

Abstract
Here, we report a novel method to produce microencapsulated enzymes using Saccharomyces cerevisiae spores. In sporulating cells, soluble secreted proteins are transported to the spore wall. Previous work has shown that the spore wall is capable of retaining soluble proteins because its outer layers work as a diffusion barrier. Accordingly, a red fluorescent protein (RFP) fusion of the α-galactosidase, Mel1, expressed in spores was observed in the spore wall even after spores were subjected to a high-salt wash in the presence of detergent. In vegetative cells, however, the cell wall cannot retain the RFP fusion. Although the spore wall prevents diffusion of proteins, it is likely that smaller molecules, such as sugars, pass through it. In fact, spores can contain much higher α-galactosidase activity to digest melibiose than vegetative cells. When present in the spore wall, the enzyme acquires resistance to environmental stresses including enzymatic digestion and high temperatures. The outer layers of the spore wall are required to retain enzymes but also decrease accessibility of the substrates. However, mutants with mild spore wall defects can retain and stabilize the enzyme while still permitting access to the substrate. In addition to Mel1, we also show that spores can retain the invertase. Interestingly the encapsulated invertase has significantly lower activity toward raffinose than toward sucrose. This suggests that substrate selectivity could be altered by the encapsulation.
INTRODUCTION
When diploid cells of the budding yeast Saccharomyces cerevisiae are incubated in the absence of nitrogen and the presence of a nonfermentable carbon source, they cease vegetative growth and enter the sporulation program (1). This process includes meiosis and spore morphogenesis so that each nucleus produced by meiosis is eventually incorporated into an individual spore. As a result, four spores are created inside the mother cell, and the mother cell plasma membrane becomes the ascal membrane. Thus, during sporulation the spore plasma membrane and spore wall are formed de novo in the cytoplasm of the mother cell (2). To accomplish this dynamic morphological change, sporulation requires a reorganization of the intracellular vesicular trafficking pathway to produce double membranes, called prospore membranes, around each nucleus (3). After the haploid nucleus is completely engulfed by the prospore membrane, deposition of spore wall materials is started inside the double membrane (4). The mature spore wall has a multilaminar structure consisting of, from the inside to the outside, mannoprotein, β-glucan, chitosan, and dityrosine layers (5, 6). Spore wall materials are deposited in a sequential manner. For example, the dityrosine layer is only synthesized after the chitosan layer is formed, and in mutants that have defects in chitosan layer formation, the dityrosine layer is not deposited (7, 8). Although the prospore membrane is initially formed as a double membrane, after enclosure of the nucleus, it is resolved into two distinct bilayers, the spore plasma membrane and an outer membrane. During the course of spore wall assembly, this outer membrane lyses by an unknown mechanism before the chitosan layer is created (9).
The chitosan and dityrosine layers are unique structures of the spore wall, whereas β-glucan and mannoprotein are common components of both spore and vegetative cell walls (10). In sporulating cells, chitosan is synthesized in two steps: chitin is first synthesized by the chitin synthase, Chs3 (7), and then it is deacetylated by sporulation-specific chitin deacetylases (11, 12). Synthesis and transport of dityrosine molecules are mediated by Dit1, Dit2, and Dtr1 (13–15). DIT1 is involved in the first step to produce dityrosine from l-tyrosine in the cytosol, and its deletion causes a lack of the dityrosine layer (16). Because of these outer two layers, especially the dityrosine layer, yeast spores are resistant to various environmental stresses, such as digestive enzymes, heat, and ether (1). To fully acquire resistance, however, it seems that deposition of dityrosine is not enough. For example, osw2Δ spores show ether sensitivity in spite of the presence of the dityrosine layer (9). Thus, Osw2 is required to properly organize the spore wall although the molecular basis for this is totally unknown.
S. cerevisiae has been extensively used as a host to produce biologically active proteins (17). If soluble proteins with secretory signal peptides are expressed in yeast cells, they are secreted out of the cell and collected from the growth medium. However, in sporulating cells, because the secretory pathway is reorganized to produce prospore membranes, secretory proteins are transported to the prospore membrane and then retained in the spore wall (18, 19). Although the outer membrane of the prospore membrane disappears during spore wall maturation, mature spores are still capable of retaining soluble proteins in their walls. This is due to the presence of the dityrosine layer. In fact, a previous report has shown that a secretory form of green fluorescent protein (GFP) expressed in wild-type spores is retained in the spore wall but not in dit1Δ spores (18). Thus, in the spore wall, the dityrosine layer works as a barrier to prevent diffusion of soluble proteins.
If secretory forms of soluble enzymes are expressed in sporulating cells, they should be entrapped in the spore wall. Although the dityrosine layer prevents diffusion of the proteins, since yeast germination is implemented by incorporation of glucose (20), smaller molecules should pass through it. Thus, in this study, we assessed whether spores can be used as microcapsules to incorporate soluble enzymes.
MATERIALS AND METHODS
Yeast strains and growth media.
Unless otherwise noted, standard media and genetic techniques were used (21). Yeast strains and oligonucleotide primers used in this study are listed in Tables 1 and 2, respectively. All strains used in this study are in the fast-sporulating SK-1 strain background. To disrupt DIT1, a DNA fragment was amplified by PCR using pFA6a-HIS3MX6 (22) as the template and HXO34 and HXO35 as the primers. The PCR fragment was integrated into haploid AN117-4B and AN117-16D cells, and the resulting strains were mated to generate the diploid dit1Δ disruptant. Other mutants were constructed in the same way. For deletion of OSW2 or SUC2, either HXO183 and HXO184 or LBO1 and LBO2 were used as the primers to generate PCR cassettes, respectively. pFA6a-HIS3MX6 was used as a template to delete OSW2. pFA6a-TRP1 (22) was used as a template to delete SUC2. suc2Δ osw2Δ and suc2Δ dit1Δ double mutants were constructed based on the suc2Δ haploid cells.
TABLE 1.
S. cerevisiae strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| AN117-4B | MATα ura3 leu2 trp1 his3Δsk arg4-NspI lys2 ho::LYS2 rme1::LEU2 | 27 |
| AN117-16D | MATa ura3 leu2 trp1 his3Δsk lys2 ho::LYS2 | 27 |
| AN120 | MATα/MATa ARG4/arg4-NspI his3ΔSK/his3ΔSK ho::LYS2/ho::LYS2 leu2/leu2 lys2/lys2 RME1/rme1::LEU2 trp1::hisG/trp1::hisG ura3/ura3 | 27 |
| AN262 (chs3Δ) | MATα/MATa ARG4/arg4-NspI his3ΔSK/his3ΔSK ho::LYS2/ho::LYS2 leu2/leu2 lys2/lys2 RME1/rme1::LEU2 trp1::hisG/trp1::hisG ura3/ura3 chs3Δ::his5+/chs3Δ::his5+ | 39 |
| HW3 (dit1Δ) | MATα/MATa ARG4/arg4-NspI his3ΔSK/his3ΔSK ho::LYS2/ho::LYS2 leu2/leu2 lys2/lys2 RME1/rme1::LEU2 trp1::hisG/trp1::hisG ura3/ura3 dit1Δ::his5+/dit1Δ::his5+ | H. Zhang, H. Tachikawa, X.-D. Gao, and H. Nakanishi, submitted for publication |
| HW83 (osw2Δ) | MATα/MATa ARG4/arg4-NspI his3ΔSK/his3ΔSK ho::LYS2/ho::LYS2 leu2/leu2 lys2/lys2 RME1/rme1::LEU2 trp1::hisG/trp1::hisG ura3/ura3 osw2Δ::his5+/osw2Δ::his5+ | This study |
| HS4 (suc2Δ) | MATα/MATa ARG4/arg4-NspI his3ΔSK/his3ΔSK ho::LYS2/ho::LYS2 leu2/leu2 lys2/lys2 RME1/rme1::LEU2 trp1::hisG/trp1::hisG ura3/ura3 suc2Δ::TRP1/suc2Δ::TRP1 | This study |
| HS5 (suc2Δ osw2Δ) | MATα/MATa ARG4/arg4-NspI his3ΔSK/his3ΔSK ho::LYS2/ho::LYS2 leu2/leu2 lys2/lys2 RME1/rme1::LEU2 trp1::hisG/trp1::hisG ura3/ura3 suc2Δ::TRP1/suc2Δ::TRP1 osw2Δ::his5+/osw2Δ::his5+ | This study |
| HS6 (suc2Δ dit1Δ) | MATα/MATa ARG4/arg4-NspI his3ΔSK/his3ΔSK ho::LYS2/ho::LYS2 leu2/leu2 lys2/lys2 RME1/rme1::LEU2 trp1::hisG/trp1::hisG ura3/ura3 suc2Δ::TRP1/suc2Δ::TRP1 dit1Δ::his5+/dit1Δ::his5+ | This study |
TABLE 2.
Oligonucleotide primers used in this study
| Name | Sequence |
|---|---|
| HXO34 | AATTTGTTAATATCCTAATTCGGTAAAGCTTTGTCGAGACATTAACAAAACGGATCCCCGGGTTAATTAA |
| HXO35 | TGTTTAAGTAAAAGAACAAAAAGGTAGACCAATGTAGCGCTCTTACTTTAGAATTCGAGCTCGTTTAAAC |
| HXO43 | GTGCGAGCTCCGAAGGTGACGTAGCAATCC |
| HXO44 | TGAATCTAGATATATATCTAAAAATGGCTA |
| HXO153 | TAAAACTAGTAAGTAAATGGTTTCGTTCAG |
| HXO170 | GTGGCTCGAGTCAGCACTGAGCAGCGTAAT |
| HXO183 | TATTCCTAAGCCTTTCTTTCTTTTTTTGAAGGCAAGAACTCGCATTAGTTCGGATCCCCGGGTTAATTAA |
| HXO184 | AATTTTGCGCATCCCACCCCTTATTAACAATCACATTTTTTTTTTTAATAGAATTCGAGCTCGTTTAAAC |
| HXO412 | TTCTCCTTGATCAGCTCAGAGGAAACAGGATTACAGTTTA |
| HXO446 | GTGTACTAGTATGTTTGCTTTCTACTTTCT |
| HXO447 | GTGTCTCGAGTCAAGAAGAGGGTCTCAACC |
| HXO486 | ATAGGTTGAGACCCTCTTCTGCTGGTGCTGGTTACCCATACGATGTTCCTGA |
| LBO1 | CTCAGAGAAACAAGCAAAACAAAAAGCTTTTCTTTTCACTAACGTATATGCGGATCCCCGGGTTAATTAA |
| LBO2 | CTTTAGAATGGCTTTTGAAAAAAATAAAAAGACAATAAGTTTTATAACCTGAATTCGAGCTCGTTTAAAC |
| LBO7 | TGCCGAATTCTCTGAGCTGATCAAGGAGAA |
| LBO8 | AACGCTCGAGAATTCCTTTGTCATCGTCAT |
| LBO9 | TGCCGAATTCAGAAGAGGGTCTCAACCTAT |
Plasmids.
Plasmids used in this study are listed in Table 3. The α-galactosidase expression vector, pRS424TEF-MEL1, was constructed as follows. First, the MEL1 gene was amplified by PCR using the primers HXO446 and HXO447. Yeast genomic DNA obtained from strain Y187, which was included in Matchmaker gold yeast two-hybrid system (Clontech, Mountain View, CA), was used as the template. The PCR fragment was digested with SpeI and XhoI and cloned into similarly digested pRS424TEF (23). pRS424TEF-MEL1-RFP was used to express a monomeric red fluorescent protein (mRFP) fusion to Mel1. To construct this, MEL1 without the stop codon was amplified by using the primers HXO446 and LBO9 and Y187 genomic DNA as the template. After digestion with SpeI and EcoRI, the PCR fragment was ligated into similarly digested pRS424TEF. Then, mRFP without the start codon was ligated into EcoRI/XhoI sites of the resulting plasmid. The mRFP fragment was amplified by PCR using LBO7 and LBO8 as the primers and TOP10-mRFP as a template. TOP10-mRFP was a gift from J. Voglmeir (Nanjing Agricultural University, Nanjing, China). pRS424TEF-spRFP was used to express mRFP fused to the signal peptide from Spr1. To construct this plasmid, first the SPR1 signal sequence (first 24 amino acids) was amplified by PCR using HXO153 and HXO412 as the primers and AN120 genomic DNA as the template. Then, using the resulting PCR fragment and LBO8 as the primers and TOP10-mRFP as a template, PCR was performed. The fragment was digested with SpeI and XhoI and cloned into similarly digested pRS424TEF. YEp352GAP-II-SUC2 was used to express SUC2 from the TDH2 promoter. In this plasmid SUC2 is cloned into EcoRI/SacI sites of YEp352GAP-II (24). pRS426-SPO20pr-SUC2 was used to express SUC2 from the SPO20 promoter. To construct this plasmid, first the promoter region of SPO20 was amplified by PCR using the primers HXO43 and HXO44 and AN120 genomic DNA as the template. The PCR fragment digested by SacI and XbaI was cloned into the similarly digested pRS426 (25). The resulting plasmid was digested with EcoRI and SalI, and the EcoRI/SalI fragment from YEp352GAP-II-SUC2 was ligated. pRS424TEF-MEL1-3HA was used to express Mel1 with three copies of a hemagglutinin (3×HA) tag. To construct this plasmid, first the 3×HA gene was amplified by PCR using HXO486 and HXO170 as the primers and pFA6a-3HA-His3MX6 as a template. The MEL1-3×HA fragment was then amplified by using the 3×HA PCR fragment and HXO446 as the primers and pRS424TEF-MEL1 as a template. The resulting fragment was digested by SpeI and XhoI and cloned into similarly digested pRS424TEF.
TABLE 3.
Plasmids used in this studya
| Name | Description |
|---|---|
| pRS424TEF-MEL1 | TRP1 2μ, for expression of Mel1 from constitutive TEF2 promoter |
| pRS424TEF-MEL1-RFP | TRP1 2μ, for expression of Mel1-RFP from constitutive TEF2 promoter |
| pRS424TEF-spRFP | TRP1 2μ, for expression of mRFP fused to the signal peptide from constitutive TEF2 promoter |
| pRS426-SPO20pr-SUC2 | URA3 2μ, for expression of Suc2 from sporulation specific SPO20 promoter |
| YEp352GAP-II-SUC2 | URA3 2μ, for expression of Suc2 from constitutive TDH2 promoter |
| pRS424TEF-MEL1-3HA | TRP1 2μ, for expression of Mel1-3×HA from constitutive TEF2 promoter |
All plasmids originated from this study.
Yeast culture, sporulation, and spore purification.
Yeast spores and vegetative cells were prepared as follows. First, yeast cells derived from a single colony were grown overnight in 5 ml of synthetic dextrose (SD) liquid medium with appropriate supplemental amino acids. For sporulation, 1 ml of the culture was then shifted to 30 ml of YPA (1% yeast extract, 2% peptone, 2% potassium acetate) medium and grown for 24 h. The cells were harvested by centrifugation, washed with H2O, resuspended in 30 ml of 2% potassium acetate medium, and cultured for 24 h. Sporulation efficiency was determined by light microscopy. Vegetative cells were similarly cultured in YPA medium: 1 ml of SD culture was shifted to 30 ml of YPA medium and grown for 24 h and harvested.
To release spores from asci, the ascal wall was first digested by β-glucanase (lyticase; Sigma-Aldrich, Shanghai, China). For this, spores prepared as described above were resuspended in 1 ml of spheroplast buffer (50 mM potassium phosphate buffer, pH 7.5, 1.4 M sorbitol, 40 mM β-mercaptoethanol) and mixed with 50 μl of β-glucanase stock solution (1 mg of β-glucanase was dissolved in 500 μl of 50% glycerol). After 3 h of incubation at 37°C, spores were washed twice with spheroplast buffer. Then, they were resuspended in spheroplast buffer and sonicated to disrupt the ascal membrane.
Spores were purified by Percoll gradient centrifugation based on a previously described method (26). Spores were washed three times with 0.5% Triton-X. After the washes, the resulting pellet was resuspended in 1 ml of 0.5% Triton-X and layered on top of Percoll (Sigma-Aldrich, Shanghai, China) gradients (50 to 80% Percoll, 10% 2.5 M sucrose, and 0.5% Triton-X). After centrifugation at 15,000 × g at 4°C for 1 h, the top of three layers, which consisted of vegetative cells and debris, were removed. The remaining layer containing spores was washed with 0.5% Triton-X. Purified spores were freeze-dried as follows. First, spores were frozen in a −20°C freezer for more than 2 h. They were then freeze-dried in an Eyela FD-1000 freeze-dryer (Tokyo Rikakikai, Tokyo, Japan) at −50°C for 72 h under the pressure of 25 Pa.
α-Galactosidase assay.
About 5 mg (for repetitive wash assay) or 2 mg (all other experiments) of spores or vegetative cells was washed with either water or water containing 0.6 M NaCl and 0.1% Triton X-100 and then suspended in 1 ml of acetate buffer (0.2 M acetic acid-sodium acetate, pH 4.6). To prevent germination, cycloheximide was added at a final concentration of 40 μg/ml. The reaction was started by the addition of 1 ml of 5% melibiose dissolved in the same buffer. The mixtures were incubated at 37°C for 10 min, and the reaction was stopped by boiling. After cells were removed by centrifugation, the supernatant was diluted, and the glucose concentration was measured by a glucose assay kit (Sigma-Aldrich, Shanghai, China). One unit of activity was defined as the amount (mg) of glucose released in 10 min at 37°C. For wet cells, cell numbers were calculated based on the chart available at http://www.pangloss.com/seidel/Protocols/ODvsCells.html. To prepare wet vegetative cells, YPA liquid medium was used as described above. To measure the activity in culture medium, 108 vegetative cells were centrifuged to remove cells. The supernatant was concentrated into 200 μl by using an Amicon-Ultra filtration instrument (molecular-mass cutoff of 10 kDa; Millipore, Shanghai, China), and the activity was assayed. The soluble α-galactosidase used for the proteinase resistance and temperature and pH stability assays was prepared as follows. Wild-type cells expressing MEL1 were cultured in 30 ml of YPA medium. The culture was centrifuged to remove cells and concentrated into 300 μl as described above (final protein concentration, 6 mg/ml). From the concentrated medium, 10 μl was used for each assay.
Invertase assay.
About 2 mg of freeze-dried spores or vegetative cells was suspended in 1 ml of acetate buffer (0.2 M acetic acid-sodium acetate, pH 4.6). To prevent germination, cycloheximide was added at a final concentration of 40 μg/ml. The reaction was started by the addition of 1 ml of 5% sucrose or 8.7% raffinose dissolved in the same buffer (final concentrations were 73 mM for both). The mixtures were incubated at 37°C for 10 min. To measure the concentration of reducing sugars, the reaction mixture was mixed with 1 ml of DNS solution (1% 3,5-dinitrosalicylic acid, 0.2% phenol, 0.05% NaSO3, 1% NaOH, 20% Rochelle salt) and boiled for 2 min. The mixture was then diluted with water to 10 ml, and the optical density at 520 nm (OD520) was measured by spectrophotometer (Ultrospec 2100 Pro; Amersham Biosciences, USA). One unit of activity was defined as the amount of reducing sugars (μmol) released in 10 min at 37°C.
Spore protection assay.
β-Glucanase treatment was performed as follows. Two milligrams of freeze-dried spores suspended in 1 ml of spheroplast buffer was mixed with 10 μl of the β-glucanase stock solution (see above) and incubated at 30°C for 3 h. Spores were then washed with 0.6 M NaCl solution containing 0.1% Triton X-100, and α-galactosidase activity was assayed as described above.
Proteinase K treatment was performed as follows. About 2 mg of freeze-dried spores was first suspended in 1 ml of proteinase buffer (50 mM Tris-HCl, pH 7.5, 10 mM CaCl2). Then, proteinase K (Sigma-Aldrich, Shanghai, China) was added to the solution at a final concentration of 500 μl/ml. After overnight incubation at 30°C, spores were washed with 0.6 M NaCl solution containing 0.1% Triton X-100, and α-galactosidase activity was assayed.
Microscopy.
Microscopy images were obtained using a Nikon Eclipse Ti-E inverted microscope equipped with a DS-Ri1 camera and NIS-Element AR software (Nikon, Tokyo, Japan).
Western blotting.
Yeast cells harboring pRS424TEF-MEL1-3HA were first cultured in 5 ml of SD (lacking tryptophan) medium overnight, and 1 ml of the cultures was shifted into 30 ml of YPA medium and cultured for 24 h. For sporulation, cells were then shifted to 2% potassium acetate medium, and spores were purified as described above. For the vegetative cell sample, cells were harvested after the YPA culture. Spores and vegetative cells were washed with water, suspended in 500 μl of 8 M urea, and lysed by sonication for 1 h on ice. The cell lysates were then centrifuged at 4,000 × g for 5 min, and 50 μg of the supernatants was subjected to SDS-PAGE (5% stacking gel and 10% separating gel). Protein concentration was determined by a bicinchoninic acid (BCA) protein assay kit (Beyotime, Jiangsu, China). Rabbit anti-HA antibodies (Sigma-Aldrich, Shanghai, China) were used as primary antibodies at dilutions of 1:3,000. Goat anti-rabbit IgG-horseradish peroxidase (HRP) (Life Science, Shanghai, China) was used as the secondary antibody at a 1:2,000 dilution. Signals were visualized by Clarity Western ECL Substrate (Bio-Rad, Shanghai, China), and images were obtained by using ImageQuant LAS4000 (GE Healthcare Bio-Science, Uppsala, Sweden).
Statistics.
Data presented are the means ± standard errors (SE) of three or six independent samples obtained from different cultures. Statistical significance was determined with Student's t test (two-tail, heteroscedastic) using Microsoft Excel software. Differences between the analyzed samples were considered significant at a P value of <0.05.
RESULTS
Spores are capable of entrapping α-galactosidase.
To examine whether spores can be used as a carrier to encapsulate enzymes, MEL1, which encodes an α-galactosidase, was expressed in an efficient sporulating strain, AN120 (27). MEL1 is a yeast gene, but it is carried by only restricted S. cerevisiae strains (28). AN120 has no or very low intrinsic α-galactosidase activity to digest melibiose (Fig. 1). α-Galactosidase is a soluble secretory protein, and previous work has shown that, in Saccharomyces carlsbergensis, a certain fraction of the enzyme was retained in the periplasmic space of the cell wall but part was also secreted in the medium (29). When MEL1 was expressed under the control of a constitutive promoter from a multicopy vector in vegetatively growing AN120, α-galactosidase activity in the growth medium was about twice that detected on the intact cells (Fig. 1). This suggests that the vegetative cell wall is leaky so that a large amount of α-galactosidase is secreted into the medium. Next, we measured the activity associated with spores. In our experiment, sporulation efficiency was greater than 90%. Yeast spores are covered with an ascal membrane and ascal wall, and the activity detected from intact asci was less than half of that in vegetative cells (Fig. 1). To prepare the spore sample, asci were first treated with β-glucanase to digest the ascal wall. After β-glucanase was washed out, the ascal membrane was broken by sonication to release spores from the ascus. The activity detected from the spores was more than twice as high as that detected from vegetative cells (Fig. 1). In this experiment, cell amounts were adjusted by turbidity, and, for the spore sample, it was measured before asci were broken. Cycloheximide was added to the reaction mixture to prevent spore germination (20).
FIG 1.
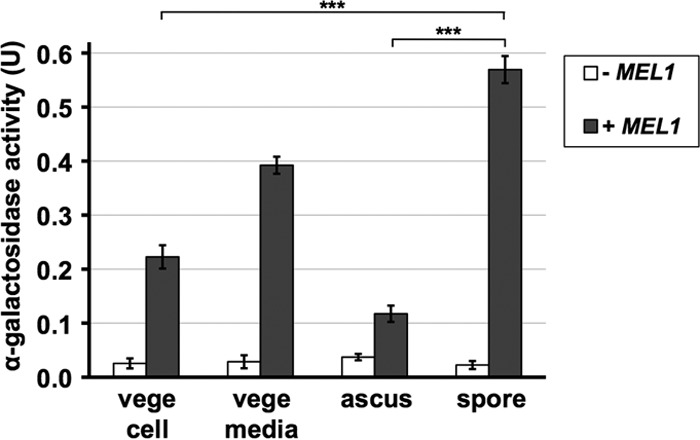
Yeast spores can retain α-galactosidase. Wild-type S. cerevisiae strain AN120 was transformed with the Mel1 expression vector (+ MEL1) or empty vector (− MEL1), and α-galactosidase activity was detected from vegetative cells (vege cell), intact asci (ascus), or spores (spore). A total of 108 vegetative cells or asci were used. Vegetative cells were cultured in YPA medium. The activity was also measured in the supernatant of the culture containing 108 vegetative cells (vege media). The spore sample was prepared by treating the asci with β-glucanase and sonication. Melibiose was used as a substrate. One unit of activity was defined as the amount (mg) of glucose released in 10 min at 37°C. Data presented are the means ± SE of three independent samples. ***, P < 0.001.
Entrapment of the α-galactosidase is dependent on the chitosan layer.
The above results indicate that the spore wall is able to entrap more α-galactosidase than the vegetative cell wall. Consistent with this interpretation, a monomeric red fluorescent protein (mRFP) fusion to Mel1 (Mel1-RFP) was observed at the periphery of the spores, whereas a clear fluorescence signal from this reporter was not detected in vegetative cell walls (Fig. 2A). Although it has been reported that yeast spores show auto-fluorescence (30), the fluorescent signal observed in spores with an empty plasmid was much fainter than that observed in spores expressing the RFP fusion (Fig. 2A and B). The RFP fusion persists in the spore wall even after spores are subjected to a high-salt wash in the presence of detergent though the intensity of the signal is reduced (Fig. 2B). It should be noted that, besides the cell periphery, the RFP signal was also observed in vacuole-like structures after the washing step (Fig. 2B). Thus, it maybe that periplasmic proteins can be incorporated into endocytic vesicles under certain conditions, such as a hyperosmotic treatment. Previous work has shown that the outermost dityrosine layer works as a diffusion barrier for soluble proteins (18). To test whether the dityrosine layer is required to hold the α-galactosidase, Mel1-RFP was expressed in dit1Δ spores, which lack the dityrosine layer (13, 16). However, we found that the RFP fusion still localized to the spore wall in dit1Δ mutants (Fig. 2A and B). Then, we expressed the fusion in chs3Δ spores in which both chitosan and dityrosine layers are absent. In this mutant, the RFP fusion was not retained in the spore wall (Fig. 2A and B). In contrast, and consistent with previous studies of GFP (18), when RFP alone was fused to a signal peptide, the protein was observed in the spore wall in wild-type cells but not in dit1Δ mutants (Fig. 2C). These results suggest that, because of some property of Mel1, the chitosan layer is sufficient to entrap Mel1-RFP.
FIG 2.
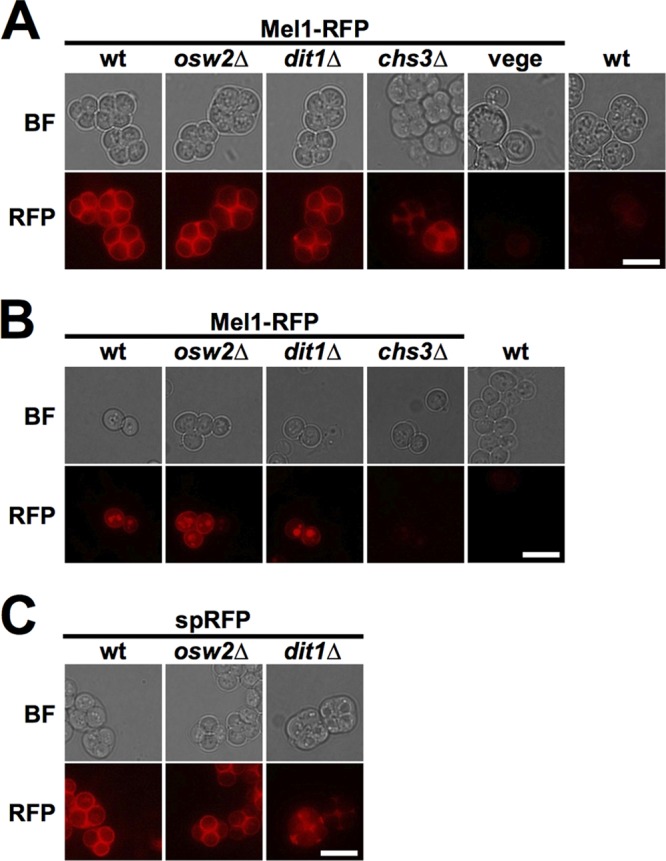
The chitosan layer is required to hold Mel1 in the spore wall. (A) Indicated spores and wild-type (wt) vegetative cells expressing Mel1-RFP were observed under fluorescent (RFP) or bright-field (BF) microscopy. Images of wild-type spores with empty plasmid are also shown as a control. (B) Indicated asci, with or without expression of Mel1-RFP, were first lysed with β-glucanase and sonicated. After a washing step with 0.6 M NaCl solution containing 0.1% Triton X-100, spores were observed using fluorescent (RFP) or bright-field (BF) microscopy. (C) A monomeric RFP fused only to the signal peptide (spRFP) was expressed in the indicated cells, and intact asci were observed by fluorescent (RFP) or bright-field (BF) microscopy. Scale bar, 5 μm. All fluorescence microscopy images were obtained under the same imaging conditions.
Activity of the encapsulated enzyme is affected by spore wall integrity.
We next assayed α-galactosidase activity in spores from the different mutants. Since it is inconvenient to adjust the amount of wet cells precisely, here, we used purified and freeze-dried spores, and their amounts were measured by weight. Freeze-drying does not kill the α-galactosidase activity held in spores. In fact, freeze-dried wild-type spores showed significantly higher activity than vegetative cells (Fig. 3A). Our α-galactosidase activity assay is based on the released amount of glucose. Because vegetative cells have higher metabolic activity than spores (31), one might expect that α-galactosidase activities measured in vegetative cells are underestimated. However, this is unlikely for the assays using freeze-dried cells because the vegetative cells cannot survive through our freeze-drying treatment (Fig. 3B). In contrast, freeze-dried spores, even the chs3Δ mutants, are viable (Fig. 3B).
FIG 3.
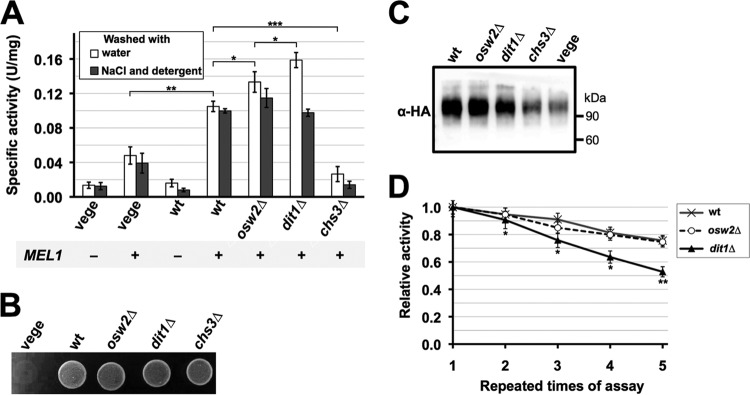
Integrity of the spore wall affects the activity and stability of the encapsulated enzyme. (A) About 2 mg of the indicated freeze-dried spores and wild-type vegetative cells (vege) with (+) or without (−) MEL1 expression were washed with either water (open bars) or 0.6 M NaCl solution containing 0.1% Triton X-100 (solid bars), and α-galactosidase activities were assayed. Data presented are the means ± SE of three independent samples. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (B) Two milligrams of freeze-dried vegetative cells (vege) or indicated spores were suspended in 500 μl of YPAD (1% yeast extract, 2% peptone, 30 mg/liter adenine, 2% glucose) liquid medium, and 5 μl of the cell suspensions was spotted onto a YPAD plate. The plate was incubated at 30°C for 2 days. (C) Fifty micrograms of lysates from indicated spores and wild-type vegetative cells (vege) expressing Mel1-HA were subjected to Western blot analysis using anti-HA (α-HA) antibodies. The lysates were prepared from purified spores and vegetative cells cultured in YPA medium. (D) For about 5 mg of wild-type (wt), osw2Δ, and dit1Δ spores expressing MEL1, α-galactosidase activity was first assayed (first assay). After a washing step with 0.6 M NaCl solution containing 0.1% Triton X-100, the activities were assayed again (second assay). This cycle was repeated four times. For each sample, the activity obtained at the first assay was determined as 1.0, and relative activities are shown. Data presented are the means ± SE of three independent samples. Results of the t test between wild-type and dit1Δ spores are shown. *, P < 0.05; **, P < 0.01.
In dit1Δ spores, we found that the activity is higher than that in wild-type spores (Fig. 3A). This is probably because the substrates can more easily access the enzymes due to the lack of the dityrosine layer. This result led us to speculate that activity of encapsulated enzyme might be improved in mutants that have minor defects in proper spore wall assembly but are still capable of holding soluble proteins. osw2Δ spores show ether sensitivity but otherwise retain resistance to stresses (9). Suda et al. (18) have shown that mutation of OSW2 causes a modest increase in the release of secreted GFP from the spore wall. These phenotypes suggest that osw2Δ mutants might be more permeable to enzyme substrates without extensively compromising the enzyme retention and stress resistance properties of the spore wall. Fluorescence microscopy results showed that osw2Δ spores are able to retain Mel1-RFP in the spore wall (Fig. 2A and B). Consistent with the hypothesis that osw2Δ mutants are more permeable than wild-type spores, we found that the activity of osw2Δ spores is higher than that of the wild type though still less than that in dit1Δ spores (Fig. 3A). To verify that comparable levels of α-galactosidase were expressed in wild-type, dit1Δ, and osw2Δ spores, we performed Western blot analysis of spores expressing a hemagglutinin (HA)-tagged Mel1 (Mel1-HA). As shown in Fig. 3C, similar levels of Mel1-HA were detected in wild-type and osw2Δ spores. The level of the HA fusion detected in dit1Δ spores was slightly less than that in the other spores, probably because the spore wall is leaky in this mutant. These results show that increased α-galactosidase activity in dit1Δ and osw2Δ spores is not due to increased amounts of the enzyme. The Mel1-HA band is higher than its predicted molecular mass (54 kDa). This is probably because Mel1 is a glycoprotein (32). In chs3Δ spores and vegetative cells, the Mel1-HA signal was significantly weaker than that in the other spores (Fig. 3C). This result further supports the idea that chs3Δ spores and vegetative cells cannot retain the enzyme.
The above results seemingly show that the dit1Δ spore is the best option to retain the α-galactosidase activity. However, because of the absence of the dityrosine layer, it may be that the enzyme in dit1Δ spores is leaky or more susceptible to damage than wild-type or osw2Δ spores. To test this possibility, spores were subjected to a high-salt wash in the presence of detergent (0.6 M NaCl and 0.1% Triton X-100), and then the activity was measured. As shown in Fig. 3A, α-galactosidase activity in dit1Δ decreased more than the activities of the wild-type and osw2Δ spores. After the wash and activity assay cycle were repeated four times, the activity in dit1Δ spores decreased by about 50%, whereas that in wild-type and osw2Δ spores decreased by about 25% (Fig. 3D). Taken together, these results indicate that the dityrosine layer can prevent diffusion of the encapsulated enzyme from the spore wall even in the presence of high salt and the detergent; however, the dityrosine layer may be an obstacle to some extent for the substrates to access the encapsulated enzymes.
The encapsulated protein is protected from environmental stresses.
Due to the presence of the spore wall, especially the dityrosine layer, yeast spores show resistance to cell wall-degrading enzymes. For example, wild-type spores can survive β-glucanase treatment, whereas spore wall mutants lacking the dityrosine layer are susceptible to digestion (9). Thus, we speculated that the enzymes encapsulated in spores are also protected from environmental stresses. To test this possibility, we incubated freeze-dried spores containing α-galactosidase with β-glucanase. As shown in Fig. 4A, we found that the α-galactosidase activity associated with wild-type and osw2Δ spores was not affected by β-glucanase treatment. In contrast, α-galactosidase activity in dit1Δ spores was markedly decreased by the treatment (Fig. 4A). It should be noted that although β-glucanase was also used for the purification of spores, in this procedure, only the ascal wall is digested because spores are enclosed by the ascal membrane.
FIG 4.
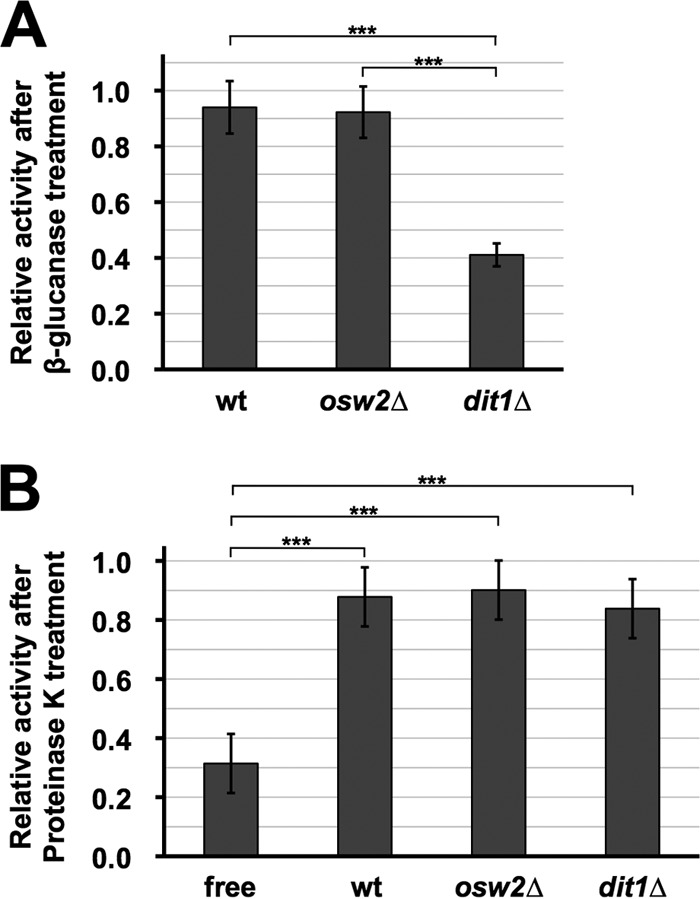
The encapsulated α-galactosidase is resistant to digestive enzymes. About 2 mg of the indicated freeze-dried spores containing the α-galactosidase was first treated with β-glucanase (A) or proteinase K (B), and then the α-galactosidase activities were assayed. For each sample, the activity obtained before treatment with the digestive enzymes was determined as 1.0, and relative activities are shown. Culture medium containing the soluble α-galactosidase (free) was used as a control for the proteinase K treatment. Data presented are the means ± SE of three independent samples. ***, P < 0.001.
In the above-described experiment, the α-galactosidase activity associated with dit1Δ spores was decreased by β-glucanase treatment, probably due to destruction of the wall and release of the enzyme. Next, we tested if the encapsulated enzyme is resistant to attack by proteinase K. The α-galactosidase secreted from vegetative cells was used as a control that is susceptible to the proteinase K digestion (Fig. 4B). As in the case of the β-glucanase assay, the encapsulated enzymes in wild-type and osw2Δ spores were significantly resistant to the proteinase treatment. The enzyme retained in the dit1Δ spores also showed proteinase resistance, suggesting that the chitosan layer to some extent works as a protective barrier.
Furthermore, we found that at higher temperatures (40 and 45°C), stability of α-galactosidase is improved by being retained in spores (Fig. 5A). In contrast, probably due to restricted mobility of substrates and the enzyme, the activity is markedly reduced at lower temperatures (below 38°C) (Fig. 5A). For pH stability, we found that the encapsulated enzyme has a higher activity than soluble α-galactosidase at pH 7 (Fig. 5B). Thus, properties of the enzyme can be altered by the encapsulation.
FIG 5.
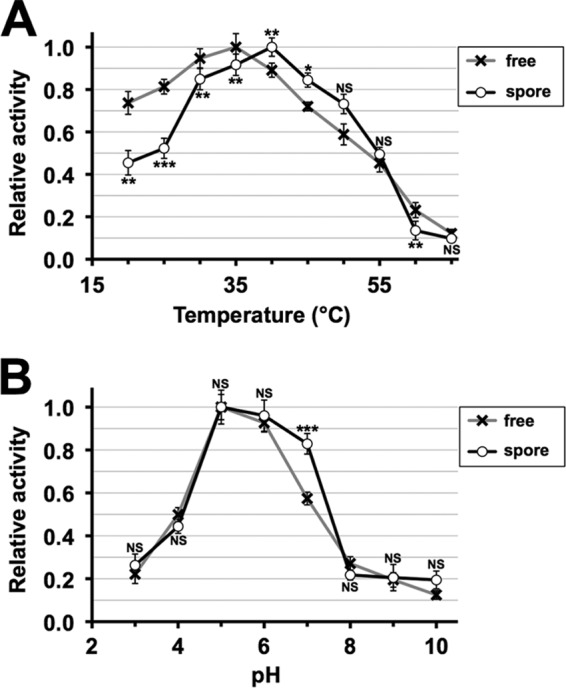
Temperature and pH sensitivities of the encapsulated α-galactosidase. Temperature (A) and pH (B) sensitivities were assessed for freeze-dried wild-type spores containing Mel1 (spore) and the soluble α-galactosidase (free). α-Galactosidase activity was assayed at various temperatures (20°C to 65°C) and pH values (3 to 10). As the soluble enzyme, culture medium containing the secreted α-galactosidase was used. For each assay, the maximum activity obtained was determined as 1.0, and relative activities are shown. Data presented are the means ± SE of six independent samples. *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, not significant.
Spore encapsulation can alter the substrate preference of invertase.
Finally, we expressed the yeast invertase encoded by the SUC2 gene (33) in spores because the enzyme can hydrolyze different sizes of nonreducing sugars, such as raffinose (trisaccharide) and sucrose (disaccharide). Since invertase is normally found in the periplasmic space of the vegetative cell wall (34), we used suc2Δ cells, and SUC2 was expressed from the sporulation-inducible SPO20 promoter (3) so that the background derived from vegetative cells was reduced. Although almost no invertase activity was detected in purified spores, significantly high activity toward sucrose was detected in those expressing SUC2 (Fig. 6). Intriguingly, we found that invertase activity toward raffinose is markedly lower than toward sucrose. Since invertase is located in the periplasmic space of the vegetative cell wall, the activity was also assayed from vegetative cells in which SUC2 was expressed from the constitutive TDH2 promoter (Fig. 6). In these cells, the specific activities toward raffinose and sucrose were very similar. Thus, as a result of the encapsulation, invertase activity toward raffinose is specifically decreased. SUC2 was also expressed in double mutants in which SUC2 and DIT1 or OSW2 were deleted. In contrast to the case where the α-galactosidase was expressed in corresponding mutants, the activity toward sucrose in dit1Δ spores was lower than that in the wild type (Fig. 6). This shows that dit1 mutation is not always beneficial for enzyme encapsulation. In osw2Δ spores, the activities toward both sucrose and raffinose were higher than those in the wild type (Fig. 6), suggesting that the mutation generally improved the activity of encapsulated enzymes.
FIG 6.
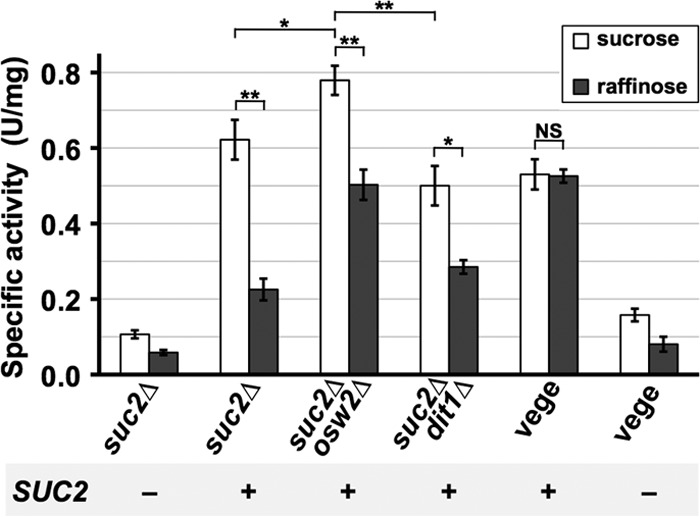
Activity of the encapsulated invertase toward sucrose and raffinose. suc2Δ, suc2Δ osw2Δ, or suc2Δ dit1Δ cells were transformed with the SUC2 expression vector (+) or empty vector (−), and invertase activities toward sucrose or raffinose were assayed for the freeze-dried spores. Freeze-dried vegetative suc2Δ cells (vege) were also prepared, and activity was measured. SPO20 or TDH2 promoters were used to express SUC2 in spores or vegetative cells, respectively. One unit of activity was defined as the amount of reducing sugars (μmol) released in 10 min at 37°C. Data presented are the means ± SE of three independent samples. *, P < 0.05; **, P < 0.01; NS, not significant.
DISCUSSION
In this report, we describe an enzyme microencapsulation method using yeast spores. One advantage of this method is that enzymes which can be secreted from yeast cells are immobilized even in soluble form. When MEL1 was expressed from the constitutive TEF2 promoter, we detected much higher α-galactosidase activity associated with spores than with vegetative cells. Although vegetative cells retain a certain amount of the enzyme, probably in the periplasmic space, the cell wall is porous so that a large amount of the enzyme is secreted into the medium. In fact, the activity detected from the culture medium was much higher than that associated with vegetative cells. On the other hand, the spore wall is more tightly sealed, and thus it can stably retain the enzyme. The fluorescence microscopy results support this idea.
The outer two layers of the spore wall are composed of chitosan and dityrosine, which do not exist in the vegetative cell wall. A previous report has shown that the dityrosine layer works as a diffusion barrier (18), and, consistently, a secretory form of mRFP expressed in spores was observed in the spore wall in wild-type cells but not in dit1Δ mutants. Thus, it was somewhat surprising that dit1Δ spores are capable of holding Mel1-RFP. In chs3Δ spores, Mel1-RFP was not retained in the spore wall, showing that the enzyme is held in the chitosan layer. Since chitosan has positive charges in acidic solutions, it may be that the α-galactosidase associates with chitosan by electrostatic interactions. Consistent with this idea, α-galactosidase activity in dit1Δ mutants was washed out by high-salt treatment more efficiently than in wild-type spores. Alternatively, since the enzyme is not completely removed even after several high-salt washes, the pore size of the chitosan layer might be small enough to prevent diffusion of Mel1 from the spore wall. For α-galactosidase, we found that the activity in dit1Δ spores is higher than that in wild-type spores. This is probably because substrate accessibility is increased by removal of the dityrosine layer. Thus, the dityrosine layer works as a diffusion barrier for expressed enzymes but is also an obstacle for substrates to access the enzymes. Although, as described below, the activity of the encapsulated enzymes is not always improved by dit1 mutation, nonetheless the results suggest that dit1Δ spores are available as a carrier to immobilize some enzymes. In addition to dit1Δ spores, we also found that osw2 mutation can improve the enzyme activity present in the spore wall. A remarkable difference between dit1Δ and osw2Δ spores is that the latter have a dityrosine layer, so they can stably entrap proteins in the spore wall and retain resistance to stresses other than ether (9, 18). In fact, the α-galactosidase activity encapsulated in osw2Δ spores is tolerant against a high-salt wash, similar to the activity in wild-type spores. Although the exact function of Osw2 has not been understood, a previous report has shown that it is required to properly organize the spore wall (9). Thus, in osw2Δ spores, it may be that the dityrosine layer or chitosan layer is structurally loose so that substrates can pass through them smoothly.
Another benefit of spore encapsulation is that the enzymes are protected by the spore wall from environmental stresses. Due to the presence of the dityrosine layer, spores are resistant to digestive enzymes (9). Consistently, we found that the α-galactosidase activity held in wild-type and osw2Δ spores was not released by the β-glucanase treatment. However, in dit1Δ spores, because β-glucanase can reach the inside of the spore wall, the encapsulated enzyme is released by the treatment. The encapsulated enzymes in wild-type and osw2Δ spores are also resistant to proteinase K digestion. After proteinase treatment, the activity of the soluble α-galactosidase secreted from the yeast decreased to 35%. Remarkably, the enzyme contained in wild-type and osw2Δ spores is more stable, and about 90% of the activity still remained after the treatment. We found that the enzyme in dit1Δ spores is also resistant to proteinase K treatment. Since the inner spore wall of the dit1 mutant is accessible to β-glucanase and since the molecular weight of proteinase K is smaller than that of β-glucanase, it is unlikely that the protection from proteinase K is due simply to an inability of the protease to diffuse into the wall. Rather, the chitosan layer may also provide a protective environment, at least for the α-galactosidase.
We found that temperature and pH sensitivities of α-galactosidase are altered by encapsulation. The encapsulated enzyme has a slightly higher optimum temperature than the soluble α-galactosidase, and its thermotolerance is improved. At lower temperatures, however, the activity is significantly decreased by the encapsulation. This is probably because mobility of the enzyme and substrate is restricted in the spore wall. We also assessed pH sensitivity and found that the activity is improved by encapsulation at pH 7.
In addition to α-galactosidase, we also showed that spores are capable of encapsulating invertase. Since invertase is a periplasmic enzyme (33), the activity was detected even in vegetative cells expressing SUC2 from the TDH2 promoter. In wild-type spores expressing the enzyme from the SPO20 promoter, we detected higher activity than in vegetative cells when sucrose was used as a substrate. Interestingly, the activity in the spores was significantly lower toward raffinose than toward sucrose. The enzyme in vegetative cells digests these substrates to comparable levels. One possible reason for this is that because raffinose is larger than sucrose, the former may not able to pass through the pore of the dityrosine layer smoothly. However, considering that the same difference was observed in dit1Δ spores, the size of the substrates may not be the cause. It could be that environmental conditions in the spore wall also affect the invertase activity. As in the case of α-galactosidase, we found that invertase activity in osw2Δ spores is higher than that in wild-type spores. This suggests that in osw2Δ cells, activities of the encapsulated enzymes are generally improved without loss of the beneficial properties. In contrast, dit1Δ spores are not quite as beneficial for invertase immobilization because in dit1Δ spores the activity toward sucrose was lower than in wild-type spores even though that toward raffinose was slightly higher.
In summary, we demonstrated that yeast spores can be used as microcapsules to hold enzymes. The enzyme encapsulated in yeast spores acquires resistance to environmental stresses such as enzymatic digestion and high temperatures. Moreover, through encapsulation, substrate selectivity can be altered. This property may be useful for selective digestion of substrates. Thus, by the encapsulation, enzymes acquire several beneficial properties. However, it should be noted that there are several points to be improved for practical applications. First, spores germinate in the presence of glucose, which causes a disorganization of the spore wall (20, 35). In this study, we used cycloheximide to prevent this from occurring, but it would be better to find a convenient method to handle this process. Use of germination mutants is an intriguing possibility to solve this issue though no mutation which can specifically and uniformly arrest cells at an earlier stage of the germination process has been found so far (20, 26, 36). Second, encapsulated enzymes can be incorporated into a cytosolic compartment under certain conditions. We observed that Mel1-RFP is localized to cytosolic foci after high-salt and detergent washes. Since the internal foci were not observed in intact spores, it is likely that the phenomenon is induced by certain stimuli, such as hyperosmotic shock. Nevertheless, this result suggests that endocytosis is active in spores, and thus prevention of the process is required to maintain activity of the encapsulated enzymes.
Microencapsulation has a long history, and it is used in a variety of fields (37, 38). Although many methods have been reported for production of microcapsules, in most, if not all, cases this is done by chemical and physical procedures. Our method is unique in that enzymes are encapsulated in a biological way. Thus, spores are an intriguing option to produce microcapsules.
ACKNOWLEDGMENTS
We are grateful to A.M. Neiman (Stony Brook University, Stony Brook, NY) for comments on the manuscript and to J. Voglmeir for providing plasmid TOP10-mRFP.
This work was supported by grants from the National 111 project (111-2-06) and Self-Determined Research Program of Jiangnan University (JUSRP311A02) to H.N. and the 2012 Key project of the Chinese Ministry of Education to X.-D.G.
Footnotes
Published ahead of print 16 May 2014
REFERENCES
- 1.Kupiec M, Byers B, Esposito RE, Mitchell AP. 1997. Meiosis and sporulation in Saccharomyces cerevisiae, p 889–1036 In Pringle JR, Broach JR, Jones EW. (ed), The molecular and cell biology of the yeast Saccharomyces. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 2.Neiman AM. 2005. Ascospore formation in the yeast Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 69:565–584. 10.1128/MMBR.69.4.565-584.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neiman AM. 1998. Prospore membrane formation defines a developmentally regulated branch of the secretory pathway in yeast. J. Cell Biol. 140:29–37. 10.1083/jcb.140.1.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lynn RR, Magee PT. 1970. Development of the spore wall during ascospore formation in Saccharomyces cerevisiae. J. Cell Biol. 44:688–692. 10.1083/jcb.44.3.688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Briza P, Ellinger A, Winkler G, Breitenbach M. 1988. Chemical composition of the yeast ascospore wall. The second outer layer consists of chitosan. J. Biol. Chem. 263:11569–11574 [PubMed] [Google Scholar]
- 6.Briza P, Winkler G, Kalchhauser H, Breitenbach M. 1986. Dityrosine is a prominent component of the yeast ascospore wall. A proof of its structure. J. Biol. Chem. 261:4288–4294 [PubMed] [Google Scholar]
- 7.Pammer M, Briza P, Ellinger A, Schuster T, Stucka R, Feldmann H, Breitenbach M. 1992. DIT101 (CSD2, CAL1), a cell cycle-regulated yeast gene required for synthesis of chitin in cell walls and chitosan in spore walls. Yeast 8:1089–1099. 10.1002/yea.320081211 [DOI] [PubMed] [Google Scholar]
- 8.Wagner M, Briza P, Pierce M, Winter E. 1999. Distinct steps in yeast spore morphogenesis require distinct SMK1 MAP kinase thresholds. Genetics 151:1327–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coluccio A, Bogengruber E, Conrad MN, Dresser ME, Briza P, Neiman AM. 2004. Morphogenetic pathway of spore wall assembly in Saccharomyces cerevisiae. Eukaryot. Cell 3:1464–1475. 10.1128/EC.3.6.1464-1475.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smits GJ, van den Ende H, Klis FM. 2001. Differential regulation of cell wall biogenesis during growth and development in yeast. Microbiology 147:781–794 [DOI] [PubMed] [Google Scholar]
- 11.Christodoulidou A, Bouriotis V, Thireos G. 1996. Two sporulation-specific chitin deacetylase-encoding genes are required for the ascospore wall rigidity of Saccharomyces cerevisiae. J. Biol. Chem. 271:31420–31425. 10.1074/jbc.271.49.31420 [DOI] [PubMed] [Google Scholar]
- 12.Mishra C, Semino CE, McCreath KJ, de la Vega H, Jones BJ, Specht CA, Robbins PW. 1997. Cloning and expression of two chitin deacetylase genes of Saccharomyces cerevisiae. Yeast 13:327–336. [DOI] [PubMed] [Google Scholar]
- 13.Briza P, Breitenbach M, Ellinger A, Segall J. 1990. Isolation of two developmentally regulated genes involved in spore wall maturation in Saccharomyces cerevisiae. Genes Dev. 4:1775–1789. 10.1101/gad.4.10.1775 [DOI] [PubMed] [Google Scholar]
- 14.Briza P, Eckerstorfer M, Breitenbach M. 1994. The sporulation-specific enzymes encoded by the DIT1 and DIT2 genes catalyze a two-step reaction leading to a soluble ll-dityrosine-containing precursor of the yeast spore wall. Proc. Natl. Acad. Sci. U. S. A. 91:4524–4528. 10.1073/pnas.91.10.4524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Felder T, Bogengruber E, Tenreiro S, Ellinger A, Sa-Correia I, Briza P. 2002. Dtrlp, a multidrug resistance transporter of the major facilitator superfamily, plays an essential role in spore wall maturation in Saccharomyces cerevisiae. Eukaryot. Cell 1:799–810. 10.1128/EC.1.5.799-810.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin CPC, Kim C, Smith SO, Neiman AM. 2013. A highly redundant gene network controls assembly of the outer spore wall in S. cerevisiae. PLoS Genet. 9:e1003700. 10.1371/journal.pgen.1003700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mattanovich D, Branduardi P, Dato L, Gasser B, Sauer M, Porro D. 2012. Recombinant protein production in yeasts. Methods Mol. Biol. 824:329–358. 10.1007/978-1-61779-433-9_17 [DOI] [PubMed] [Google Scholar]
- 18.Suda Y, Rodriguez RK, Coluccio AE, Neiman AM. 2009. A screen for spore wall permeability mutants identifies a secreted protease required for proper spore wall assembly. PLoS One 4:e7184. 10.1371/journal.pone.0007184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neiman AM. 2011. Sporulation in the budding yeast Saccharomyces cerevisiae. Genetics 189:737–765. 10.1534/genetics.111.127126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herman PK, Rine J. 1997. Yeast spore germination: a requirement for Ras protein activity during re-entry into the cell cycle. EMBO J. 16:6171–6181. 10.1093/emboj/16.20.6171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rose MD, Winston FM, Hieter P. 1990. Methods in yeast genetics: a laboratory course manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 22.Longtine MS, McKenzie A, III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953–961. [DOI] [PubMed] [Google Scholar]
- 23.Mumberg D, Muller R, Funk M. 1995. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156:119–122. 10.1016/0378-1119(95)00037-7 [DOI] [PubMed] [Google Scholar]
- 24.Abe H, Shimma Y, Jigami Y. 2003. In vitro oligosaccharide synthesis using intact yeast cells that display glycosyltransferases at the cell surface through cell wall-anchored protein Pir. Glycobiology 13:87–95. 10.1093/glycob/cwg014 [DOI] [PubMed] [Google Scholar]
- 25.Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P. 1992. Multifunctional yeast high-copy-number shuttle vectors. Gene 110:119–122. 10.1016/0378-1119(92)90454-W [DOI] [PubMed] [Google Scholar]
- 26.Kloimwieder A, Winston F. 2011. A screen for germination mutants in Saccharomyces cerevisiae. G3 (Bethesda) 1:143–149. 10.1534/g3.111.000323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neiman AM, Katz L, Brennwald PJ. 2000. Identification of domains required for developmentally regulated SNARE function in Saccharomyces cerevisiae. Genetics 155:1643–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naumov G, Turakainen H, Naumova E, Aho S, Korhola M. 1990. A new family of polymorphic genes in Saccharomyces cerevisiae: alpha-galactosidase genes MEL1-MEL7. Mol. Gen. Genet. 224:119–128 [DOI] [PubMed] [Google Scholar]
- 29.Lazo PS, Ochoa AG, Gascon S. 1977. α-Galactosidase from Saccharomyces carlsbergensis. Cellular localization and purification of the external enzyme. Eur. J. Biochem. 77:375–382. 10.1111/j.1432-1033.1977.tb11677.x [DOI] [PubMed] [Google Scholar]
- 30.Joseph-Strauss D, Zenvirth D, Simchen G, Barkai N. 2007. Spore germination in Saccharomyces cerevisiae: global gene expression patterns and cell cycle landmarks. Genome Biology 8:R241. 10.1186/gb-2007-8-11-r241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brengues M, Pintard L, Lapeyre B. 2002. mRNA decay is rapidly induced after spore germination of Saccharomyces cerevisiae. J. Biol. Chem. 277:40505–40512. 10.1074/jbc.M206700200 [DOI] [PubMed] [Google Scholar]
- 32.Fernandez-Leiro R, Pereira-Rodriguez A, Cerdan ME, Becerra M, Sanz-Aparicio J. 2010. Structural analysis of Saccharomyces cerevisiae alpha-galactosidase and its complexes with natural substrates reveals new insights into substrate specificity of GH27 glycosidases. J. Biol. Chem. 285:28020–28033. 10.1074/jbc.M110.144584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carlson M, Taussig R, Kustu S, Botstein D. 1983. The secreted form of invertase in Saccharomyces cerevisiae is synthesized from mRNA encoding a signal sequence. Mol. Cell. Biol. 3:439–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burger M, Bacon EE, Bacon JS. 1961. Some observations on the form and location of invertase in the yeast cell. Biochem. J. 78:504–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kono K, Matsunaga R, Hirata A, Suzuki G, Abe M, Ohya Y. 2005. Involvement of actin and polarisome in morphological change during spore germination of Saccharomyces cerevisiae. Yeast 22:129–139. 10.1002/yea.1205 [DOI] [PubMed] [Google Scholar]
- 36.Deutschbauer AM, Williams RM, Chu AM, Davis RW. 2002. Parallel phenotypic analysis of sporulation and postgermination growth in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 99:15530–15535. 10.1073/pnas.202604399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Powell LW. 1984. Developments in immobilized-enzyme technology. Biotechnol. Genet. Eng. Rev. 2:409–438. 10.1080/02648725.1984.10647807 [DOI] [PubMed] [Google Scholar]
- 38.Jyothi NV, Prasanna PM, Sakarkar SN, Prabha KS, Ramaiah PS, Srawan GY. 2010. Microencapsulation techniques, factors influencing encapsulation efficiency. J. Microencapsul. 27:187–197. 10.3109/02652040903131301 [DOI] [PubMed] [Google Scholar]
- 39.Coluccio A, Neiman AM. 2004. Interspore bridges: a new feature of the Saccharomyces cerevisiae spore wall. Microbiology 150:3189–3196. 10.1099/mic.0.27253-0 [DOI] [PubMed] [Google Scholar]