

Abstract
Actinobacteria in the genus Streptomyces are critical players in microbial communities that decompose complex carbohydrates in the soil, and these bacteria have recently been implicated in the deconstruction of plant polysaccharides for some herbivorous insects. Despite the importance of Streptomyces to carbon cycling, the extent of their plant biomass-degrading ability remains largely unknown. In this study, we compared four strains of Streptomyces isolated from insect herbivores that attack pine trees: DpondAA-B6 (SDPB6) from the mountain pine beetle, SPB74 from the southern pine beetle, and SirexAA-E (SACTE) and SirexAA-G from the woodwasp, Sirex noctilio. Biochemical analysis of secreted enzymes demonstrated that only two of these strains, SACTE and SDPB6, were efficient at degrading plant biomass. Genomic analyses indicated that SACTE and SDPB6 are closely related and that they share similar compositions of carbohydrate-active enzymes. Genome-wide proteomic and transcriptomic analyses revealed that the major exocellulases (GH6 and GH48), lytic polysaccharide monooxygenases (AA10), and mannanases (GH5) were conserved and secreted by both organisms, while the secreted endocellulases (GH5 and GH9 versus GH9 and GH12) were from diverged enzyme families. Together, these data identify two phylogenetically related insect-associated Streptomyces strains with high biomass-degrading activity and characterize key enzymatic similarities and differences used by these organisms to deconstruct plant biomass.
INTRODUCTION
Plant biomass is the most abundant terrestrial carbon source, and its deconstruction and subsequent catabolism are key components of global carbon cycling (1, 2). The majority of the energy in plant biomass is stored in plant cell walls, primarily in the recalcitrant polysaccharides cellulose and hemicellulose. Efficient breakdown of these insoluble polymers is difficult, and only a limited number of bacteria and fungi have this capability (3). Microbial decomposition in the soil is the primary pathway for carbon stored in terrestrial plant biomass to be returned to the atmosphere (1). Symbiotic cellulolytic microbes associated with a wide variety of herbivores also make significant contributions to global carbon flux (4). However, our molecular and biochemical understanding of how microbes deconstruct plant biomass is largely limited to a few model cellulolytic organisms (3, 5–9).
Actinobacteria in the genus Streptomyces are well represented in both culture-based and culture-independent studies of soil biodiversity (10). These bacteria are generally thought to make important contributions to the carbon cycle through the decomposition of plant biomass and other biopolymers. However, individual cellulose-degrading enzymes have been isolated and biochemically tested for only a few strains (11–15). Genome-wide transcriptomic analyses have been completed for Streptomyces coelicolor and S. griseus, but not for these organisms grown on plant biomass as the carbon source (16–18). Very few Streptomyces strains have been shown to efficiently degrade crystalline cellulose (11), and fewer still have been analyzed at the molecular, biochemical, or system-wide level to identify key biomass-degrading enzymes.
Streptomyces organisms associated with a variety of insects that feed on living trees and dead wood have been demonstrated to deconstruct plant biomass and pure cellulose with high specific activity (19–22). Two distinct digestive models have emerged for insect-associated Streptomyces. In the internal digestive model, Streptomyces bacteria in the insect gut secrete enzymes to aid in digestion of recalcitrant plant biomass (21, 22). In the external digestive system model, Streptomyces, likely in combination with other bacteria and fungi, is inoculated directly into living pine trees during oviposition; bark beetles and woodwasps are insects that use this model (20, 23–25). The external microbial community inoculated into the pine tree by the insect secretes biomass-degrading enzymes that deconstruct plant cell walls and thus facilitate energy accumulation by the developing larvae inside the tree (26). Streptomyces sp. strain SirexAA-E (SACTE) is a cellulolytic strain that was isolated from the pine-boring woodwasp Sirex noctilio and fits into the external digestive model of insect-Streptomyces interactions (20). SACTE produces and secretes a suite of enzymes that efficiently deconstruct lignocellulosic plant biomass into small polysaccharides and free sugars (19). Genome-wide transcriptomic and proteomic analyses showed that coordinated expression and secretion of select enzymes were tailored to the carbon sources present in the growth medium (19). In contrast to previous microarray-based transcriptomic analysis of SACTE, in this study, we used RNA-sequencing techniques and data analysis to compare gene expression between multiple strains of Streptomyces.
In this study, we characterized four strains of Streptomyces isolated from insects which, in combination with their host's symbiotic microbial community, feed on pine trees. These four strains come from insects that span two taxonomic orders and have a broad geographic distribution: the mountain pine beetle (Dendroctonus ponderosae), the southern pine beetle (Dendroctonus frontalis), and the Sirex woodwasp (Sirex noctilio) (Fig. 1A; see also Fig. S1 in the supplemental material). Isolation of three of these Streptomyces stains has been previously reported: SirexAA-E (SACTE) and SirexAA-G (SACTG) from the Sirex woodwasp (20) and SPB74 from the southern pine beetle (23). A fourth Streptomyces strain, DpondAA-B6 (SDPB6), was isolated from an adult mountain pine beetle collected from a naturally attacked Pinus contorta tree near Mackenzie, British Columbia, Canada (Fig. 1A). Comparative genomics provided insight into the phylogenetic relationships of the Streptomyces strains from these different insect hosts and their genomic potential to degrade plant polymers. Microbiology, biochemistry, and next-generation sequencing were used to characterize the molecular responses of these organisms during growth on biomass and plant polysaccharides. Genes that were differentially expressed in response to growth on plant biomass versus glucose as a sole carbon source in the medium were identified by transcriptome sequencing (RNA-Seq), and proteins that were secreted during growth were identified using mass spectrometry. Together, these results provide new insight into how highly cellulolytic Streptomyces strains contribute to deconstruction of plant biomass in insect-herbivore systems.
FIG 1.

(A) General range of pine-boring insects D. ponderosae, S. noctilio, and D. frontalis in North America based on survey maps in the NAPIS database. Collection sites for individual insects and their associated Streptomyces strains are indicated. (B) Streptomyces organisms from these pine-boring insects were grown in minimal medium with cellulose filter paper as the sole carbon source. (C) Quantification of cellulose filter paper deconstruction in cultures grown for 10 days. Data represent the means ± SDs from at least three experiments; lowercase letters represent statistically significant differences between strains (Tukey-Kramer method; P < 0.01).
MATERIALS AND METHODS
Isolation of Streptomyces.
The isolation of Streptomyces strains SirexAA-E (SACTE; NCBI Taxonomy identification number [ID] 862751), SirexAA-G (SACTG; NCBI Taxonomy ID 683219) (20), and SPB74 (NCBI Taxonomy ID 465543) (23) was previously reported. Streptomyces sp. DpondAA-B6 (SDPB6; NCBI Taxonomy ID 682311) was isolated from samples of D. ponderosae adults collected from naturally attacked P. contorta trees near Mackenzie, British Columbia, Canada (27). Adult insects were surface sterilized in 95% ethanol for 1 min, rinsed twice in sterile phosphate-buffered solution (PBS), and homogenized in PBS, and dilutions were spread onto agar plates supplemented with chitin. Bacteria were isolated and maintained on yeast malt extract agar (YMEA) plates (Difco).
Culturing of organisms.
Strains were grown in M63 minimal medium at pH 6.8 containing, per 1 liter, 10.72 g of K2HPO4, 5.24 g of KH2PO4, 2 g of (NH4)2SO4, 0.5 ml of an iron solution (1 mg of FeSO4 dissolved in 1 ml of 0.01 M HCl), 1 ml of 1 M MgSO4, 1 ml of thiamine (1 mg/ml), and 5 ml of trace element solution (SPV-4 with FeIII-citrate substituted for FeII-chloride [28]). The sole carbon source (0.5%, wt/vol) in the medium was glucose, crystalline cellulose, Whatman no. 1 filter paper (GE Healthcare Life Sciences, Pittsburgh, PA), Sigmacell-20 (Sigma-Aldrich, St. Louis, MO), birch wood xylan (Sigma-Aldrich), or sterile AFEX-pretreated corn stover (GLBRC Corn Stover UW 2009 AFEX, AFEX ID 712-192). Cultures were incubated with shaking at 250 rpm at 30°C for the number of days indicated below.
Quantification of residual cellulose.
Strains were grown in test tubes containing 8 ml of M63 medium with ∼0.07 g of Whatman no. 1 filter paper (GE Healthcare Life Sciences, Pittsburgh, PA) as the sole carbon source. After 10 days of growth, residual cellulose concentrations were determined using an acid-detergent fiber method modified from the work of Weimer et al. (29). Briefly, 16 ml of acid detergent (30) was added to each culture. Tubes were crimp sealed with flanged rubber stoppers and autoclaved for 45 min at 121°C. One at a time, the tubes were vented, decapped, and immediately vacuum filtered through preweighed glass microfiber filters (Whatman GF/D, 2.7-μm pore size [GE Healthcare Life Sciences, Pittsburgh, PA]). Then, filters were washed liberally with hot water. Filters were dried at 105°C overnight and reweighed immediately after removal from the oven. Net cellulose loss was calculated by subtracting the final residual cellulose mass from initial cellulose mass. This value was then divided by the initial cellulose mass to calculate percent cellulose degraded and corrected to account for cellulose and water loss in uninoculated samples.
Genomic sequencing and phylogenetic analysis.
Genome sequences were previously described for SACTE (Gold ID Gc01941) and SACTG (Gold ID Gi06834) (19, 20). A high-quality draft genome sequence for Streptomyces sp. DpondAA-B6 was determined at the Joint Genome Institute (JGI) using Illumina HiSeq2000 (Gold ID Gi0022546). All general aspects of library construction and sequencing performed at the Joint Genome Institute can be found at http://www.jgi.doe.gov/. The genome of SPB74 was generated at the Broad Institute (Gold ID Gi02993). Genome annotations were generated at JGI and the Broad Institute. Putative carbohydrate-active enzyme (CAZy) gene annotations were generated (19). Putative antiSMASH annotations were generated by uploading genomes to the antiSMASH online annotation server (31).
For multilocus phylogenetic analysis, proteins from all genomes included in the phylogeny were predicted using Prodigal and annotated using HMMer 3.0 models provided in TIGRfam 13.0, curated using the provided score cutoffs (32–34). A total of 646 TIGRfam families were found to be present in all 17 genomes (see Data Set S1 in the supplemental material). The protein sequences of each family from each genome were aligned using the MAFFT 7.029 FFT-NS-i algorithm (35). If more than one protein from a single family was found in a genome, the protein with the highest bit score, i.e., best match to the respective HMMer model, was chosen as the representative ortholog. The protein sequences were then reverse translated to their respective DNA sequence, and all alignments were concatenated. The phylogeny was generated using RAxML 7.2.6 with the GTRGAMMA substitution model and 100 rapid bootstrap replicates (36).
Preparation of secretomes.
Supernatants were prepared from growing cultures by centrifuging the culture medium for 15 min at 5,125 × g to remove insoluble polysaccharides and cells. The supernatant fraction was then passed through a 0.22-μm polyethersulfone (PES) membrane filter (GE Healthcare Life Sciences, Pittsburgh, PA) to remove remaining cells. For enzymatic assays, the supernatants were concentrated using a 10-kDa-cutoff PES membrane (Sartorius Stedim, NY). The concentration of secreted protein was determined by Bio-Rad protein assay.
Enzyme activity measurements.
Reducing-sugar assays were carried out by mixing concentrated (1 mg/ml) Streptomyces secretome preparations with polysaccharide-containing substrates. Control secretomes were prepared with Spezyme CP (Genencor), an industrial cellulolytic enzyme cocktail. Polysaccharide substrates include Whatman no. 1 filter paper (GE Healthcare Life Sciences, Pittsburgh, PA), Sigmacell-20 (Sigma-Aldrich, St. Louis, MO), cellulose III (a generous gift from Bruce Dale at Michigan State University, East Lansing, MI [37]), phosphoric acid-swollen cellulose (PASC) (38), birch wood xylan (Sigma-Aldrich), d-mannan (Megazyme, Ireland), corn stover (GLBRC 2009 Corn Stover), AFEX-pretreated corn stover (GLBRC Corn Stover UW 2009 AFEX, AFEX ID 712-192), and ionic-liquid pretreated switchgrass (a generous gift from Department of Energy Joint Bioenergy Institute, CA). Enzyme hydrolysis reactions were carried out using 0.1 mg/ml of secretome protein in 0.1 M sodium phosphate, pH 6, containing the different substrates (10 mg/ml) at 40°C for 20 h. Reducing-sugar content was determined by the dinitrosalicylic acid (DNS) assay (39). Glucose was used as the standard to quantify the amount of reducing ends produced.
Extracellular proteomics.
Extracellular proteins from culture supernatants were precipitated by trichloroacetic acid (TCA) precipitation of 100 ng of total secreted protein from 7-day-old culture supernatants. Protein samples were digested with trypsin (sequencing grade; Promega, Madison, WI) and desalted using C18 pipette tips (Millipore, Billerica, MA). High-energy collision dissociation mass spectrometry employing capillary liquid chromatography-tandem mass spectrometry (LC-MS/MS) was performed on an electrospray ionization FT/ion-trap mass spectrometer (LTQ Orbitrap XL; Thermo Fisher Scientific, San Jose, CA) in the University of Wisconsin—Madison Biotechnology Center (Madison, WI) and database searched with Scaffold (Scaffold_3_00_06; Proteome Software, Portland, OR). The spectra from SACTE were searched against the SACTE predicted proteome (translation of all protein coding sequences); a protein false-discovery rate of 0.0% (probabilistic method) was reported. The spectra from SDPB6 were searched against the SDPB6 predicted proteome; a protein false-discovery rate of 0.0% (probabilistic method) was reported. Homology between SACTE and SDPB6 proteins was generated by BLAST comparison. Homology was reported when E values were less than 1e−50 for more than 75% of the protein sequence.
RNA extraction and preparation.
SDPB6 and SACTE were grown in M63 minimal medium plus either glucose or AFEX corn stover (0.5%, wt/vol) for 7 days with shaking at 30°C. The cell pellet was separated from the culture supernatant by centrifugation for 10 min at 4,500 × g. Total RNA extraction was adapted as previously described (19, 40). Briefly, 700 μl of cells in 50 mM sodium acetate, pH 5.1, containing 10 mM EDTA was combined with 100 μl of 10% (wt/vol) SDS, 700 μl of saturated phenol (pH 4.3; Ameresco), 100 μl of glass beads (400 μm), and 100 μl of zirconia-silica beads (1.0 mm). Cells were shaken in a Mini-Beadbeater-96 for 2 min to lyse the cells. Samples were heat shocked at 65°C for 10 min, followed by 2 more min in the beadbeater. Then, samples were centrifuged at 12,000 × g for 5 min, and the aqueous layer was transferred to a new tube. Samples were extracted once with phenol (pH 4.5), twice with phenol-chloroform (1:1, vol/vol), and once with chloroform. RNA was precipitated from solution by adding 0.2 volume of 8 M LiCl to the tube and incubating the tube overnight at −80°C. RNA pellets were collected by centrifugation at 12,000 × g for 20 min and washed with 70% ethanol. Pellets were dried and resuspended in 40 μl of RNase-free water. DNA contamination was removed from samples by incubation with 5 μl of RQ1 RNase-free DNase (Promega, Madison, WI) for 60 min and then extracted once with phenol (pH 4.5) and twice with phenol-chloroform (1:1, vol/vol), followed by chloroform extraction. RNA was precipitated by addition of 0.1 volume of 3 M sodium acetate and 2 volumes of 100% ethanol and incubation overnight at −80°C. Samples were then centrifuged at 12,000 × g for 20 min, washed twice with 70% ethanol, and air dried. Samples were resuspended in RNase-free water and checked for quality and quantity with a NanoDrop spectrophotometer (Thermo Scientific, Waltham, MA). High-quality RNA preparations had a total RNA yield greater than 5 μg and an A260/A280 ratio greater than 1.7. RNA stability was also checked by Agilent RNA PicoChip analysis. Finally, bacterial rRNA was removed from 5 μg of total RNA with the Ribo-Zero magnetic kit for bacteria (Epicentre, Madison, WI).
RNA sequencing and analysis.
cDNA library construction and Illumina HiSeq2000 sequencing were performed at the University of Wisconsin Biotechnology Center (Madison, WI). Single-ended 100-bp sequencing runs were performed, and data were demultiplexed using the CASAVA software package. Our analysis pipeline began with trimming the fastq sequences to remove low-quality bases, adapter sequences, and reads with fewer than 60 bases (41). Trimmed fastq files were then aligned to their respective genomes using the Burrows Wheeler Aligner BWA-mem (42). Data were then converted to BAM files using Samtools, and alignment statistics were calculated with Bamtools (43). Reads with overlapping genes were then counted to quantify expression level using the R package Rsamtools. Statistical analysis was performed with the R package DEseq to identify differentially expressed genes and generate heatmaps of expression data. Scatter plots were generated with the R plot function (44). Homology networks were created by BLAST comparison of all CAZy annotated genes in SACTE and SDPB6. Proteins with a BLAST E value of <1e−50 were connected with an edge, as were CAZy family annotations. A network was generated with Cytoscape (45).
Nucleotide sequence accession numbers.
Genomic sequences are available at NCBI RefSeq as follows: SACTE, PRJNA72627; SACTG, PRJNA61899; SDPB6, PRJNA195846; and SPB74, PRJNA48415.
RESULTS
Cellulolytic activity screen.
Three strains of Streptomyces isolated from pine tree-associated insects have been previously reported: SirexAA-E (SACTE) and SirexAA-G (SACTG) from the Sirex woodwasp (20) and SPB74 from the southern pine beetle (23). A fourth Streptomyces strain, DpondAA-B6 (SDPB6), was isolated from a surface-sterilized adult mountain pine beetle collected from a naturally attacked Pinus contorta tree near Mackenzie, British Columbia, Canada (Fig. 1A) (27). Cellulolytic activities were significantly different among these four strains (Fig. 1). Only two of the four strains, SACTE and SDPB6, grew well in liquid medium containing filter paper as the only carbon source (Fig. 1B and C). Quantitative cellulose utilization assays were performed, and these two strains deconstructed 48.9% ± 2.3% and 44.3% ± 3.6% of crystalline cellulose in 10 days, respectively (Fig. 1C). The other two strains, SPB74 and SACTG, showed little to no growth in the qualitative assay, and essentially no filter paper deconstruction was determined by quantitative analysis.
Biochemical analysis of secreted enzymes.
Polysaccharide-hydrolyzing activities of the secreted proteins from these four strains supported significant differences between the two cellulolytic strains, SACTE and SDPB6, and the two noncellulolytic strains, SPB74 and SACTG. While all strains grew visibly on ammonia fiber expansion-pretreated corn stover (AFEX-CS [37]) as the sole carbon source, there were significant differences in the amount of total protein secreted by each strain. SACTE gave the highest concentration of total secreted protein (167 ± 5 μg/ml), followed by SPB74 (75 ± 11 μg/ml), then SDPB6 (69 ± 12 μg/ml), and SACTG (43 ± 9 μg/ml). When normalized to total protein concentration, the secretomes from SACTE and SDPB6 had significantly higher specific activity for hydrolysis than SACTG and SPB74 in reactions with cellulosic substrates (filter paper, Sigmacell, PASC, and lichenan), hemicellulosic substrates (xylan and mannan), and plant biomass (AFEX-CS) (P < 0.01 for all; Tukey-Kramer method) (Fig. 2). The secretomes from SDPB6 had significantly higher activity than those of SACTE when reacted against crystalline cellulose (24%; P = 0.0012) and cellulose III (83%; P = 0.0168). In contrast, secretomes from SACTE were significantly more active than those of SDPB6 when reacted against birch wood xylan (P < 0.0001), lichenan (P = 0.0409), and AFEX-CS (P = 0.0160). High-performance liquid chromatography (HPLC) analysis of the products produced by the reaction of both SACTE and SDPB6 secretomes with crystalline cellulose showed that cellobiose was the primary product.
FIG 2.
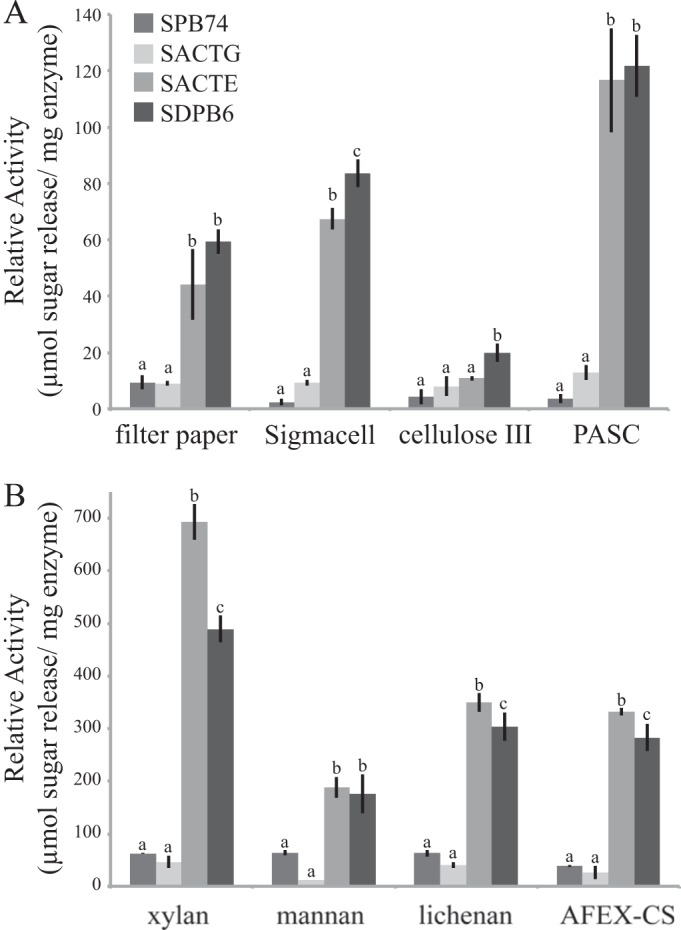
Reactions of secretomes from 4 Streptomyces strains with polysaccharide substrates. (A) Reducing sugars released from the reaction of the secretomes of 4 Streptomyces strains with filter paper, crystalline cellulose (Sigmacell-20), cellulose III, and amorphous cellulose (PASC). (B) Reducing sugars released from reaction with hemicellulosic polymers xylan, mannan, lichenan, and AFEX-pretreated corn stover. Data represent the means ± SDs from three experiments; lowercase letters represent statistically significant differences within each substrate (Tukey-Kramer method; P < 0.05).
Phylogenetic and genomic analyses.
Given the significant differences in biomass deconstruction activities of the four strains, we compared their genomes to gain insight into genetic differences that contribute to the phenotypes. A finished genome sequence was previously described for SACTE, and a draft sequence was described for SACTG (19, 20). Draft genomes for SPB74 and SDPB6 were generated at the Broad Institute and Joint Genome Institute, respectively (Table 1). Multilocus phylogenetic analysis of these four insect-associated Streptomyces strains, in addition to 13 strains with publicly available genome sequences, demonstrated that SDPB6 and SACTE are closely related species that form a monophyletic clade, as are SPB74 and SACTG. However, these two clades occur in separate lineages of Streptomyces (Fig. 3).
TABLE 1.
Genome statistics
| Parameter | SPB74 | SACTG | SACTE | SDPB6 |
|---|---|---|---|---|
| Genome size (bp) | 6,970,553 | 7,443,083 | 7,414,440 | 7,295,516 |
| No. of scaffolds | 2 | 254 | 1 | 36 |
| G+C (%) | 68.13 | 73.32 | 71.75 | 72.14 |
| No. of predicted ORFs | 5,808 | 6,612 | 6,647 | 6,362 |
| No. of protein-coding genes | 5,742 | 6,547 | 6,563 | 6,272 |
| No. of RNA genes | 66 | 65 | 84 | 90 |
| No. of CAZy proteinsa | 130 | 166 | 185 | 141 |
| % CAZy | 2.3 | 2.5 | 2.9 | 2.3 |
| No. of antiSMASH clustersb | 16 | 20 | 22 | 28 |
FIG 3.

Multilocus phylogenetic tree of the genus Streptomyces. The maximum-likelihood tree was generated from a concatenated alignment of 646 orthologous genes conserved in all genomes of 16 strains of Streptomyces plus the closely related outgroup Kitasatospora setae. Bootstrap support for the maximum-likelihood analysis is given at each node.
Genome analyses for the presence of carbohydrate-active enzymes (CAZy) revealed that SACTE had the highest percentage of CAZy genes in its genome, followed by SACTG, then SDPB6, and finally SPB74 (Table 1; see also Data Set S2 in the supplemental material). Genomic differences between the cellulolytic and noncellulolytic strains included specific families of glycoside hydrolases (GHs) (Table 2; see also Data Set S2). Notably, SPB74 and SACTG were missing genes that encode the important cellulose-degrading families GH5, GH9, GH74, and GH48 as well as the primary xylanase families GH10 and GH11. Furthermore, SPB74 and SACTG had significantly fewer carbohydrate binding modules (CBMs) in their genomes. The CBM2 family was expanded in the SACTE and SDPB6 genomes, with 10 copies per genome, while the genomes of SPB74 and SACTG contain only two CBM2 domains each. Interestingly, the two cellulolytic strains, SACTE and SDPB6, also had the most secondary metabolite gene clusters, as predicted through antiSMASH genome-based analyses (31). Given the dissimilarities observed in these strain's phenotypic, biochemical, and genomic analyses, we chose to focus further molecular analyses on the two cellulolytic strains, SACTE and SDPB6, to gain insights into how these organisms successfully degrade plant biomass.
TABLE 2.
Genomic compositions of putative biomass-degrading enzymes and accessory proteins
| Family | No. of genes in indicated strain |
Predicted functiona | |||
|---|---|---|---|---|---|
| SPB74 | SACTG | SACTE | SDPB6 | ||
| GH5 | 0 | 0 | 3 | 2 | Endoglucanase |
| GH8 | 0 | 0 | 0 | 0 | Endoglucanase |
| GH9 | 0 | 0 | 1 | 1 | Endoglucanase |
| GH12 | 1 | 1 | 1 | 1 | Endoglucanase |
| GH74 | 0 | 0 | 1 | 2 | Endoglucanase |
| GH6 | 2 | 3 | 1 | 2 | Cellobiohydrolase |
| GH48 | 0 | 0 | 1 | 1 | Cellobiohydrolase |
| GH1 | 3 | 3 | 6 | 6 | Beta-glucosidase |
| GH3 | 7 | 8 | 7 | 5 | Beta-glucosidase |
| AA10 | 1 | 1 | 6 | 4 | LPMO |
| GH10 | 0 | 0 | 1 | 1 | Xylanase |
| GH11 | 0 | 0 | 1 | 2 | Xylanase |
| GH13 | 9 | 9 | 12 | 11 | Amylase |
| GH18 | 4 | 4 | 7 | 7 | Chitinase |
| GH19 | 1 | 1 | 3 | 2 | Chitinase |
| CBM1 | 0 | 0 | 0 | 0 | Cellulose binding |
| CBM2 | 2 | 2 | 10 | 9 | Cellulose binding |
| CBM3 | 1 | 1 | 0 | 0 | Cellulose binding |
| CBM4 | 0 | 0 | 1 | 1 | Cellulose binding |
| CBM5 | 1 | 1 | 3 | 6 | Chitin binding |
| CBM12 | 0 | 0 | 2 | 7 | Chitin binding |
Predicted function based on CAZy database (26).
Secretome analysis on pure polysaccharides.
We used LC-MS/MS to identify proteins in the secretomes from SACTE and SDPB6 cultures grown in minimal medium containing cellulose, xylan, or glucose as the sole carbon source (see Data Sets S3 and S4 in the supplemental material). Relatively simple secretomes were found for both strains during growth on cellulose; 23 and 30 proteins constitute 95% of the total identified mass spectral counts from SACTE and SDPB6, respectively. In contrast, both SACTE and SDPB6 secreted a more diverse set of proteins in response to growth on either glucose (122 and 162 proteins, respectively) or xylan (92 and 125 proteins, respectively). Other than a constitutively secreted putative glucosylceramidase (SDPB6_04637) in SDPB6, the dominant proteins observed in the secretomes from xylan- and cellulose-grown cultures were absent in the secretomes of the glucose-grown cultures. In xylan-grown cultures, putative xylan-degrading enzymes were highly abundant: GH10 xylanases (SACTE_0265 and SDPB6_04909), putative xylose isomerase proteins (SACTE_5230, SDPB6_02230, and SDPB6_05756), and a xylanase from the GH11 family in SACTE (SACTE_0358) (see Data Sets S3 and S4).
In both the SACTE and SDPB6 cellulose secretomes, four homologous proteins represented 76% of the total spectral counts (Fig. 4A) (the threshold for counting proteins was greater than 1% of total spectral counts; the homology threshold was a BLAST E value of <1e−50). All four proteins were annotated as CAZymes and include the nonredundant set of enzymes necessary to deconstruct crystalline cellulose to cellobiose and a beta-mannanase: reducing- and nonreducing-end 1,4-beta cellobiohydrolases (GH6 and GH48, respectively), a lytic polysaccharide monooxygenase (LPMO; AA10), and a beta-mannanase (GH5). When the threshold was relaxed to include proteins with more than 0.1% of the total spectral counts, the core secretome contained 14 homologous proteins that accounted for 87% of the total counts (Fig. 4D).
FIG 4.

Composition of secretomes from cellulose-grown cultures of SACTE and SDPB6 identified by LC-MS/MS. (A) Homologous proteins identified in secretomes with more than 1% of the total spectral counts. (B and C) Proteins unique to secretomes from SDPB6 (B) and SACTE (C) with more than 1% of total spectral counts. (D) Homologous and unique proteins when threshold is lowered to proteins with more than 0.1% of the spectral counts.
One major difference between the SACTE and SDPB6 secretomes was the predominant type of enzyme produced for endocellulolytic hydrolysis of internal bonds in the cellulose chain (Fig. 4B and C). SACTE secreted a GH5 enzyme (SACTE_0482), while SDPB6 secreted two enzymes with predicted endoglucanase activity from the GH9 and GH12 families (SDPB6_01939 and SDPB6_04855, respectively). Furthermore, the percentage of the secretome representing endoglucanase activity was 10-fold greater in SDPB6 than in SACTE (23.1% and 2%, respectively). There were also significant differences in the detected abundance of putative cellulolytic LPMOs in each strain, 20% in SDPB6 and 7% in SACTE (SDPB6_03538 and SACTE_3159, respectively). When added to the endo-1,4-beta-glucosidase activity, 43% of the total spectral counts in cellulose-grown SDPB6 were from enzymes that cleave cellulose internally, versus 9% in SACTE. Conversely, the cellulose-grown SACTE secretome was enriched in xylan-degrading enzymes (GH10 SACTE_0265 and acetylxylan esterase SACTE_0357). The SDPB6 secretome also contained proteins with putative 1,3-beta-glucosidase and peptidase functions and a protein with unknown function (SDPB6_03112, SDPB6_02147, and SDPB6_02490, respectively). The absence of detectable spectra does not always correlate with the absence of a protein in a sample. Thus, we investigated mRNA expression to gain further insight into these differences.
RNA sequencing.
To gain further insight into how SACTE and SDPB6 deconstruct and metabolize plant biomass, we analyzed the differential expression of mRNA in these strains when grown in medium containing either glucose or AFEX-CS as the sole carbon source. In total, approximately 800 Mbp and 1 Gbp were sequenced for SACTE and SDPB6, respectively. When these reads were mapped to their respective genomes, gene coverage was approximately 500 to 2,000 reads per open reading frame (ORF) (see Table S1 in the supplemental material). Variation between biological replicates was measured by pairwise comparison of normalized reads for all protein-coding genes (see Fig. S2 in the supplemental material). Linear models (slope and r2 value) of the pairwise data demonstrate the actual fit of the data versus the theoretical perfect fit. SACTE glucose had the closest fit to the theoretical, followed by SDPB6 AFEX-CS, then SACTE AFEX-CS, and then SDPB6 glucose. Sample clustering of variance-stabilized data reflected our experimental design (i.e., glucose samples cluster together and AFEX-CS samples cluster together [see Fig. S3 in the supplemental material]). Together, these data quality assessments indicate that our experiment produced biologically relevant data.
Differential expression analysis.
Comparison of transcript abundance between growth in media containing either glucose or AFEX-CS as the sole carbon source identified 684 and 840 genes that were differentially expressed for SACTE and SDPB6, respectively (P < 0.05) (Fig. 5; see also Data Sets S3 and S6 in the supplemental material). SDPB6 differentially expressed 84 genes with >2-fold changes and 15 with >4-fold changes (Fig. 5, green and red circles, respectively; all fold change descriptions are log2 fold change). Similarly, in SACTE, 90 genes were differentially expressed with >2-fold changes and 23 with >4-fold changes. Interestingly, 22% of genes in the >4-fold differential expression (DE) category were CAZy annotated genes (Fig. 5, bold annotations). To confirm this observation, we categorized all genes from SDPB6 and SACTE into broad KEGG and CAZy categories; the CAZy functional category had the greatest number of >4-fold differentially expressed genes in both strains (see Data Set S7 in the supplemental material). ABC transporters were also significantly upregulated in both strains, as were a number of metabolic pathways (carbon, starch, and benzoate) and a number of nonpolar and basic amino acid biosynthetic pathways (Val, Ile, Leu, Lys, Pro, and Arg). Lastly, SACTE upregulated five transcription factors 4-fold or greater, while SDPB6 did not have any transcription factors with a >4-fold change.
FIG 5.
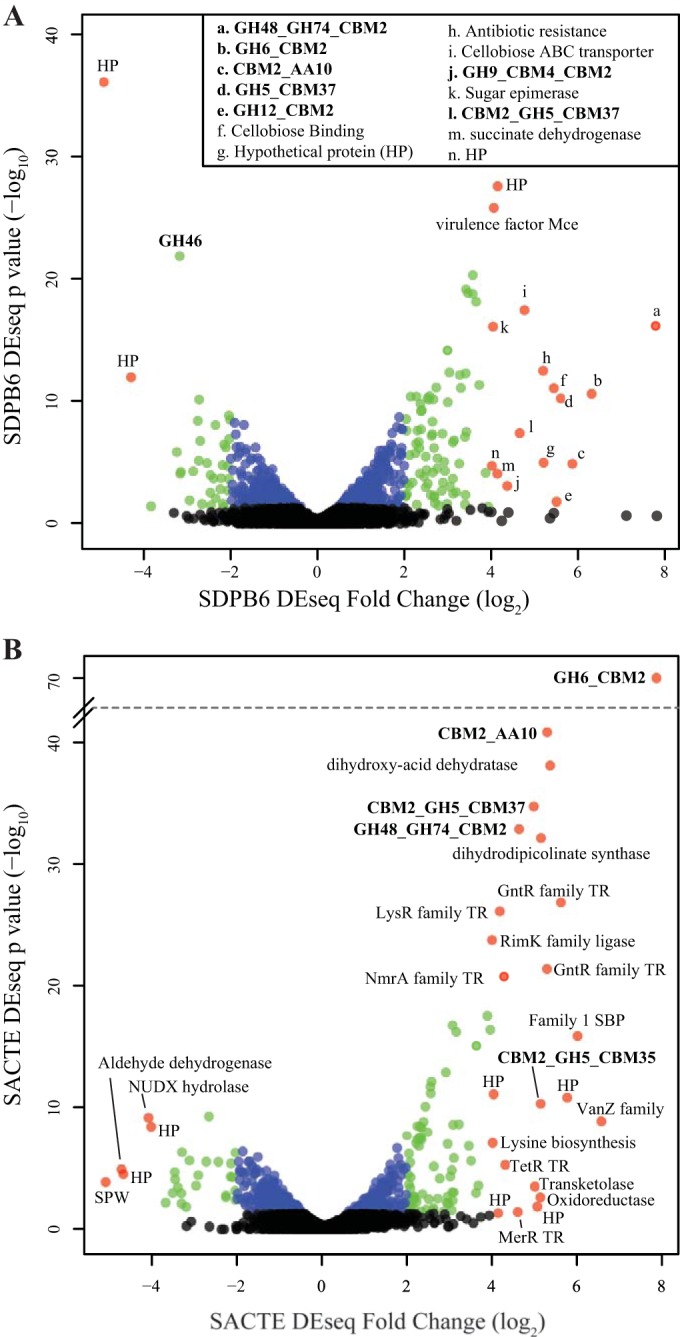
Differential expression of genes from SDPB6 (A) and SACTE (B) grown on glucose versus AFEX-pretreated corn stover. Each point represents a gene; the x axis shows the log2 fold change between carbon sources, and the y axis shows the −log10 of the P value for the measured fold change. Red points represent genes with more than a 4-fold change, green points represent genes with a 2- to 4-fold change, blue points represent genes with statistically significant changes less than 2-fold, and black points represent non-statistically significantly induced genes (P > 0.05). Annotations are shown for genes with more than a 4-fold change; bold indicates a CAZy gene family (HP, hypothetical protein).
To further examine the genes in the CAZy family that may contribute to the deconstruction of plant biomass, we built a homology network of all CAZy proteins from SDPB6 and SACTE (Fig. 6). Nodes in this network represent proteins (circles) or CAZy families (triangles), and edges represent annotations or homologous proteins (BLAST E value less than 1e−50). The size of protein nodes corresponds to the fold change observed in the RNA-Seq data. Only a small number of the total CAZy genes were significantly differentially expressed (>2-fold; SDPB6, 12/131, and SACTE, 17/166 [Fig. 6, inset; see also Data Sets S5 and S6 in the supplemental material]). However, a majority of genes that contain a CBM2 domain were significantly induced (SDPB6, 7/10, and SACTE, 8/11). Similar to the proteomic results, the RNA-Seq data identified a small combination of genes that encode enzymes to degrade crystalline cellulose to cellobiose (endoglucanase [GH5, GH9, and GH12], reducing-end exoglucanase [GH48], nonreducing-end exoglucanase [GH6], and polysaccharide monooxygenase [AA10]). The most highly induced genes in these two strains were both genes for exoglucanases: in SDPB6, the reducing-end GH48 family enzyme SDPB6_04936, and in SACTE, the nonreducing-end GH6 family enzyme SACTE_0237. Both strains also induced the gene homologous to each of these exoglucanase genes, SACTE_0236 and SDPB6_04935, respectively. Interestingly, each pair of nonreducing- and reducing-end exocellulases was concurrent in their respective genomes, which suggests coregulated expression. These two Streptomyces strains also induced homologous AA10 LPMOs in response to growth on plant biomass (SDPB6_03538 and SACTE_3159).
FIG 6.

Protein similarity network of CAZymes present in SDPB6 and SACTE. Nodes are proteins (circles) or CAZy functional categories (yellow triangles); edges indicate that the gene belongs to the respective CAZy family or a BLAST similarity with an E value less than 1e−50. Node size represents the fold change in RNA abundance between AFEX-CS and glucose. The inset shows all CAZy proteins; the expanded region shows highly induced genes.
While the two strains induced homologous GH9 endoglucanases (SDPB6_01939 and SACTE_3717), the most highly induced endoglucanases were from different GH families. SDPB6 expressed a GH12 enzyme (SDPB6_04855), while SACTE expressed a GH5 enzyme (SACTE_0482). The homolog of SDPB6_04855 in SACTE (SACTE_5809) does not contain a CBM2 domain and was not significantly expressed. Moreover, SDPB6 does not contain a homolog of the endocellulolytic SACTE GH5 enzyme (SACTE_0482).
DISCUSSION
Streptomyces strains are found in a wide variety of terrestrial and marine ecosystems. Classically, they have been viewed as free-living organisms, but recently they also have been found to be associated with eukaryotic hosts (20, 21, 23, 26, 46). Despite the ubiquitous presence of Streptomyces in most ecosystems (10), their capability to degrade plant biomass remains largely uncharacterized. In this study, we compared four strains of Streptomyces isolated from pine-boring insect herbivores. Using a combination of biochemical and molecular tools, we have provided insight into the biomass-deconstructing capability of these Streptomyces and the similarities and differences in enzymes they use to break down plant biomass.
Phenotypic and biochemical data showed that two of the strains, SACTE and SDPB6, are highly efficient at degrading both plant biomass and pure polysaccharides, whereas SPB74 and SACTG have limited activity on all polysaccharide substrates tested (Fig. 1 and 2). Our phylogenetic analysis showed that these two pairs of Streptomyces strains are in separate lineages of the genus. Analysis of the content of CAZy genes identified important genetic differences between the cellulolytic and noncellulolytic strains. While CAZy genes are abundant in the genomes of all four strains (2.3% to 2.9%), the SPB74 and SACTG genomes are missing genes encoding critical endoglucanase, cellobiohydrolase, and xylanase families and are also depleted in CBM2 domains. The absence of these enzymes and binding modules likely impacts the ability of these organisms to efficiently deconstruct the diverse sugar linkages found in plant cell walls. From an evolutionary perspective, it is interesting to consider whether the abundance of CAZy genes in SACTE and SDPB6 represents selection toward enhanced biomass-degrading activity or whether the absence of important CAZymes in SPB74 and SACTG genomes represents specialization for another purpose, such as secondary-metabolite production. SPB74 has been shown to produce a novel secondary metabolite, mycangimycin, which inhibits growth of an antagonistic fungus of the beetle system (23, 47). These insect-associated strains may have coevolved with their hosts to play either a nutritional or protective role. Further microbiology and genomic analyses are needed to understand the evolution of these strains and their putative symbiotic roles in these insect-microbe systems.
Proteomic and transcriptomic analyses of SACTE and SDPB6 revealed homologous and relatively simple secreted enzyme cocktails used to deconstruct plant biomass. The major exocellulases, LPMOs, and mannanases were conserved in both organisms. In contrast, the molecular data from SACTE and SDPB6 showed that these strains use endocellulases from different enzyme families (SACTE_0482, GH5, and SDPB6_04855, GH12). The genome of SDPB6 does not contain a homolog to SACTE_0482. Conversely, while SACTE has a homologous GH12 enzyme in its genome, this gene does not contain a CBM2 (Fig. 6). We hypothesize that together with their evolutionary relatedness, the endocellulolytic activities of the GH5 and GH12 enzymes have been selected by the insect-host and environment for slightly different functional activities. The SDPB6 secretome contains twice as many cellulolytic LPMOs as does that of SACTE based on mass spectral counts (Fig. 4), suggesting differential reliance on oxidative cellulose deconstruction in the two strains. This variation in endocellulolytic and LPMO enzymes may contribute to the small but significant differences observed in the secretome activity assays for hydrolysis of crystalline cellulose (Fig. 2).
All highly expressed enzymes in SACTE and SDPB6 shared conserved enzyme domain architectures; i.e., they all contained a CBM2 domain linked to a catalytic domain, even though the majority of CAZy genes in both genomes are not linked to a CBM2 domain. In contrast, the SPB74 and SACTG genomes contain only two CBM2 domains (GH6_CBM2 and GH18_CBM2). This conservation of enzyme domain architecture suggests that highly cellulolytic Streptomyces strains have converged on the use of secreted enzymes fused to an individual CBM2 domain. This pattern is similar to that for fungal secreted cellulases, which primarily consist of enzymes fused to CBM1 domains (48). It is possible that the similar, filamentous cellular morphology of fungi and Streptomyces may have selected for secretion of free enzymes with this similar domain architecture. Additionally, non-CAZy genes likely play a role in plant biomass deconstruction. Genes classified as ABC transporter genes were significantly upregulated; the products of these genes may be involved in transport of solubilized sugars into the cell. Genes in the benzoate degradation pathway were also upregulated; these proteins may deconstruct aromatic rings in lignin (49).
This work identified two phylogenetically related strains of cellulolytic Streptomyces isolated from insect herbivores and established an overall similarity in the enzymatic apparatus they use for cellulose deconstruction. Further sampling from insect herbivore systems will help clarify how these Streptomyces and their relatively simple external aerobic enzyme complexes help insects efficiently unlock the vast amount of energy stored in the cell walls of plants. The simplicity of these cellulolytic enzyme complexes may also provide insights into how to modify the currently complex enzyme cocktails needed for the production of sustainable lignocellulosic biofuels.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science; grant DE-FC-02-07ER64494). Funding for J.C. and C.R.C. was provided by the National Institutes of Health (grant GM096347). Funding for G.R.L. was provided by the National Science Foundation (grant GRFP DGE-1256259).
We thank Irene Ong for help with developing our RNA-Seq analysis pipeline.
Footnotes
Published ahead of print 16 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01133-14.
REFERENCES
- 1.Schlesinger WH, Andrews JA. 2000. Soil respiration and the global carbon cycle. Biogeochemistry 48:7–20. 10.1023/A:1006247623877 [DOI] [Google Scholar]
- 2.Klemm D, Heublein B, Fink HP, Bohn A. 2005. Cellulose: fascinating biopolymer and sustainable raw material. Angew. Chem. Int. Ed. Engl. 44:3358–3393. 10.1002/anie.200460587 [DOI] [PubMed] [Google Scholar]
- 3.Lynd LR, Weimer PJ, van Zyl WH, Pretorius IS. 2002. Microbial cellulose utilization: fundamentals and biotechnology. Microbiol. Mol. Biol. Rev. 66:506–577. 10.1128/MMBR.66.3.506-577.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh BK, Bardgett RD, Smith P, Reay DS. 2010. Microorganisms and climate change: terrestrial feedbacks and mitigation options. Nat. Rev. Microbiol. 8:779–790. 10.1038/nrmicro2439 [DOI] [PubMed] [Google Scholar]
- 5.Glass NL, Schmoll M, Cate JHD, Coradetti S. 2013. Plant cell wall deconstruction by ascomycete fungi. Annu. Rev. Microbiol. 67:477–498. 10.1146/annurev-micro-092611-150044 [DOI] [PubMed] [Google Scholar]
- 6.Desvaux M. 2005. Clostridium cellulolyticum: model organism of mesophilic cellulolytic clostridia. FEMS Microbiol. Rev. 29:741–764. 10.1016/j.femsre.2004.11.003 [DOI] [PubMed] [Google Scholar]
- 7.Brunecky R, Alahuhta M, Xu Q, Donohoe BS, Crowley MF, Kataeva IA, Yang SJ, Resch MG, Adams MWW, Lunin VV, Himmel ME, Bomble YJ. 2013. Revealing nature's cellulase diversity: the digestion mechanism of Caldicellulosiruptor bescii CelA. Science 342:1513–1516. 10.1126/science.1244273 [DOI] [PubMed] [Google Scholar]
- 8.Dam P, Kataeva I, Yang SJ, Zhou FF, Yin YB, Chou WC, Poole FL, Westpheling J, Hettich R, Giannone R, Lewis DL, Kelly R, Gilbert HJ, Henrissat B, Xu Y, Adams MWW. 2011. Insights into plant biomass conversion from the genome of the anaerobic thermophilic bacterium Caldicellulosiruptor bescii DSM 6725. Nucleic Acids Res. 39:3240–3254. 10.1093/nar/gkq1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fontes CMGA, Gilbert HJ. 2010. Cellulosomes: highly efficient nanomachines designed to designed to deconstruct plant cell wall complex carbohydrates. Annu. Rev. Biochem. 79:655–681. 10.1146/annurev-biochem-091208-085603 [DOI] [PubMed] [Google Scholar]
- 10.Janssen PH. 2006. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 72:1719–1728. 10.1128/AEM.72.3.1719-1728.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wachinger G, Bronnenmeier K, Staudenbauer WL, Schrempf H. 1989. Identification of mycelium-associated cellulase from Streptomyces reticuli. Appl. Environ. Microbiol. 55:2653–2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schlochtermeier A, Walter S, Schroder J, Moorman M, Schrempf H. 1992. The gene encoding the cellulase (Avicelase) Cel1 from Streptomyces reticuli and analysis of protein domains. Mol. Microbiol. 6:3611–3621 [DOI] [PubMed] [Google Scholar]
- 13.Walter S, Schrempf H. 1996. Physiological studies of cellulase (Avicelase) synthesis in Streptomyces reticuli. Appl. Environ. Microbiol. 62:1065–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forsberg Z, Vaaje-Kolstad G, Westereng B, Bunaes AC, Stenstrom Y, MacKenzie A, Sorlie M, Horn SJ, Eijsink VGH. 2011. Cleavage of cellulose by a CBM33 protein. Protein Sci. 20:1479–1483. 10.1002/pro.689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marushima K, Ohnishi Y, Horinouchi S. 2009. CebR as a master regulator for cellulose/cellooligosaccharide catabolism affects morphological development in Streptomyces griseus. J. Bacteriol. 191:5930–5940. 10.1128/JB.00703-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yagüe P, Rodriguez-Garcia A, Lopez-Garcia MT, Martin JF, Rioseras B, Sanchez J, Manteca A. 2013. Transcriptomic analysis of Streptomyces coelicolor differentiation in solid sporulating cultures: first compartmentalized and second multinucleated mycelia have different and distinctive transcriptomes. PLoS One 8(3):e60665. 10.1371/journal.pone.0060665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodríguez-Garcia A, Barreiro C, Santos-Beneit F, Sola-Landa A, Martin JF. 2007. Genome-wide transcriptomic and proteomic analysis of the primary response to phosphate limitation in Streptomyces coelicolor M145 and in a ΔphoP mutant. Proteomics 7:2410–2429. 10.1002/pmic.200600883 [DOI] [PubMed] [Google Scholar]
- 18.Hara H, Ohnishi Y, Horinouchi S. 2009. DNA microarray analysis of global gene regulation by A-factor in Streptomyces griseus. Microbiology 155:2197–2210. 10.1099/mic.0.027862-0 [DOI] [PubMed] [Google Scholar]
- 19.Takasuka TE, Book AJ, Lewin GR, Currie CR, Fox BG. 2013. Aerobic deconstruction of cellulosic biomass by an insect-associated Streptomyces. Sci. Rep. 3:1030. 10.1038/srep01030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adams AS, Jordan MS, Adams SM, Suen G, Goodwin LA, Davenport KW, Currie CR, Raffa KF. 2011. Cellulose-degrading bacteria associated with the invasive woodwasp Sirex noctilio. ISME J. 5:1323–1331. 10.1038/ismej.2011.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pasti MB, Belli ML. 1985. Cellulolytic activity of actinomycetes isolated from termites (Termitidae) gut. FEMS Microbiol. Lett. 26:107–112 [Google Scholar]
- 22.Watanabe Y, Shinzato N, Fukatsu T. 2003. Isolation of actinomycetes from termites' guts. Biosci. Biotechnol. Biochem. 67:1797–1801. 10.1271/bbb.67.1797 [DOI] [PubMed] [Google Scholar]
- 23.Scott JJ, Oh DC, Yuceer MC, Klepzig KD, Clardy J, Currie CR. 2008. Bacterial protection of beetle-fungus mutualism. Science 322:63. 10.1126/science.1160423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slippers B, Coutinho TA, Wingfield BD, Wingfield MJ. 2003. A review of the genus Amylostereum and its association with woodwasps. S. Afr J. Sci. 99:70–74 [Google Scholar]
- 25.Vasanthakumar A, Handelsman J, Schloss PD, Bauer LS, Raffa KF. 2008. Gut microbiota of an invasive subcortical beetle, Agrilus planipennis Fairmaire, across various life stages. Environ. Entomol. 37:1344–1353. 10.1603/0046-225X(2008)37[1344:GMOAIS]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- 26.Seipke RF, Kaltenpoth M, Hutchings MI. 2012. Streptomyces as symbionts: an emerging and widespread theme? FEMS Microbiol. Rev. 36:862–876. 10.1111/j.1574-6976.2011.00313.x [DOI] [PubMed] [Google Scholar]
- 27.Adams AS, Aylward FO, Adams SM, Erbilgin N, Aukema BH, Currie CR, Suen G, Raffa KF. 2013. Mountain pine beetles colonizing historical and naive host trees are associated with a bacterial community highly enriched in genes contributing to terpene metabolism. Appl. Environ. Microbiol. 79:3468–3475. 10.1128/AEM.00068-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balows A. 1992. The prokaryotes: a handbook on the biology of bacteria. Ecophysiology, isolation, identification, applications. Springer-Verlag, New York, NY [Google Scholar]
- 29.Weimer PJ, Lopezguisa JM, French AD. 1990. Effect of cellulose fine-structure on kinetics of its digestion by mixed ruminal microorganisms in vitro. Appl. Environ. Microbiol. 56:2421–2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goering HK, Van Soest PJ. 1970. Forage fiber analyses (apparatus, reagents, procedures, and some applications). US Agricultural Research Service, Washington, DC [Google Scholar]
- 31.Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R. 2011. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 39:W339–W346. 10.1093/nar/gkr466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eddy SR. 2011. Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. 10.1371/journal.pcbi.1002195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haft DH, Selengut JD, White O. 2003. The TIGRFAMs database of protein families. Nucleic Acids Res. 31:371–373. 10.1093/nar/gkg128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30:772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teymouri F, Laureano-Perez L, Alizadeh H, Dale BE. 2004. Ammonia fiber explosion treatment of corn stover. Appl. Biochem. Biotechnol. 113:951–963. 10.1385/ABAB:115:1-3:0951 [DOI] [PubMed] [Google Scholar]
- 38.Schülein M. 1997. Enzymatic properties of cellulases from Humicola insolens. J. Biotechnol. 57:71–81. 10.1016/S0168-1656(97)00090-4 [DOI] [PubMed] [Google Scholar]
- 39.Miller GL. 1959. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Chem. 31:426–428. 10.1021/ac60147a030 [DOI] [Google Scholar]
- 40.Chen JQ, Weimer PJ. 2001. Competition among three predominant ruminal cellulolytic bacteria in the absence or presence of non-cellulolytic bacteria. Microbiology 147:21–30 [DOI] [PubMed] [Google Scholar]
- 41.Lohse M, Bolger AM, Nagel A, Fernie AR, Lunn JE, Stitt M, Usadel B. 2012. RobiNA: a user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Res. 40:W622–W627. 10.1093/nar/gks540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol. 11(10):R106. 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13:2498–2504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Poulsen M, Oh DC, Clardy J, Currie CR. 2011. Chemical analyses of wasp-associated Streptomyces bacteria reveal a prolific potential for natural products discovery. PLoS One 6(2):e16763. 10.1371/journal.pone.0016763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oh DC, Scott JJ, Currie CR, Clardy J. 2009. Mycangimycin, a polyene peroxide from a mutualist Streptomyces sp. Org. Lett. 11:633–636. 10.1021/ol802709x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37:D233–D238. 10.1093/nar/gkn663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bianchetti CM, Harmann CH, Takasuka TE, Hura GL, Dyer K, Fox BG. 2013. Fusion of dioxygenase and lignin-binding domains in a novel secreted enzyme from cellulolytic Streptomyces sp. SirexAA-E. J. Biol. Chem. 288:18574–18587. 10.1074/jbc.M113.475848 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.