. P.G. was funded by a Medical Research Council UK clinical research training fellowship grant (G1000288), S.R. and H.W. were supported by the Chief Scientist’s Office, Scotland grant (CAF/13/09), A.D.W. was funded by Leukemia and Lymphoma Research Program grant (13005), H.G.J. was funded by the Friends of Paul O’Gorman Leukemia Research Centre, R.B. was funded by the National Institutes of Health, National Cancer Institute grants (R01 CA095684 and R01 CA172447), and L.H. and T.L.H. were supported by Cancer Research UK Programme grant (C11074/A11008).
Footnotes
The online version of this article contains a data supplement.
There is an Inside Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: P.G. designed and performed research, analyzed and interpreted data, and wrote the manuscript; A.C., S.R., H.G.J., L.H., and H.W. performed research, analyzed data, and reviewed the manuscript; and A.D.W., H.G.J., R.B., and T.L.H. designed research, interpreted data, and reviewed the manuscript.
Conflict-of-interest disclosure: P.G. has previously received travel grants from Bristol-Myers Squibb; R.B. has previously served in advisory boards and received honoraria from Novartis, Bristol-Myers Squibb, Pfizer, and Teva; and T.L.H. has previously received research funding from Novartis and Bristol-Myers Squibb. The remaining authors declare no competing financial interests.
The current affiliation for P.G. is the Cambridge Institute for Medical Research, University of Cambridge, Cambridge, United Kingdom.
Correspondence: Tessa L. Holyoake, Paul O’Gorman Leukaemia Research Centre, College of Medical, Veterinary & Life Sciences, Institute of Cancer Sciences, University of Glasgow, 21 Shelley Rd, Glasgow G12 0ZD, Scotland, United Kingdom; e-mail: tessa.holyoake@glasgow.ac.uk.
References
- 1.Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5(3):172–183. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- 2.Druker BJ, Guilhot F, O’Brien SG, et al. IRIS Investigators. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 3.Cortes J, Hochhaus A, Hughes T, Kantarjian H. Front-line and salvage therapies with tyrosine kinase inhibitors and other treatments in chronic myeloid leukemia. J Clin Oncol. 2011;29(5):524–531. doi: 10.1200/JCO.2010.31.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chomel JC, Bonnet ML, Sorel N, et al. Leukemic stem cell persistence in chronic myeloid leukemia patients with sustained undetectable molecular residual disease. Blood. 2011;118(13):3657–3660. doi: 10.1182/blood-2011-02-335497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chu S, McDonald T, Lin A, et al. Persistence of leukemia stem cells in chronic myelogenous leukemia patients in prolonged remission with imatinib treatment. Blood. 2011;118(20):5565–5572. doi: 10.1182/blood-2010-12-327437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ross DM, Branford S, Seymour JF, et al. Patients with chronic myeloid leukemia who maintain a complete molecular response after stopping imatinib treatment have evidence of persistent leukemia by DNA PCR. Leukemia. 2010;24(10):1719–1724. doi: 10.1038/leu.2010.185. [DOI] [PubMed] [Google Scholar]
- 7.Sobrinho-Simões M, Wilczek V, Score J, Cross NC, Apperley JF, Melo JV. In search of the original leukemic clone in chronic myeloid leukemia patients in complete molecular remission after stem cell transplantation or imatinib. Blood. 2010;116(8):1329–1335. doi: 10.1182/blood-2009-11-255109. [DOI] [PubMed] [Google Scholar]
- 8.Mahon FX, Réa D, Guilhot J, et al. Intergroupe Français des Leucémies Myéloïdes Chroniques. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11(11):1029–1035. doi: 10.1016/S1470-2045(10)70233-3. [DOI] [PubMed] [Google Scholar]
- 9.Hamilton A, Helgason GV, Schemionek M, et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood. 2012;119(6):1501–1510. doi: 10.1182/blood-2010-12-326843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121(1):396–409. doi: 10.1172/JCI35721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ward AC, Touw I, Yoshimura A. The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood. 2000;95(1):19–29. [PubMed] [Google Scholar]
- 12.Neubauer H, Cumano A, Müller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998;93(3):397–409. doi: 10.1016/s0092-8674(00)81168-x. [DOI] [PubMed] [Google Scholar]
- 13.Grundschober E, Hoelbl-Kovacic A, Bhagwat N, et al. Acceleration of Bcr-Abl(+) leukemia induced by deletion of JAK2. [published online ahead of print May 5, 2014]. Leukemia. doi: 10.1038/leu.2014.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park SO, Wamsley HL, Bae K, et al. Conditional deletion of Jak2 reveals an essential role in hematopoiesis throughout mouse ontogeny: implications for Jak2 inhibition in humans. PLoS ONE. 2013;8(3):e59675. doi: 10.1371/journal.pone.0059675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chai SK, Nichols GL, Rothman P. Constitutive activation of JAKs and STATs in BCR-Abl-expressing cell lines and peripheral blood cells derived from leukemic patients. J Immunol. 1997;159(10):4720–4728. [PubMed] [Google Scholar]
- 16.Carlesso N, Frank DA, Griffin JD. Tyrosyl phosphorylation and DNA binding activity of signal transducers and activators of transcription (STAT) proteins in hematopoietic cell lines transformed by Bcr/Abl. J Exp Med. 1996;183(3):811–820. doi: 10.1084/jem.183.3.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie S, Wang Y, Liu J, et al. Involvement of Jak2 tyrosine phosphorylation in Bcr-Abl transformation. Oncogene. 2001;20(43):6188–6195. doi: 10.1038/sj.onc.1204834. [DOI] [PubMed] [Google Scholar]
- 18.Chen M, Gallipoli P, DeGeer D, et al. Targeting primitive chronic myeloid leukemia cells by effective inhibition of a new AHI-1-BCR-ABL-JAK2 complex. J Natl Cancer Inst. 2013;105(6):405–423. doi: 10.1093/jnci/djt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Samanta AK, Lin H, Sun T, Kantarjian H, Arlinghaus RB. Janus kinase 2: a critical target in chronic myelogenous leukemia. Cancer Res. 2006;66(13):6468–6472. doi: 10.1158/0008-5472.CAN-06-0025. [DOI] [PubMed] [Google Scholar]
- 20.Samanta A, Perazzona B, Chakraborty S, et al. Janus kinase 2 regulates Bcr-Abl signaling in chronic myeloid leukemia. Leukemia. 2011;25(3):463–472. doi: 10.1038/leu.2010.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoelbl A, Schuster C, Kovacic B, et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol Med. 2010;2(3):98–110. doi: 10.1002/emmm.201000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walz C, Ahmed W, Lazarides K, et al. Essential role for Stat5a/b in myeloproliferative neoplasms induced by BCR-ABL1 and JAK2(V617F) in mice. Blood. 2012;119(15):3550–3560. doi: 10.1182/blood-2011-12-397554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye D, Wolff N, Li L, Zhang S, Ilaria RL., Jr STAT5 signaling is required for the efficient induction and maintenance of CML in mice. Blood. 2006;107(12):4917–4925. doi: 10.1182/blood-2005-10-4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Warsch W, Kollmann K, Eckelhart E, et al. High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood. 2011;117(12):3409–3420. doi: 10.1182/blood-2009-10-248211. [DOI] [PubMed] [Google Scholar]
- 25.Nelson EA, Walker SR, Weisberg E, et al. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood. 2011;117(12):3421–3429. doi: 10.1182/blood-2009-11-255232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ilaria RL, Jr, Van Etten RA. P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J Biol Chem. 1996;271(49):31704–31710. doi: 10.1074/jbc.271.49.31704. [DOI] [PubMed] [Google Scholar]
- 27.Hantschel O, Warsch W, Eckelhart E, et al. BCR-ABL uncouples canonical JAK2-STAT5 signaling in chronic myeloid leukemia. Nat Chem Biol. 2012;8(3):285–293. doi: 10.1038/nchembio.775. [DOI] [PubMed] [Google Scholar]
- 28.Zhou T, Georgeon S, Moser R, Moore DJ, Caflisch A, Hantschel O. Specificity and mechanism-of-action of the JAK2 tyrosine kinase inhibitors ruxolitinib and SAR302503 (TG101348). Leukemia. 2014;28(2):404–407. doi: 10.1038/leu.2013.205. [DOI] [PubMed] [Google Scholar]
- 29.Quintás-Cardama A, Vaddi K, Liu P, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115(15):3109–3117. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. doi: 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. doi: 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- 32.Copland M, Hamilton A, Elrick LJ, et al. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood. 2006;107(11):4532–4539. doi: 10.1182/blood-2005-07-2947. [DOI] [PubMed] [Google Scholar]
- 33.Bhatia R, McGlave PB, Dewald GW, Blazar BR, Verfaillie CM. Abnormal function of the bone marrow microenvironment in chronic myelogenous leukemia: role of malignant stromal macrophages. Blood. 1995;85(12):3636–3645. [PubMed] [Google Scholar]
- 34.Wood AD, Chen E, Donaldson IJ, et al. ID1 promotes expansion and survival of primary erythroid cells and is a target of JAK2V617F-STAT5 signaling. Blood. 2009;114(9):1820–1830. doi: 10.1182/blood-2009-02-206573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jankovic V, Ciarrocchi A, Boccuni P, DeBlasio T, Benezra R, Nimer SD. Id1 restrains myeloid commitment, maintaining the self-renewal capacity of hematopoietic stem cells. Proc Natl Acad Sci USA. 2007;104(4):1260–1265. doi: 10.1073/pnas.0607894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perry SS, Zhao Y, Nie L, Cochrane SW, Huang Z, Sun XH. Id1, but not Id3, directs long-term repopulating hematopoietic stem-cell maintenance. Blood. 2007;110(7):2351–2360. doi: 10.1182/blood-2007-01-069914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hurtz C, Hatzi K, Cerchietti L, et al. BCL6-mediated repression of p53 is critical for leukemia stem cell survival in chronic myeloid leukemia. J Exp Med. 2011;208(11):2163–2174. doi: 10.1084/jem.20110304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shilling AD, Nedza FM, Emm T, et al. Metabolism, excretion, and pharmacokinetics of [14C]INCB018424, a selective Janus tyrosine kinase 1/2 inhibitor, in humans. Drug Metab Dispos. 2010;38(11):2023–2031. doi: 10.1124/dmd.110.033787. [DOI] [PubMed] [Google Scholar]
- 39.Holyoake TL, Jiang X, Jorgensen HG, et al. Primitive quiescent leukemic cells from patients with chronic myeloid leukemia spontaneously initiate factor-independent growth in vitro in association with up-regulation of expression of interleukin-3. Blood. 2001;97(3):720–728. doi: 10.1182/blood.v97.3.720. [DOI] [PubMed] [Google Scholar]
- 40.Hiwase DK, White DL, Powell JA, et al. Blocking cytokine signaling along with intense Bcr-Abl kinase inhibition induces apoptosis in primary CML progenitors. Leukemia. 2010;24(4):771–778. doi: 10.1038/leu.2009.299. [DOI] [PubMed] [Google Scholar]
- 41.Traer E, MacKenzie R, Snead J, et al. Blockade of JAK2-mediated extrinsic survival signals restores sensitivity of CML cells to ABL inhibitors. Leukemia. 2012;26(5):1140–1143. doi: 10.1038/leu.2011.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nair RR, Tolentino JH, Argilagos RF, Zhang L, Pinilla-Ibarz J, Hazlehurst LA. Potentiation of Nilotinib-mediated cell death in the context of the bone marrow microenvironment requires a promiscuous JAK inhibitor in CML. Leuk Res. 2012;36(6):756–763. doi: 10.1016/j.leukres.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quintarelli C, De Angelis B, Errichiello S, et al. Selective strong synergism of Ruxolitinib and second generation tyrosine kinase inhibitors to overcome bone marrow stroma related drug resistance in chronic myelogenous leukemia. Leuk Res. 2014;38(2):236–242. doi: 10.1016/j.leukres.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 44.He Y, Wertheim JA, Xu L, et al. The coiled-coil domain and Tyr177 of bcr are required to induce a murine chronic myelogenous leukemia-like disease by bcr/abl. Blood. 2002;99(8):2957–2968. doi: 10.1182/blood.v99.8.2957. [DOI] [PubMed] [Google Scholar]
- 45.Million RP, Van Etten RA. The Grb2 binding site is required for the induction of chronic myeloid leukemia-like disease in mice by the Bcr/Abl tyrosine kinase. Blood. 2000;96(2):664–670. [PubMed] [Google Scholar]
- 46.Zhang X, Subrahmanyam R, Wong R, Gross AW, Ren R. The NH(2)-terminal coiled-coil domain and tyrosine 177 play important roles in induction of a myeloproliferative disease in mice by Bcr-Abl. Mol Cell Biol. 2001;21(3):840–853. doi: 10.1128/MCB.21.3.840-853.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dawson MA, Bannister AJ, Göttgens B, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461(7265):819–822. doi: 10.1038/nature08448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xie S, Lin H, Sun T, Arlinghaus RB. Jak2 is involved in c-Myc induction by Bcr-Abl. Oncogene. 2002;21(47):7137–7146. doi: 10.1038/sj.onc.1205942. [DOI] [PubMed] [Google Scholar]
- 49.Neviani P, Harb JG, Oaks JJ, et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J Clin Invest. 2013;123(10):4144–4157. doi: 10.1172/JCI68951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heidel FH, Bullinger L, Feng Z, et al. Genetic and pharmacologic inhibition of β-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell. 2012;10(4):412–424. doi: 10.1016/j.stem.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao C, Blum J, Chen A, et al. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007;12(6):528–541. doi: 10.1016/j.ccr.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu Y, Chen Y, Douglas L, Li S. beta-Catenin is essential for survival of leukemic stem cells insensitive to kinase inhibition in mice with BCR-ABL-induced chronic myeloid leukemia. Leukemia. 2009;23(1):109–116. doi: 10.1038/leu.2008.262. [DOI] [PubMed] [Google Scholar]
- 53.Gallipoli P, Pellicano F, Morrison H, et al. Autocrine TNF-α production supports CML stem and progenitor cell survival and enhances their proliferation. Blood. 2013;122(19):3335–3339. doi: 10.1182/blood-2013-02-485607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kobayashi CI, Takubo K, Kobayashi H, et al. The IL-2/CD25 axis maintains distinct subsets of chronic myeloid leukemia-initiating cells. Blood. 2014;123(16):2540–2549. doi: 10.1182/blood-2013-07-517847. [DOI] [PubMed] [Google Scholar]
- 55.Cook AM, Li L, Ho Y, et al. Role of altered growth factor receptor-mediated JAK2 signaling in growth and maintenance of human acute myeloid leukemia stem cells. Blood. 2014;123(18):2826–2837. doi: 10.1182/blood-2013-05-505735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keil E, Finkenstädt D, Wufka C, et al. Important scaffold function of the Janus kinase 2 uncovered by a novel mouse model harboring a Jak2 activation-loop mutation. Blood. 2014;123(4):520–529. doi: 10.1182/blood-2013-03-492157. [DOI] [PubMed] [Google Scholar]