

Background: Hyaluronan (HA)-mediated angiogenesis has been implicated in tumor progression.
Results: LMW-HA-mediated transactivation of EphA2 is required for PATJ and Dbs membrane recruitment and subsequent RhoA activation required for angiogenesis.
Conclusion: EphA2 plays a crucial role in HA-mediated angiogenesis.
Significance: Targeting downstream effectors of LMW-HA could be a useful therapeutic intervention for angiogenesis-associated diseases including various malignancies.
Keywords: Angiogenesis; CD44; Endothelial Cell; Hyaluronan; Hyaluronate; Ras Homolog Gene Family, Member A (RhoA); Dbs; EphA2; PATJ; Anginex
Abstract
Angiogenesis or the formation of new blood vessels is important in the growth and metastatic potential of various cancers. Therefore, understanding the mechanism(s) by which angiogenesis occurs can have important therapeutic implications in numerous malignancies. We and others have demonstrated that low molecular weight hyaluronan (LMW-HA, ∼2500 Da) promotes endothelial cell (EC) barrier disruption and angiogenesis. However, the mechanism(s) by which this occurs is poorly defined. Our data indicate that treatment of human EC with LMW-HA induced CD44v10 association with the receptor-tyrosine kinase, EphA2, transactivation (tyrosine phosphorylation) of EphA2, and recruitment of the PDZ domain scaffolding protein, PATJ, to the cell periphery. Silencing (siRNA) CD44, EphA2, PATJ, or Dbs (RhoGEF) expression blocked LMW-HA-mediated angiogenesis (EC proliferation, migration, and tubule formation). In addition, silencing EphA2, PATJ, Src, or Dbs expression blocked LMW-HA-mediated RhoA activation. To translate our in vitro findings, we utilized a novel anginex/liposomal targeting of murine angiogenic endothelium with either CD44 or EphA2 siRNA and observed inhibition of LMW-HA-induced angiogenesis in implanted Matrigel plugs. Taken together, these results indicate LMW-HA-mediated transactivation of EphA2 is required for PATJ and Dbs membrane recruitment and subsequent RhoA activation required for angiogenesis. These results suggest that targeting downstream effectors of LMW-HA could be a useful therapeutic intervention for angiogenesis-associated diseases including tumor progression.
Introduction
CD44, initially described as an adhesion molecule involved in leukocyte migration, has since emerged as an important family of adhesion receptors expressed on multiple cell types (1, 2). CD44 is involved in many different cell processes including cytokine secretion, chemokine gene expression, and cell proliferation and differentiation (3). Alternative splicing of the CD44 transcript can give rise to a number of CD44 variants that express an extra insert in their extracellular membrane proximal domain. CD44 has previously been reported to interact with a wide range of cytoskeletal and signaling molecules including cytoskeletal proteins (4), growth factor receptors including ErbB2 (5) and EGF receptor (6), and Rho/Rac nucleotide exchange factors such as Tiam1, Vav-2, and LARG (7) along with Src family tyrosine kinases (8). CD44 is the primary cell surface receptor for hyaluronan (HA),2 an integral component of the extracellular matrix (9, 10).
HA is a non-sulfated glycosaminoglycan with a molecular mass of up to 104 kDa and is made up of repeating disaccharide subunits of β-1,3-N-acetyl-d-glucosamine in linkage to β-1,4-d-glucuronic acid (11). HA is an abundant part of the extracellular matrix (ECM) where it is involved in a number of processes including organization of the ECM, tissue hydration, lubrication, and filtration. HA can also play a more active biological role and is involved in cell proliferation, differentiation, and migration by acting as a ligand for specific cell surface receptors including CD44, RHAMM, and LYVE-1 (12–15) Lower molecular weight forms of HA, which result from degradation of the native high molecular form via hyaluronidase enzymes or reactive oxygen species, are also found in the body. Increased HA concentrations are observed in bronchoalveolar lavage fluid and/or plasma from patients with lung disorders such as acute lung injury/acute respiratory distress syndrome (ALI/ARDS), pulmonary fibrosis, chronic obstructive pulmonary disease (16), allergic alveolitis, asthma, interstitial lung disease, sarcoidosis, and idiopathic pulmonary arterial hypertension (17). Increased HA content has been detected in the tumor microenvironment of most human malignancies and is associated with an aggressive phenotype and correlates with a poor clinical prognosis (18, 19).
The Eph family of receptor-tyrosine kinases (the largest subfamily of tyrosine kinases) and their ligands, the ephrins, have emerged as important regulators of endothelial cell function, particularly during angiogenesis and in barrier function (20, 21) The Eph receptors and their ligands are expressed in adult tissues and during embryonic development (22, 23). The Eph receptor family is subdivided into two classes based on homology and ligand binding. EphA receptors bind mainly to ephrin-A ligands, which are associated with the cell membrane via a glycosylphosphatidylinositol linkage, whereas EphB receptors bind ephrin-B ligands, which are transmembrane proteins. The intracellular domain of the Eph receptors contains two autophosphorylation sites and the kinase domain along with a sterile alpha motif and PDZ domain-binding motif which can serve as docking sites for downstream signaling molecules (23). Another important aspect of the Eph-Ephrin interaction is bidirectional signaling. Although activation of the kinase domain within the Eph receptor facilitates “forward” signaling, “reverse” signaling may also be initiated via the membrane-bound ephrin ligands (22). Binding of ephrin-A1 by the EphA2 receptor leads to trans/autophosphorylation of its cytoplasmic domains, although it may also act as a substrate for Src family kinases (24). These phosphorylated sites then act as binding sites for downstream signaling molecules including Vav2/3, Shc, Cbl, and SHP-2. The EphA2 receptor and its ligand ephrin-A1 have recently gained further attention as their role in regulating angiogenesis and endothelial barrier function, promoting tumor metastasis and resistance to tyrosine kinase inhibitors has emerged (25). EphA2 receptor activation by ephrin-A1 increases endothelial permeability in cultured pulmonary aortic endothelial cells as measured using transendothelial electrical resistance (21). EphA2 activation can lead to an increase in active RhoA, resulting in the destabilization of adherens and tight junctions and a decrease in VE-cadherin localization at endothelial cell-cell contacts. Ephrin-A1 treatment also leads to disrupted epithelial cell-cell contacts (26).
Angiogenesis is an essential phenotype in a number of physiologic and pathologic processes including growth and development (27), wound healing (28), and reproduction (29). Inadequate angiogenesis contributes to ulcer formation (30), whereas excessive angiogenesis contributes to the pathology of arthritis (31), psoriasis (32), and neoplasia (33). In a series of now classical experiments, Folkman and colleagues demonstrated that solid tumors cannot grow larger than 3–4 mm in diameter unless they induce their own blood supply (34). In the current study we investigated the mechanism of LMW-HA-mediated human pulmonary microvascular endothelial cell angiogenesis. We have identified a novel Src-mediated transactivation of the EphA2 receptor that occurs upon LMW-HA binding to the CD44 variant, CD44v10. Furthermore, we demonstrated the involvement of the PATJ (Pals-1-associated tight junction protein) scaffolding protein and its role in recruiting specific RhoGEFs in response to LMW-HA or ephrin-A1 activation of CD44 or EphA2, respectively. These events culminated in RhoA activation, a crucial step in LMW-HA-mediated angiogenesis. Our findings demonstrate a critical interaction between two key signaling pathways regulating endothelial function. This insight may identify novel targets in the treatment of numerous angiogenesis-related pathologies.
EXPERIMENTAL PROCEDURES
Antibodies and Reagent
Antibodies utilized in this study include anti-EphA2, Dbs, LARG, RhoGEF p115, Syx (Santa Cruz Biotechnology, Santa Cruz, CA), anti-phospho-EphA2Tyr594 (Cell Applications), anti-CD44 (IM7 clone) (BD Biosciences), anti-CD44v10 (Novus Biologicals), anti-PATJ (GeneTex Inc), anti-EphA2, anti-Src (Cell Signaling), anti-phospho-Src (Novus Biologicals), and anti-actin (Sigma). Recombinant ephrin-A1-Fc protein was purchased from R&D Systems. Hyaluronic acid (sodium salt from Streptococcus zooepidemicus) was purchased from Sigma. Anginex was purchased from Phoenix Pharmaceuticals, Inc. (Burlingame, CA). Reagents for SDS-PAGE electrophoresis were purchased from Bio-Rad, and Immobilon-P transfer membrane was purchased from Millipore (Millipore Corp., Bedford, MA).
Preparation of LMW-HA
LMW-HA was prepared similar to that as previously described (8). Briefly 500 mg of hyaluronan sodium salt from S. zooepidemicus was digested with 20,000 units of bovine testicular hyaluronidase (Type VI-S, lyophilized powder, 3,000–15,000 units/mg (Sigma, H3631) in digestion buffer (0.1 m sodium acetate, pH 5.4, 0.15 m NaCl) for 24 h, and the reaction was stopped with 10% trichloroacetic acid. The resulting solution was centrifuged in an Ultrafree-MCTM Millipore 5-kDa molecular mass cutoff filter, and the flow-through was dialyzed against distilled water for 24 h at 4 °C in 500-Da cutoff Spectra-Por tubing (Pierce-Warriner, Chester, UK). LMW-HA was quantitated using an ELISA-like competitive binding assay with a known amount of fixed HA and biotinylated HA-binding peptide as the indicator (Echelon Inc). LMW-HA solutions were filtered through 0.22-μm filters and kept in sterile tubes. In some cases both low and high molecular weight HAs were subject to boiling, proteinase K (50 μg/ml) digestion, hyaluronidase SD digestion (Streptococcus dysgalactiae, NorthStar Bioproducts Associates of Cape Cod Inc., East Falmouth, MA (100741-1A), 100 milliunits/ml utilized), or the addition of boiled (inactivated) hyaluronidase SD to test for possible protein/lipid contaminants (35). To test for endotoxin contamination of HA, a lipopolysaccharides (LPS) bioAssay ELISA kit (USBiological Life Sciences) was used. LMW-HA with HA standards (Sigma and Enzo Life Sciences) was run on 4–20% Tris borate-EDTA gels and stained with Stains-All (Sigma) to confirm LMW-HA purity and size.
Human Pulmonary Microvascular Endothelial Cell (HPMVEC) Culture
Primary human pulmonary microvascular cells were purchased from Lonza and maintained in EBM-2 growth media supplemented with EGM-2-MV Bulletkit and 10% fetal bovine serum. Cells were maintained at 37 °C in a humidified atmosphere of 5% CO2, 95% air and used for experimentation at passages 3–6.
Inhibition of Protein Expression in Human EC Using siRNA
Human lung microvascular EC were transfected with siRNA against specific mRNA (Santa Cruz Biotechnology) using siPORTamineTM as the transfection reagent (Ambion) according to the protocol provided by Ambion. Cells (∼40% confluent) were serum-starved for 1 h followed by incubated with 250 nm concentrations of target siRNA (or scramble siRNA or no siRNA) for 6 h in serum-free media. The serum-containing media was then added (10% serum final concentration) for 42 h before biochemical experiments, and/or functional assays were conducted. Effective silencing of target protein expression was determined with immunoblot analysis of siRNA-transfected EC lysates using specific antibodies.
Immunoprecipitation and Immunoblotting
Cellular materials from treated or untreated HPMVEC were incubated with immunoprecipitation buffer (50 mm HEPES, pH 7.5, 150 mm NaCl, 20 mm MgCl2, 1% Nonidet P-40, 0.4 mm Na3VO4, 40 mm NaF, 50 μm okadaic acid, 0.2 mm phenylmethylsulfonyl fluoride, and a 1:250 dilution of Calbiochem protease inhibitor mixture 3). The samples were then immunoprecipitated with either anti-EphA2 or anti-PATJ IgG followed by SDS-PAGE in 4–15% polyacrylamide gels, transferred onto ImmobilonTM membranes, and developed with specific primary and secondary antibodies. Visualization of immunoreactive bands was achieved using enhanced chemiluminescence (Amersham Biosciences). In some instances computer-assisted densitometry was used to quantitate immunoreactive bands.
HLMVEC Proliferation Assay
Measurement of in vitro EC growth was performed as we have previously described (36). Control, VEGF (200 pg/ml) or LMW-HA (0.1–1000 nm)-pretreated EC (5 × 103 cells/well) were incubated with 0.2 ml of serum-free media for 72 h at 37 °C in 5%CO2, 95% air in 96-well culture plates. The in vitro cell proliferation assay was analyzed by measuring increases in cell number using the CellTiter96TM MTS assay (Promega, Madison, WI) and read at 492 nm. Each assay was set up in triplicate and repeated at least five times.
RhoA Activation Assay
After agonist and/or inhibitor treatment, ECs are solubilized in solubilization buffer and incubated with Rho-bonding domain-conjugated beads for 30 min at 4 °C. The supernatant was removed, and the Rho-bonding domain beads with the GTP-bound form of RhoA bound were washed extensively. The Rho-bonding domain beads were boiled in SDS-PAGE sample buffer, and the bound RhoA material was run on SDS-PAGE, transferred to ImmobilonTM, and immunoblotted with anti-RhoA antibody.
Matrigel Tubule Formation Assay
The tubule formation assay was adapted from the method described here (37). Briefly Matrigel (BD Biosciences) was mixed with 2% serum EBM-2 media in a 1:1 ratio, used to coat 12 well plates (500 μl per well), and allowed to polymerize at 37 °C for 30 min. Control or silenced endothelial cells were then seeded into each well (5000 cells/cm2) in 2% serum EBM-2. LMW-HA (100 nm) or an equivalent volume of PBS was then added to the appropriate wells. Cells were then incubated for 6 h to allow for tubule formation. Tubule formation was then recorded. 10 images were recorded per well, and ImageJ was used to measure total tubule length per image. Each treatment was performed in triplicate, and experiments were repeated three times. Results are expressed as tubule length per treatment.
Transwell Migration Assay
Transwell filters (8-μm pore size) were purchased from Corning Costar. Control or silenced endothelial cells were plated in triplicate in the upper chamber of the transwell filter in serum-free media, and serum-free media with LMW-HA (100 nm) was added to the lower well. Cells were incubated and allowed to migrate for 24 h. For the inhibitor studies cells were pretreated for 1 h before seeding on the transwell filter. Cell migration was then quantified by counting the number of cells that had migrated to the bottom well and expressing as a percentage of the total number of cells initially plated.
In Vivo LMW-HA-mediated Matrigel Plug Assay Using Anginex-conjugated Liposome Delivery of CD44 and EphA2 siRNA
Animal protocols were approved by the University of Chicago Institutional Animal Care and Use Committee, and all animals were cared for according to the national Institute of Health guidelines for the care and use of laboratory animals. Matrigel was purchased from BD Biosciences. Briefly, C57BL/6J mice were lightly anesthetized with ketamine (100 mg/kg) and xylazine (8 mg/kg) and were injected subcutaneously with 500 μl of Matrigel, which was premixed with vehicle or LMW-HA. The injections were made rapidly with a 30-gauge 1/2-inch needle to ensure the entire content was delivered as a single plug. The mice were allowed to recover, and 7 days later the silencing RNA complexes were delivered via liposome injection. Liposomes were prepared as we have previously described (38) using a 1:1 molar ratio of DOTAP to DOPE dissolved in chloroform to a concentration of 10 mg/ml. The solvent was evaporated in a water bath set at 50 °C under nitrogen. The resulting dry lipid film was immediately suspended in 100 μl of PBS (pH 7.4, final concentration 20 mg/ml). The cationic lipid dispersion was combined with no siRNA (control), siSTABLE CD44, EphA2, or control siRNA (ThermoScientific Dharmacon), or pCMV6 empty vector or GFP-expression vector (pCMV6-A-GFP Vector, Origene, Rockville, MD) (1 μg per 10 μg lipid) in a glass container. The liposome-siRNA mixture was sonicated in a water bath sonicator (Fisher) to clarity. For the anginex peptide (39, 40), primary amines were blocked with sulfo-NHS acetate in PBS, pH 7.4, and incubated for 1 h at room temperature. The solution was then filtered with a 5-kDa filter (Ultrafree-MC) and adjusted to a final concentration of 0.2 mg/ml. The modified peptide was cross-linked to liposomes containing siRNAs or expression vectors by covalent linking of the carboxyl groups on the anginex peptide with the amine groups on liposomes using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloric acid reagent (Pierce). Labeled liposomes were purified by dialysis in a Slide-A-Lyzer (20-kDa cutoff; Pierce) against a 1000-fold excess volume of sterile PBS, pH 7.4, overnight. Sterile anginex-conjugated liposomes containing no siRNA (control), CD44, EphA2, or control siRNA (100 μl) or pCMV6 empty vector or GFP expression vector (pCMV6-A-GFP Vector, Origene) were injected into the internal jugular vein of C57B6/6J mice. Animals were treated for 7 days with siRNA (10 mg/kg). The animals were then sacrificed, and the Matrigel plugs were harvested and fixed in 10% buffered formalin PBS solution. Matrigel plugs were then either: 1) depolymerized by a BD Cell Recovery Solution (BD Biosciences) and each plug dissolved in 300 μl of recovery solution as described in manufacturer's protocol, run on SDS-PAGE, and immunoblotted for specific proteins; 2) placed in OCT embedding medium, cut in 12-μm-thick sections and processed for GFP staining with anti-turboGFP antibody (OriGene); Alexa 488-conjugated secondary antibodies (Molecular Probes, Eugene, OR) were then used to visualize GFP staining. Nuclear staining was performed with DAPI; or 3) processed for Masson's Trichrome histological staining to identify invaded cells. The prepared slides were then scanned using a Pannoramic Scan (3DHistech) whole slide scanner. Ten fields of view were selected per slide, and the number of cells per field was counted using ImageJ. These experiments were repeated three times with five animals per treatment group. Results are expressed as the number of cells/plug.
Statistical Analysis
Results are expressed as the mean ± S.D. of three independent experiments. For data analysis, experimental samples were compared with controls by unpaired Student's t test. For multiple-group comparisons, a one-way variance analysis and post hoc multiple comparisons tests were used. Differences between groups were considered statistically significant when the p value was less than 0.05. All statistical analyses were performed using the GraphPad Prism program (GraphPad Software Inc.).
RESULTS
Characterization of LMW-HA and Its Effects on EC Proliferation
Previous reports have described the angiogenic effects of various sizes of LMW-HA in numerous angiogenesis models (13, 41, 42). To evaluate the molecular weight and purity of our LMW-HA preparation, we ran a 4–20% Tris borate-EDTA gel with HA molecular weight standards and our purified LMW-HA (see “Experimental Procedures”). Development of the resulting gel was achieved with Stains-All (Sigma). Based on our isolation procedure (8), we anticipated our LMW-HA to have a size range of 500–5000 Da (∼2500 Da average, which we used to calculate molar concentrations). The results of Fig. 1A indicate that our purified LMW-HA migrates to a position similar to small HA (1.5–3.0 kDa, Enzo Life Sciences) and OligoHATM10 (∼1.9 kDa, Hyalose). Furthermore, the uniform bluish color (with lack of other bands and colors) of our prep and the HA standards suggests minimal contamination. We next determined the minimal molar concentration of our purified LMW-HA to induce maximal EC proliferation, a process intimately involved in angiogenesis (43). Our results indicate that 100 nm LMW-HA fits this criteria (Fig. 1B). To further test the purity of our purified LMW-HA, we subjected it to boiling and proteinase K digestion, which did not affect the ability to promote EC proliferation compared with untreated LMW-HA. Next, to determine HA itself is the active agent in our preparations, we fully digested LMW-HA with hyaluronidase SD and observed virtually no increase in EC proliferation, a process that was reversed with boiling (inactivating) hyaluronidase SD before HA digestion (Fig. 1C). In addition, using a LPS ELISA (USBiological Life Sciences) on our LMW-HA preparation revealed no LPS present in the detection range of the assay (12.35–1000 ng/ml; data not shown).
FIGURE 1.

Characterization of LMW-HA and its effects on EC proliferation. Panel A, to evaluate the molecular weight (MW) and purity of our LMW-HA preparation, a 4–20% Tris borate-EDTA (TBE) gel was run with HA molecular weight standards (Select-HATM 75–150 kDa (Sigma), Select-HATM 15–30 kDa (Sigma), small HA 1.5–3.0 kDa (Enzo Life Sciences), and OligoHATM10 ∼1.9 kDa (Hyalose, L.L.C.)) and our purified LMW-HA (see “Experimental Procedures”). Development of the resulting gel was achieved with Stains-All (Sigma). Panel B, determination of the minimal molar concentration of our purified LMW-HA to induce maximal HLMVEC proliferation. Control, VEGF (200 pg/ml), or LMW-HA (0.1–1000 nm) pretreated EC (5 × 103 cells/well) were incubated with 0.2 ml of serum-free media for 72 h at 37 °C in 5%CO2, 95% air in 96-well culture plates. The in vitro cell proliferation assay was analyzed by measuring increases in cell number using the CellTiter96TM MTS assay (Promega) and read at 492 nm. Each assay was set up in triplicate and repeated at least five times. An asterisk indicates a statistically significant difference (p < 0.05) from Control. Panel C, to further test the purity of our purified LMW-HA, it was subjected to boiling and proteinase K digestion (50 μg/ml), which did not affect the ability to promote EC proliferation compared with untreated 100 nm LMW-HA. LMW-HA was also fully digested with hyaluronidase SD (100 milliunits/ml), and there was virtually no increase in EC proliferation, a process that was reversed with boiling (inactivating) hyaluronidase SD before HA digestion. Each assay was set up in triplicate and repeated at least five times.
LMW-HA Stimulates Angiogenesis via CD44 Receptor and Src Activation
Our experiments in Fig. 2, A and B, confirm the angiogenic potential of our LMW-HA fragments of ∼2500 Da (500–5000-Da range) on HLMVEC. This is consistent with previous reports indicating low molecular weight fragments of HA (1350–4500 Da) are potent inducers of angiogenesis in vitro and in vivo (44, 45). Furthermore, we have established the involvement of CD44 in the angiogenic process (Fig. 2, B and C). Control and CD44-silenced HLMVEC were seeded on Matrigel basement membrane and stimulated with LMW-HA for 6 h (Fig. 2A). The results show that LMW-HA stimulated increased tubule formation in this model. Silencing of CD44 did not significantly affect basal levels of tubule formation (p > 0.05) by the endothelial cells but significantly inhibited LMW-HA-stimulated tubule formation (p < 0.001) (Fig. 2B). HMW-HA did not stimulate tubule formation by the endothelial cells on Matrigel (Fig. 2B, VEGF (200 pg/ml) was used as a positive control). Silencing of CD44 was confirmed by immunoblotting of siControl and siCD44-treated cells (Fig. 2C). This validates a role for CD44 in mediating angiogenesis stimulated by LMW-HA.
FIGURE 2.

Silencing of CD44 blocks LMW-HA-induced angiogenesis. Panel A, CD44 and control silenced cells were plated on Matrigel in the presence or absence of LMW-HA (100 nm) with CD44 siRNA or control siRNA. Images were captured after a 6-h incubation. Panel B, endothelial cell tubule formation on Matrigel was quantified using ImageJ to measure tubule length and graphically depicted. The asterisks (*) indicate a statistically significant difference (p < 0.05) from control. No significant difference was detected between siCD44 and siCD44 LMW-HA. Each treatment was performed in triplicate, and experiments were repeated three times. Results are expressed as tubule length per treatment. Panel C, immunoblot (IB) analysis of HLMVEC treated with CD44 siRNA or control siRNA. Cell lysates were blotted using CD44 (IM7) and actin antibodies to confirm CD44 silencing.
Previous work by ourselves and others has indicated that Src kinases play a key role in downstream signaling of CD44 after binding of LMW-HA (8, 46). We assessed Src activation in control and CD44-silenced endothelial cells as follows. Endothelial cells were serum-starved for 6 h and then incubated with LMW-HA for 0, 5 15, and 30 min. Cells lysates were prepared and immunoblotted for phospho-Src Tyr418, total Src, CD44, and actin. In siControl cells, LMW-HA stimulated an increase in phosphorylation of Src on Tyr418 (Fig. 3A). Phosphorylation of Src on Tyr418 is associated with increased kinase activity of the enzyme (47). Src phosphorylation was also decreased in the CD44-silenced cells after incubation with LMW-HA (Fig. 3). These results confirm that CD44 is required for Src phosphorylation in response to LMW-HA in HLMVEC.
FIGURE 3.

Src is activated after LMW-HA treatment of HMVEC and regulates tubule formation. Panel A, siControl and siCD44 HLMVE cells were serum-starved for 6 h before incubation for 0, 5, 15, or 30 min with LMW-HA (100 nm). Cells lysates were then prepared and immunoblotted (IB) with phospho-Src (Tyr418), total Src, CD44, and actin antibodies. Panel B, Src- and control-silenced cells were seeded on Matrigel-coated plates in the presence or absence of LMW-HA (100 nm), VEGF (200 pg/ml), or HMW-HA (100 nm). Images were captured after a 6-h incubation, and tubule formation was quantified using ImageJ image analysis software to measure tubule length and graphically depicted. The asterisks (*) indicate a statistically significant difference (p < 0.05) from control. No significant difference was detected between siControl and siSrc or siSrc and siSrc LMW-HA. Each treatment was performed in triplicate, and experiments were repeated three times. Results are expressed as tubule length per treatment. Panel C, immunoblot analysis of HLMVE cells treated with Src siRNA or control siRNA. Cell lysates were blotted using Src and actin antibodies to confirm Src silencing.
The role of Src signaling in LMW-HA-stimulated angiogenesis was confirmed via silencing of Src in HLMVEC. Control and Src silenced HLMVEC were subjected to a Matrigel tubule formation assay in the presence or absence of LMW-HA as described above. Tubule formation in response to LMW-HA was inhibited in the Src-silenced cells compared with control cells (Fig. 3, B and C).
LMW-HA Stimulates Src-mediated Phosphorylation of EphA2
Activation of Src kinase can lead to transactivation of certain cell surface receptors in endothelial cells. We have previously shown that LMW-HA/CD44 stimulated activation of Src leads to S1P3 receptor activation (8). Others have shown that Src activation is involved in EGF receptor transactivation after endothelin-1 (ET-1) binding to the ET-1 receptor in microvascular cells (48). A number of studies have demonstrated transactivation of the EphA2 receptor by various signaling pathways, including thrombin/protease-activated receptors (PARs) and VEGF/VEGF receptor pathways. The Epha2 receptor is known to be involved in the regulation of lung endothelial cell function (21, 49, 50). These findings prompted us to investigate the possibility of cross-talk between the CD44 and EphA2 signaling pathways. Previous data have illustrated the importance of EphA2 phosphorylation in regulating its functional activity (50, 51). Therefore, we investigated the effects of LMW-HA on EphA2 phosphorylation. HLMVEC were serum-starved and stimulated with LMW-HA or Ephrin-A1-Fc as a positive control. LMW-HA-stimulated cells showed a time-dependent increase in EphA2 phosphorylation on Tyr594 (Fig. 4A). A similar increase was seen when cells were stimulated with the EphA2 ligand, ephrin-A1-Fc. Pretreatment of cells with the Src kinase inhibitor, PP2 (1 h, 1 μm), inhibited the increase in EphA2 Tyr594 phosphorylation induced by either LMW-HA or ephrin-A1-Fc treatment. Src activation was also blocked by PP2 pretreatment (Fig. 4A).
FIGURE 4.

LMW-HA stimulates CD44 and Src-dependent phosphorylation of EphA2. Panel A, HPMVEC were treated with 100 nm LMW-HA or 1 μg/ml Ephrin-A1-Fc for 5, 15, or 30 min in the presence or absence of the Src inhibitor, PP2 (1 μm). Cells lysates were then prepared and immunoblotted (IB) with phospho-EphA2 (Tyr594), phosphor-EphA2 (Ser897), EphA2, Phospho-Src (Tyr418), Src, or actin antibodies. LMW-HA and Ephrin-A1 promote Src-dependent tyrosine phosphorylation of EphA2. Panel B, HPMVEC were pretreated with either control siRNA or CD44 siRNA and then treated with LMW-HA (100 nm) or Ephrin-A1-Fc (1 μg/ml) for 15 min. Cell lysates were prepared in 1% Nonidet P-40 lysis buffer and incubated with anti-EphA2-cross-linked beads for immunoprecipitation. The resulting immune precipitations (IP) were then blotted with phospho-EphA2 (Tyr594), CD44v10, Src, or EphA2 antibodies. CD44 siRNA inhibits LMW-HA and Ephrin-A1-Fc-mediated Src recruitment to EphA2. Panel C, graphical depiction of quantity of protein normalized to EphA2 staining intensity (arbitrary units) from experiments described in panel B performed in triplicate and quantitated using computer-assisted densitometry. The asterisks (*) indicate a statistically significant difference (p < 0.05) from control.
To examine the link between EphA2 and CD44, we immunoprecipitated EphA2 from control and CD44-silenced cells treated with LMW-HA or ephrin-A1-fc for 15 min. EphA2 immunoprecipitates were then immunoblotted for CD44, Src, p-tyrosine, and EphA2. Incubation of control endothelial cells with LMW-HA or ephrin-A1-fc stimulated increased association of CD44 and Src with the EphA2 receptor and increased tyrosine phosphorylation of EphA2 (Fig. 4B). Silencing of CD44 blocked both recruitment of Src to EphA2 and the phosphorylation of EphA2 in response to LMW-HA. Phosphorylation of EphA2 in response to Ephrin-A1-fc was not affected by CD44 silencing; however, recruitment of Src to Epha2 was inhibited in these cells. This was confirmed by computer-assisted densitometry of triplicate experiments and assessing the quantity of protein normalized to EphA2 staining intensity (Fig. 3C). Indirect immunofluorescence microscopy was also performed on control and LMW-HA-treated endothelial cells to investigate the association of CD44 with EphA2. Control or LMW-HA-treated cells on glass slides were fixed and co-stained for CD44v10 and EphA2 as indicated in Fig. 5. LMW-HA-treated cells show increased co-localization of CD44 and EphA2 (Fig. 5, compare d and e with i and j). The “small dots” staining pattern of colocalized EphA2 and CD44v10 is reminiscent of caveolin-1 immunocytochemical staining of these cells (38). Indeed, both CD44v10 and EphA2 can reside in caveolin-1-containing lipid rafts (8, 53). Thus LMW-HA stimulated association of CD44, EphA2, and Src in microvascular endothelial cells. LMW-HA stimulated phosphorylation of the EphA2 receptor in a CD44- and Src-dependent manner. CD44 is also required for ephrin-A1-fc-stimulated recruitment of Src to EphA2, although it is not required for ephrin-A1-fc-stimulated phosphorylation of EphA2.
FIGURE 5.
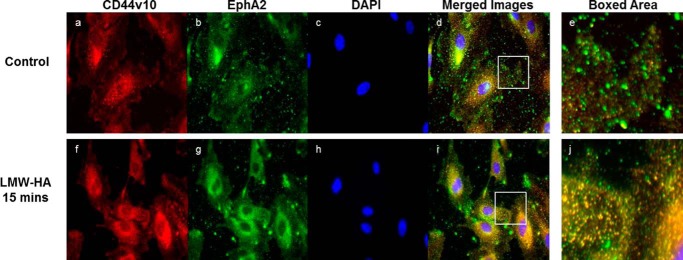
CD44 and EphA2 colocalize after stimulation of HMVEC with LMW-HA. HLMVEC were cultured on glass slides and incubated with 100 nm LMW-HA for 15 min in serum-free media. Cells were then fixed in 4% paraformaldehyde and double-stained with anti-CD44v10 coupled with Alexa-594 (a and f) and anti-EphA2 coupled with Alexa 488 (b and g) antibodies and mounted using Prolong anti-fade with DAPI (c and h). Images were taken at 20× magnification and merged (d and h) using ImageJ software. Without stimulation, there is minimal CD44v10 (red) colocalization with EphA2 (green). With LMW-HA challenge, there is much greater (yellow) colocalization (Boxed area).
The Role of EphA2 in LMW-HA-stimulated Angiogenesis
To further examine the involvement of EphA2 in LMW-HA-stimulated angiogenesis, tubule formation by control and EphA2-silenced cells in response to LMW-HA was measured. Fig. 6 shows that EphA2 silencing significantly inhibited tubule formation in response to LMW-HA (p < 0.001). It did not have a significant effect on basal levels of tubule formation (p > 0.05). These results confirm that EphA2 is required for LMW-HA stimulated in vitro angiogenesis in HLMVEC.
FIGURE 6.

Silencing of EphA2 blocks LMW-HA-induced angiogenesis. Panel A, EphA2 and control silenced cells were seeded on Matrigel-coated plates in the presence or absence of LMW-HA (100 nm). Images were captured after a 6-h incubation, and tubule formation was quantified using ImageJ image analysis software. The mean tubule length was calculated for 20 images per treatment and graphically depicted. The asterisks (*) indicate a statistically significant difference (p < 0.05) from control. No significant difference was detected between siEphA2 and siEphA2 LMW-HA. Each treatment was performed in triplicate, and experiments were repeated three times. Results are expressed as tubule length per treatment. Panel B, immunoblot (IB) analysis of HLMVEC treated with EphA2 siRNA or control siRNA. Cell lysates were blotted using EphA2 and actin antibodies to confirm EphA2 silencing.
Complex Formation with CD44v10, EphA2, and the PATJ Scaffolding Protein
The above results confirmed the central role of EphA2 in the CD44/LMW-HA signaling pathway. We next examined other potential downstream signaling targets. Directional migration is an important factor in facilitating tubule formation and angiogenic spread by endothelial cells. Correct spatial recruitment and activation of numerous signaling molecules within the migrating cell is key to controlling and directing this migration. Recently it has been reported that PATJ (Pals1-associated tight junction), a scaffold protein possessing 10 PDZ binding domains, is required for correct localization of RhoA activity at the leading edge of migrating endothelial cells (54). Immunoblotting of EphA2 immunoprecipitations from LMW-HA- or ephrin-A1-Fc-stimulated cells show PATJ is recruited to the EphA2 receptor in response to both stimuli in a time-dependent manner (Fig. 7, A and B). Immunoprecipitation of PATJ also resulted in co-precipitation of EphA2 and CD44 in response to LMW-HA and ephrin-A1-fc (Fig. 7, C and D). The results presented thus far demonstrate that upon binding of LMW-HA or ephrin-A1-Fc to their respective receptors, CD44v10 and EphA2 associate, leading to increased EphA2 phosphorylation via Src activation and recruitment of the scaffolding protein PATJ to the CD44v10-EphA2 complex.
FIGURE 7.
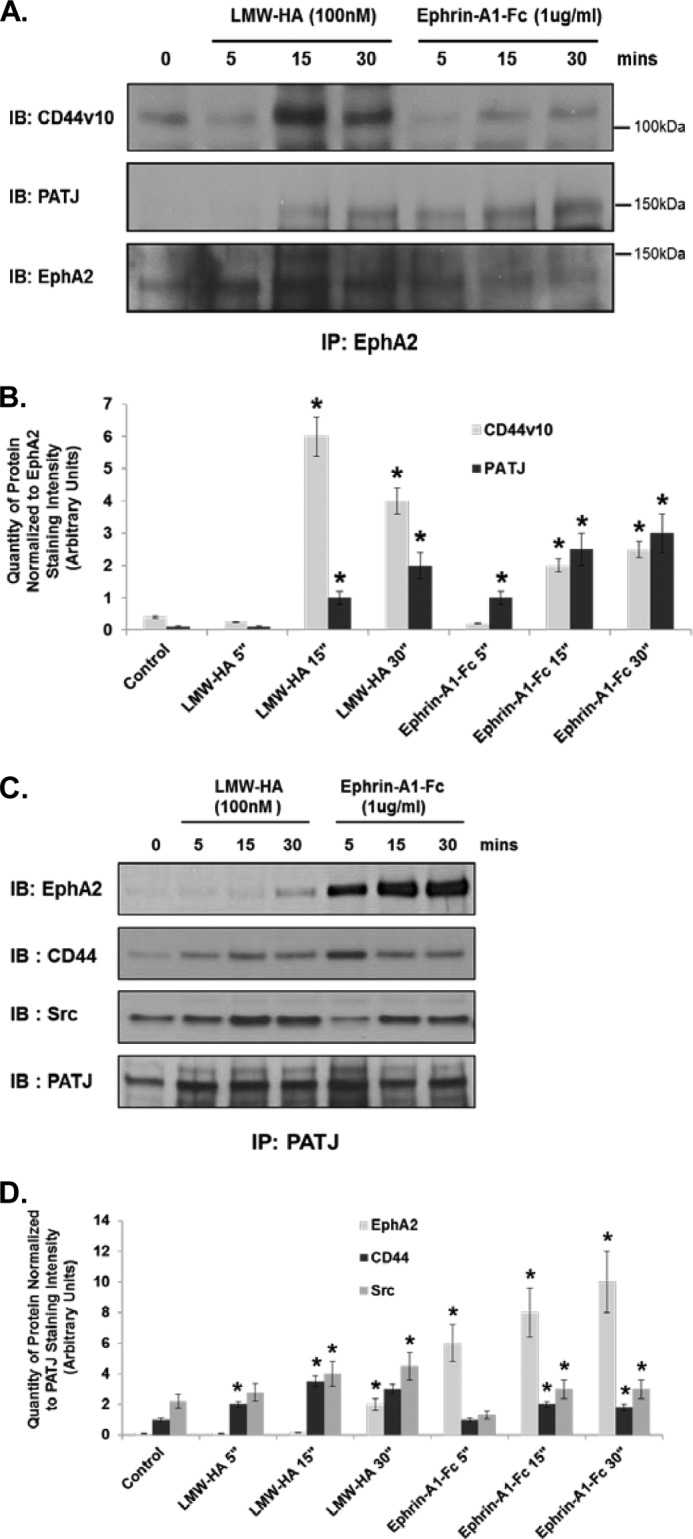
LMW-HA stimulation of HMVEC induces CD44, EphA2, PATJ, and Src complex formation. Panel A, ECs were serum-starved for 1 h in EBM-2 media and then incubated with LMW-HA (100 nm) or Ephrin-A1/Fc (10 ng/ml) for 5, 15, and 30 min. Cell lysates were prepared in 1% Nonidet P-40 lysis buffer and incubated with anti-EphA2-cross-linked beads for immunoprecipitation. The resulting immune precipitations (IP) were then blotted for CD44v10, PATJ, and EphA2. IB, immunoblot. Panel B, graphical depiction of quantity of protein normalized to EphA2 staining intensity (arbitrary units) from experiments described in panel A performed in triplicate and quantitated using computer-assisted densitometry. The asterisks (*) indicate a statistically significant difference (p < 0.05) from control. Panel C, ECs were serum-starved for 1 h in EBM-2 media and then incubated with LMW-HA (100 nm) or Ephrin-A1/Fc (10 ng/ml) for 5, 15, and 30 min. Cell lysates were prepared in 1% Nonidet P-40 lysis buffer and incubated with anti-PATJ-cross-linked beads for immunoprecipitation. The resulting immune precipitations were then blotted with EphA2, CD44, Src, and PATJ antibodies. Panel D, graphical depiction of quantity of protein normalized to PATJ staining intensity (arbitrary units) from experiments described in panel A performed in triplicate and quantitated using computer-assisted densitometry. The asterisks indicate a statistically significant difference (p < 0.05) from control.
Involvement of RhoGEFs in LMW-HA-stimulated RhoA Activation and Angiogenesis
PATJ, along with angiomotin, another PDZ-containing protein, and the RhoGEF Syx are known to have an important role in the spatial regulation of RhoA GTPase activity in randomly migrating endothelial cells (54). PATJ was reported to act as the scaffold that recruits Syx to angiomotin at the leading edge of these migrating cells. We investigated if PATJ had a similar role in regulating RhoA activity in response to LMW-HA and ephrin-A1-Fc. Immunoblotting of control-, LMW-HA-, or ephrin-A1-fc-stimulated PATJ immunoprecipitates indicated differential recruitment of RhoGEFs. First, Syx association with PATJ was not elevated in response to either LMW-HA or ephrin-A1-Fc treatment (Fig. 8, A and B). However, Dbs, another Rho-specific GEF, was recruited in response to both LMW-HA and ephrin-A1-Fc stimulation. Three other RhoGEFs, LARG, p115, and PDZ-RhoGEF, were also recruited to PATJ by LMW-HA but not ephrin-A1-fc stimulation. Angiomotin was not recruited to PATJ in response to either LMW-HA or Ephrin-A1-Fc (data not shown).
FIGURE 8.

Specific recruitment of RhoGEFs by PATJ after LMW-HA stimulation of HPMVEC. ECs were serum-starved for 1 h in EBM-2 media and then incubated with LMW-HA (100 nm) or Ephrin-A1-Fc (10 ng/ml) for 5, 15, and 30 min. Cell lysates were prepared in 1% Nonidet P-40 lysis buffer and incubated with anti-PATJ-cross-linked beads for immunoprecipitation. The resulting immune precipitations (IP) were then immunoblotted (IB) with Dbs, LARG, PDZ RhoGEF, Syx, p115 RhoGEF, and PATJ antibodies. Only Dbs was recruited to PATJ in a time-dependent manner with both LWM-HA and Ephrin-A1 treatment of HLMVEC. Panel B, graphical depiction of quantity of protein normalized to PATJ staining intensity (arbitrary units) from experiments described in panel A performed in triplicate and quantitated using computer-assisted densitometry. The asterisks (*) indicates a statistically significant difference (p < 0.05) from control.
Next we performed a RhoA activation assay to confirm its involvement along with CD44, EphA2, PATJ, Dbs, and Src in the LMW-HA-stimulated signaling pathway. HLMVEC were incubated with the appropriate siRNA complexes, and the silenced cells were incubated with LMW-HA for 15 min, the GTP-RhoA pulldown assay was then performed, and lysates immunoblotted for RhoA. The resulting immunoblot showed robust activation of RhoA in response to LMW-HA in the siControl cells, but RhoA activation was abolished in the CD44-, Src-, and EphA2-silenced cells and significantly reduced in the PATJ- and Dbs-silenced cells (p < 0.001) (Fig. 9A). The functional effects of blocking RhoA activation through silencing each of these components of the LMW-HA/CD44 pathway was concurrently examined using a transwell cell migration assay. Cells were seeded to the upper well of the transwell assay, whereas LMW-HA was added to the bottom well. The cells, which migrated though the filter, were then counted after 24 h. The bar chart in Fig. 9B shows cell migration in response to LMW-HA at 24 h was significantly reduced in each case (p < 0.05).
FIGURE 9.

Silencing of CD44, EphA2, Src, PATJ, or DBS reduces RhoA activation in response to LMW-HA and inhibits HLMVEC migration. Panel A, ECs that had been subjected to control siRNA or target protein siRNA were incubated ± LMW-HA (100 nm) for 15 min. Total cell lysates were then prepared, and a pulldown assay for GTP-bound active RhoA was performed. Panel B, ECs were incubated with the indicated siRNAs and then plated on 8-μm transwell filters in serum-free media. 10% serum media was added to the bottom wells, and cells were incubated for 24 h. Cell numbers from the upper and lower chambers were quantified using the MTS cell assay. An asterisk (*) indicates a statistically significant difference (p < 0.05) from control and scramble siRNA. n = 3 per condition.
To verify the results of the RhoA activation and migration assay as to their role in angiogenesis, we assessed the effect of silencing PATJ and Dbs on tubule formation. Control-, PATJ-, or Dbs-silenced endothelial cells were plated on a Matrigel basement membrane and stimulated with LMW-HA. Silencing of either PATJ or Dbs attenuated LMW-HA-stimulated tubule formation (Fig. 10A). Silencing was confirmed in each case by immunoblotting (Fig. 10, B and C).
FIGURE 10.
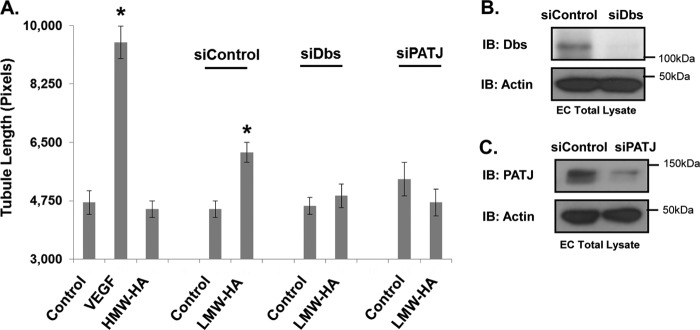
Silencing PATJ and Dbs inhibits LMW-HA-induced tubule formation. Panel A, PATJ-, Dbs-, and control-silenced EC were plated on Matrigel in the presence or absence of LMW-HA (100 nm), VEGF (200 pg/ml), or HMW-HA (100 nm). Images were captured after a 6-h incubation. Endothelial cell tubule formation on Matrigel was quantified using ImageJ to measure tubule length. An asterisk (*) indicates a statistically significant difference (p < 0.05) from control. Each treatment was performed in triplicate, and experiments were repeated three times. Results are expressed as tubule length per treatment. Panels B and C, immunoblot (IB) analysis of HLMVEC treated with PATJ siRNA, Dbs siRNA, or control siRNA. Cell lysates were blotted using PATJ, Dbs, and actin antibodies to confirm silencing.
The Role of CD44 and EphA2 in LMW-HA-mediated in Vivo Angiogenesis
Finally, to determine the effects of LMW-HA on angiogenesis in vivo, we utilized a novel anginex/liposomal targeting of murine angiogenic endothelium with either CD44 or EphA2 siRNA and observed inhibition of LMW-HA-induced angiogenesis in implanted Matrigel plugs (Fig. 11). To test angiogenic targeting of our anginex-conjugated liposomes, we introduced pCMV6 empty vector and pCMV6-A-GFP (Origene) as cargo. Briefly, Matrigel supplemented with LMW-HA or PBS was injected into mice. After 7 days, to allow for initiation of angiogenesis in the plugs, anginex-conjugated liposomes with cargo (expression vectors or siRNA) were delivered via intravenous injection. Targeting of the liposomes to the proliferating endothelium was achieved by covalent conjugation of “anginex” peptide (39, 40, 55) (Phoenix Pharmaceuticals) to the liposome surface. This peptide is bound by galetin-1, which shows increased expression on activated endothelial cells (56). The plugs were then harvested 7 days after liposome delivery (14 days after Matrigel injection). The plugs were then utilized for determination of turboGFP expression (Fig. 11A), protein assessment (Fig. 11B), gross examination (Fig. 11C), and angiogenesis (Fig. 11D). For turboGFP expression, Matrigel plugs were frozen in OCT embedding medium and cut in 12-μm-thick sections. Frozen sections were fixed, permeabilized, blocked, and incubated overnight with turboGFP antibody (OriGene). Alexa 488-conjugated secondary antibodies (Molecular Probes) were used to visualize GFP staining. Nuclear staining was performed with DAPI. The results of Fig. 11A indicate that our anginex-conjugated liposomes can successfully target cells in the Matrigel plug. For protein assessment, Matrigel plugs from mice treated with intravenous injections of anginex-conjugated liposomes containing no siRNA (control), control siRNA, EphA2 siRNA, or CD44 siRNA were depolymerized by BD Cell Recovery Solution (BD Biosciences), and each plug was dissolved in 300 μl of recovery solution as described in the manufacturer's protocol, run on SDS-PAGE, and immunoblotted for EphA2, CD44, and laminin (used as a protein constant; see Ref. 57). The results of Fig. 11B indicate that we can specifically silence the expression of proteins in Matrigel plugs. For gross examination, LMW-HA stimulated increased vascularization of the Matrigel plugs, as evidenced by increased endothelial cell numbers in the plugs and the pink appearance on the excised plugs (Fig. 11C). For angiogenesis quantitation, the plugs were fixed in formalin and processed for histological staining using Masson's Trichome stain to visualize endothelial cell invasion and vessel formation. Silencing of CD44 or EphA2 significantly reduced the number of endothelial cells per plug as expected (Fig. 11D). Overall these results indicate that EphA2 is a key regulator of LMW-HA- and CD44-mediated angiogenesis.
FIGURE 11.

Targeting of angiogenic ECs with CD44 or EphA2 siRNA blocks in vivo LMW-HA-mediated angiogenesis. Panel A, Matrigel plugs from mice with intravenous administration of anginex-conjugated liposomes containing either pCMV6 empty vector or pCMV6-A-GFP (Origene) were frozen in OCT embedding medium and cut in 12-μm-thick sections. Frozen sections were fixed, permeabilized, blocked, and incubated overnight with turboGFP antibody (OriGene). Alexa 488-conjugated secondary antibodies (Molecular Probes) were used to visualize GFP staining. Nuclear staining was performed with DAPI. The results of panel A indicate that our anginex-conjugated liposomes can successfully target cells in the Matrigel plug. Panel B, Matrigel plugs from mice with intravenous administration of anginex-conjugated liposomes containing either no siRNA (Control), control siRNA, EphA2 siRNA, or CD44 siRNA were depolymerized by BD Cell Recovery Solution (BD Biosciences), and each plug was dissolved in 300 μl of recovery solution as described in the manufacturer's protocol, run on SDS-PAGE, and immunoblotted (IB) for EphA2, CD44, and laminin. The results of panel B indicate that we can specifically silence the expression of proteins in Matrigel plugs. Panel C, Matrigel plugs ± LMW-HA (5 μg/ml) were implanted subcutaneously in mice that were subsequently injected (day 7) with anginex-labeled liposomes containing control, CD44, or EphA2 siRNA. The plugs were then harvested 14 days after the initial implantation, and images were obtained. Panel D, plugs from panel C were analyzed for angiogenesis histologically using Trichrome staining and cell counting (see “Experimental Procedures”). These experiments were repeated three times with five animals per treatment group. Results are expressed as the number of cells/plug. An asterisk indicates a statistically significant difference (p < 0.05) from Control.
DISCUSSION
In this study we present data illustrating the crucial role of the EphA2 receptor in angiogenesis stimulated by LMW-HA and the CD44 receptor. LMW-HA binding to CD44v10 induces CD44v10-EphA2 complex formation, recruitment, and activation of Src and Src-mediated EphA2 tyrosine phosphorylation. These events are required for recruitment of the scaffolding protein, PATJ, and consequent recruitment of the rhoGEF, Dbs, to induce RhoA activation and angiogenesis (Fig. 12).
FIGURE 12.
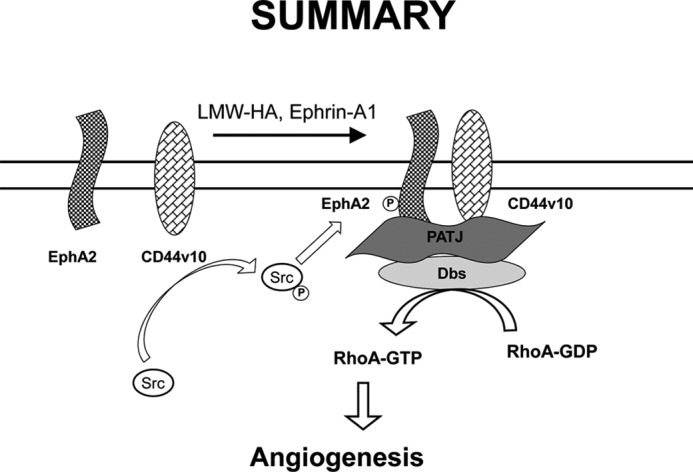
Schematic diagram indicating the role of EphA2 transactivation in LMW-HA-mediated angiogenesis. LMW-HA binding to CD44v10 induces CD44v10-EphA2 complex formation, recruitment, and activation of Src and Src-mediated EphA2 tyrosine phosphorylation. These events are required for recruitment of the scaffolding protein, PATJ, and consequent recruitment of the rhoGEF, Dbs, to induce RhoA activation and angiogenesis.
The Eph family is the largest of receptor-tyrosine kinase families, and they are involved in the regulation of a wide and diverse number of cellular processes. Most recently their role in regulating endothelial function has been investigated. The essential role of Eph receptors and EphA2 in vasculogenesis, vascular patterning, and axonal guidance during development has been known for some time but now, as their role in angiogenesis, particularly during tumor growth, has emerged interest in the Eph family of receptors has been rejuvenated (24, 58).
We previously reported that HA can regulate the permeability of the lung endothelial barrier both in vitro and in vivo (8, 38, 59). The barrier disruptive effects of LMW-HA are mediated via binding of LMW-HA to the CD44v10 receptor leading to activation of Src, RhoA, and transactivation and recruitment of the S1P3 receptor to a subset of lipid rafts called caveolin-enriched microdomains (8). The EphA2 receptor is another regulator of lung endothelium permeability. A study by Larson et al. (21) revealed that stimulation of pulmonary microvascular endothelial cells with the EphA2 receptor ligand, ephrin-A1, increased monolayer permeability by 44%, whereas intravenous injection of ephrin-A1 in rats increased albumin leakage into lungs. They observed disruption of both tight and adherens junctions, the main regulators of paracellular permeability. Disruption of the endothelial monolayer is generally regarded as one of the initial steps during angiogenesis and a key step in the metastatic process, allowing tumor cells entry to the vasculature resulting in their dissemination throughout the body. EphA2 promotes angiogenesis via regulation of tight junction formation and recruitment of GEFs Vav2/3 and the p85 phosphatidylinositol 3-kinase subunit. EphA2 function is tightly regulated by phosphorylation. Phosphorylation of EphA2 on Tyr593 has been shown to be important for the regulation of vascular assembly (50). EphA2-tyrosine kinase activity is also required for maximal VEGF-induced angiogenesis. VEGF induces expression of the ephrin-A1 ligand leading to EphA2 receptor activation via phosphorylation resulting in maximal cell migration and vessel sprouting in response to VEGF (61). Soluble EphA2-Fc can inhibit VEGF-induced endothelial migration while not effecting cell proliferation. Silencing of EphA2 via siRNA knockdown or overexpression of the kinase-null mutant inhibited basal levels of brain microvascular endothelial cell tubule formation on Matrigel while enhancing tight junction formation (62). Conversely, overexpression of EphA2 leads to destabilized adherens junctions via a RhoA-dependent mechanism (26).
Our results indicate that EphA2 plays a central role in regulating LMW-HA-stimulated angiogenesis via a Src- and RhoA-dependent pathway. Silencing of CD44 or EphA2 inhibits LMW-HA-stimulated angiogenesis both in an in vitro Matrigel tubule formation assay and in an in vivo Matrigel plug angiogenesis assay. We observed that LMW-HA treatment of HPMVEC leads to a time-dependent recruitment of CD44v10 to the EphA2 receptor, which we initially detected via immunofluorescence and confirmed via co-immunoprecipitation of the receptors.
Stimulation of endothelial cells with LMW-HA increases phosphorylation of EphA2 on Tyr593, which is blocked in CD44-silenced cells. Inhibition of Src kinase by PP2 similarly blocked LMW-HA-stimulated phosphorylation of EphA2 on Tyr593. These results indicate that CD44-mediated activation of Src is required for the LMW-HA-stimulated increase in EphA2 phosphorylation. Silencing of Src also blocked LMW-HA-mediated tubule formation by HLMVEC on Matrigel. In a study by Chan and Sukhatme (63), they reported that thrombin-induced phosphorylation of EphA2 via PAR-1 (protease-activated receptor-1) was Src-dependent in their study of EphA2 transactivation in HUVEC. In their study of Epha2 phosphorylation sites, Fang et al. (50) reported that phosphorylation on Tyr593 was required for correct vascular assembly of endothelial cells on a Matrigel matrix. Furthermore, they reported that this phosphorylated residue may act as a binding site for guanine-nucleotide exchange factors, which are necessary for cell migration.
The scaffold protein, PATJ, plays a key role in localizing RhoA GTPase activity to the leading edge of migrating epithelial and more recently endothelial cells (54, 64). PATJ is a multi-PDZ domain-containing protein, which allows it to interact with a large number of PDZ binding domain-containing signaling proteins. Although much is known about the role of PATJ in regulating epithelial cell migration and polarity, less is known about its function in endothelial cells. Ernkvist et al. (54) were the first to report that PATJ could also be involved in spatial regulation of RhoA activity in endothelial cells. Their study reported that PATJ, along with angiomotin, a membrane-associated scaffold protein, and Syx, a RhoAGEF, were crucial in maintaining correct focal RhoA activity in endothelial cells. Disruption of this complex (via morpholino-silencing of angiomotin in vivo) led to impaired migration of intersegmental vessels and disrupted vascular development in zebrafish embryos. Our studies have revealed that PATJ may have a similar role in LMW-HA-stimulated endothelial cells. Immunoblotting of EphA2 immunoprecipitates after treatment of HLMVEC with either LMW-HA or ephrin-A1-Fc indicated that PATJ along with CD44V10 was recruited to the EphA2 receptor. We did not detect any interaction of angiomotin with either CD44 or EphA2. Furthermore, we observed that silencing of PATJ inhibited LMW-HA-stimulated activation of RhoA. Silencing of PATJ also inhibited HPMVEC migration and tubule formation in response to LMW-HA. These results indicate that PATJ may not only be crucial for correct localization of RhoA in migrating cells but also for its activation. As PATJ does not possess any intrinsic RhoGEF activity, this regulation is likely mediated via recruitment of other RhoGEF proteins such as Syx.
Syx was observed to be constitutively associated with PATJ in HPMVEC, and this association was not altered after treatment with either LMW-HA or ephrin-A1-Fc. Of the other RhoGEFs examined, LARG, PDZ-RhoGEF, and p115-Rho GEF showed increased association with PATJ after treatment with LMW-HA but not ephrin-A1-Fc. Dbs (Dbl's Big Sister/MCF2L), however, showed increased association with PATJ after treatment with either LMW-HA or ephrin-A1-Fc. Silencing of Dbs attenuated RhoA activation, endothelial cell migration, and tubule formation in response to LMW-HA. This attenuation rather than complete inhibition indicates that other RhoGEFs may also be involved in this RhoA activation pathway. This is not surprising as we observed that LMW-HA could stimulate increased association of multiple RhoGEFs with PATJ. Dbs was first identified in a screen for proteins whose overexpression could deregulate the growth of fibroblasts (65). Although Dbs can regulate the activity of both Cdc42 and RhoA in vitro, its in vivo exchange activity appears to be regulated by cell type. It has previously been reported that Dbs activation may be regulated via Src kinases (66). Kostenko et al. (67) demonstrated that the scaffold protein, Ccpg1, recruited both Src kinase and Dbs to a signaling complex in COS-7 cells. The authors suggest that Ccpg1 may act as a regulatory scaffolding protein, recruiting Dbs and Src and stimulating Src phosphorylation of Dbs and consequent stimulation of GEF activity. It is tempting to speculate that PATJ may serve a similar function in endothelial cells. Because Src and Dbs do not contain a PDZ binding domain, this interaction is likely mediated via a different domain or adaptor protein.
RhoA activation is a key factor in increasing endothelial barrier permeability and the angiogenic process. LMW-HA is a known activator of RhoA in endothelial cells (8, 68). Here we have outlined a novel signaling pathway, linking CD44, EphA2, PATJ, Src, and the RhoGEF Dbs to RhoA activation and LMW-HA-stimulated angiogenesis.
To further confirm our in vitro findings in vivo we employed a targeted angiogenic endothelial delivery of CD44 and EphA2 siRNA using anginex-conjugated liposomes in the mouse Matrigel plug assay. Matrigel enriched with LMW-HA showed increased invasion by endothelial cells into the plugs. Silencing or either CD44 or EphA2 blocked this increase in angiogenesis and even decreased basal levels of angiogenesis in the plugs, although the basal decrease was not statistically significant. Our results indicate that the EphA2 receptor is a crucial partner of CD44 in the angiogenic response stimulated by LMW-HA.
CD44 and EphA2 have both been implicated in regulating endothelial cell barrier function and angiogenesis. The results presented in this study demonstrate for the first time that there is significant cross-talk between these signaling pathways and that EphA2 is required for LMW-HA-stimulated angiogenesis mediated via the CD44 receptor. We have identified a signaling pathway between CD44 and Epha2 leading to RhoA activation and ultimately angiogenesis in HPMVEC. Upon binding of either LMW-HA or ephrin-A1-Fc, CD44 and EphA2 associate, resulting in phosphorylation of EphA2 via Src activation. PATJ is then recruited to this complex along with the RhoGEF Dbs; at this point it is unclear whether Dbs directly associates with PATJ or another scaffold protein. Dbs recruitment to this complex results in RhoA activation, leading to angiogenesis.
Although CD44 appears to be the major endothelial receptor for LMW-HA in our angiogenesis models, we cannot rule out the potential involvement of other HA receptors (14). Wang et al. (70) reported that CD44 regulates EC proliferation and tubule formation induced by HA fragments in HUVEC. In telomerase-immortalized foreskin microvascular ECs, Olofsson et al. (71) demonstrated that inhibiting CD44 or Hyal2 expression prevented tubular network formation in three-dimensional matrices. In addition, CD44 and TLR4 can be physically associated in a signaling complex after exposure to HA, and endothelial TLR4 regulates certain responses to HA fragments (72, 73). Furthermore, Savani et al. (74) demonstrated that HA fragment-induced HUVEC and H5V (murine EC line) angiogenesis involved the expression of both CD44 and RHAMM. Specifically, CD44 regulated HA-mediated adhesion, proliferation, and tube formation, whereas RHAMM regulated migration and in vivo angiogenesis. Gao et al. (75) showed that RHAMM is the major receptor involved in HA fragment-induced angiogenesis during wound healing. Furthermore, Matou-Nasri et al. (60) demonstrated that both CD44 and RHAMM are involved in HA fragment-induced BAEC angiogenesis. Additional confirmation of a dual role of CD44 and RHAMM in HA fragment-mediated HUVEC angiogenesis was determined by Park et al. (69), who demonstrated that HA induced RHAMM-TGFβ receptor complexes through a CD44-PKCδ mechanism. Interestingly, in post-mortem human coronary arteries that have endothelial injury and angiogenesis associated with atherosclerotic plaques, there is increased expression of RHAMM and Hyal-1 but not CD44 in the neovessels, suggesting differential roles of RHAMM and CD44 in angiogenesis from small versus large blood vessels (52). Therefore, the size of HA utilized, the differential expression of CD44 and RHAMM in various endothelial cell types, and the model system employed can all contribute to the relative contributions of different HA receptors.
In conclusion, this study demonstrates the crucial involvement of EphA2 in LMW-HA-mediated angiogenesis. The discovery of a novel interaction between EphA2 and CD44 signaling pathways could lead to the development of improved therapeutics in the treatment of angiogenesis-associated diseases including tumor progression.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1-HL 095723 (NHLBI; to P. A. S.). This work was also supported by the American Heart Association National Scientist Development Grant 0730277N (to P. A. S.).
- HA
- hyaluronan
- LMW-HA
- low molecular weight HA
- HPMVEC
- human pulmonary microvascular endothelial cell(s)
- GEF
- guanine nucleotide exchange factor
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- PATJ
- Pals-1-associated tight junction protein
- EC
- endothelial cell(s)
- HUVEC
- human umbilical vein endothelial cell(s).
REFERENCES
- 1. Stamenkovic I., Amiot M., Pesando J. M., Seed B. (1989) A lymphocyte molecule implicated in lymph node homing is a member of the cartilage link protein family. Cell 56, 1057–1062 [DOI] [PubMed] [Google Scholar]
- 2. Stoolman L. M. (1989) Adhesion molecules controlling lymphocyte migration. Cell 56, 907–910 [DOI] [PubMed] [Google Scholar]
- 3. Puré E., Cuff C. A. (2001) A crucial role for CD44 in inflammation. Trends Mol. Med. 7, 213–221 [DOI] [PubMed] [Google Scholar]
- 4. Morrison H., Sherman L. S., Legg J., Banine F., Isacke C., Haipek C. A., Gutmann D. H., Ponta H., Herrlich P. (2001) The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev. 15, 968–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ghatak S., Misra S., Toole B. P. (2005) Hyaluronan constitutively regulates ErbB2 phosphorylation and signaling complex formation in carcinoma cells. J. Biol. Chem. 280, 8875–8883 [DOI] [PubMed] [Google Scholar]
- 6. Wang S. J., Bourguignon L. Y. (2006) Hyaluronan and the interaction between CD44 and epidermal growth factor receptor in oncogenic signaling and chemotherapy resistance in head and neck cancer. Arch. Otolaryngol. Head Neck Surg. 132, 771–778 [DOI] [PubMed] [Google Scholar]
- 7. Bourguignon L. Y. (2008) Hyaluronan-mediated CD44 activation of RhoGTPase signaling and cytoskeleton function promotes tumor progression. Semin. Cancer Biol. 18, 251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Singleton P. A., Dudek S. M., Ma S. F., Garcia J. G. (2006) Transactivation of sphingosine 1-phosphate receptors is essential for vascular barrier regulation. Novel role for hyaluronan and CD44 receptor family. J. Biol. Chem. 281, 34381–34393 [DOI] [PubMed] [Google Scholar]
- 9. Goodison S., Urquidi V., Tarin D. (1999) CD44 cell adhesion molecules. Mol. Pathol. 52, 189–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Turley E. A., Noble P. W., Bourguignon L. Y. (2002) Signaling properties of hyaluronan receptors. J. Biol. Chem. 277, 4589–4592 [DOI] [PubMed] [Google Scholar]
- 11. Toole B. P. (2004) Hyaluronan: from extracellular glue to pericellular cue. Nat. Rev. Cancer 4, 528–539 [DOI] [PubMed] [Google Scholar]
- 12. Nandi A., Estess P., Siegelman M. H. (2000) Hyaluronan anchoring and regulation on the surface of vascular endothelial cells is mediated through the functionally active form of CD44. J. Biol. Chem. 275, 14939–14948 [DOI] [PubMed] [Google Scholar]
- 13. Cuff C. A., Kothapalli D., Azonobi I., Chun S., Zhang Y., Belkin R., Yeh C., Secreto A., Assoian R. K., Rader D. J., Puré E. (2001) The adhesion receptor CD44 promotes atherosclerosis by mediating inflammatory cell recruitment and vascular cell activation. J. Clin. Investig. 108, 1031–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Slevin M., Krupinski J., Gaffney J., Matou S., West D., Delisser H., Savani R. C., Kumar S. (2007) Hyaluronan-mediated angiogenesis in vascular disease: uncovering RHAMM and CD44 receptor signaling pathways. Matrix Biol. 26, 58–68 [DOI] [PubMed] [Google Scholar]
- 15. Termeer C., Sleeman J. P., Simon J. C. (2003) Hyaluronan: magic glue for the regulation of the immune response? Trends Immunol. 24, 112–114 [DOI] [PubMed] [Google Scholar]
- 16. Dentener M. A., Vernooy J. H., Hendriks S., Wouters E. F. (2005) Enhanced levels of hyaluronan in lungs of patients with COPD: relationship with lung function and local inflammation. Thorax 60, 114–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lennon F. E., Singleton P. A. (2011) Role of hyaluronan and hyaluronan-binding proteins in lung pathobiology. Am. J. Physiol. Lung Cell. Mol. Physiol. 301, L137–L147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Itano N., Zhuo L., Kimata K. (2008) Impact of the hyaluronan-rich tumor microenvironment on cancer initiation and progression. Cancer Sci. 99, 1720–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sironen R. K., Tammi M., Tammi R., Auvinen P. K., Anttila M., Kosma V. M. (2011) Hyaluronan in human malignancies. Exp. Cell Res. 317, 383–391 [DOI] [PubMed] [Google Scholar]
- 20. Brantley-Sieders D. M., Fang W. B., Hwang Y., Hicks D., Chen J. (2006) Ephrin-A1 facilitates mammary tumor metastasis through an angiogenesis-dependent mechanism mediated by EphA receptor and vascular endothelial growth factor in mice. Cancer Res. 66, 10315–10324 [DOI] [PubMed] [Google Scholar]
- 21. Larson J., Schomberg S., Schroeder W., Carpenter T.C. (2008) Endothelial EphA receptor stimulation increases lung vascular permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 295, L431–L439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pasquale E. B. (2008) Eph-ephrin bidirectional signaling in physiology and disease. Cell 133, 38–52 [DOI] [PubMed] [Google Scholar]
- 23. Wykosky J., Debinski W. (2008) The EphA2 receptor and ephrinA1 ligand in solid tumors: function and therapeutic targeting. Mol. Cancer Res. 6, 1795–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pasquale E. B. (2005) Eph receptor signalling casts a wide net on cell behaviour. Nat. Rev. Mol. Cell Biol. 6, 462–475 [DOI] [PubMed] [Google Scholar]
- 25. Gusenbauer S., Vlaicu P., Ullrich A. (2013) HGF induces novel EGFR functions involved in resistance formation to tyrosine kinase inhibitors. Oncogene 32, 3846–3856 [DOI] [PubMed] [Google Scholar]
- 26. Fang W. B., Ireton R. C., Zhuang G., Takahashi T., Reynolds A., Chen J. (2008) Overexpression of EPHA2 receptor destabilizes adherens junctions via a RhoA-dependent mechanism. J. Cell Sci. 121, 358–368 [DOI] [PubMed] [Google Scholar]
- 27. Flamme I., Frölich T., Risau W. (1997) Molecular mechanisms of vasculogenesis and embryonic angiogenesis. J. Cell. Physiol. 173, 206–210 [DOI] [PubMed] [Google Scholar]
- 28. Arnold F., West D. C. (1991) Angiogenesis in wound healing. Pharmacol. Ther. 52, 407–422 [DOI] [PubMed] [Google Scholar]
- 29. Shimizu T., Hoshino Y., Miyazaki H., Sato E. (2012) Angiogenesis and microvasculature in the female reproductive organs: physiological and pathological implications. Curr. Pharm. Des. 18, 303–309 [DOI] [PubMed] [Google Scholar]
- 30. Folkman J., Szabo S., Stovroff M., McNeil P., Li W., Shing Y. (1991) Duodenal ulcer: discovery of a new mechanism and development of angiogenic therapy that accelerates healing. Ann. Surg. 214, 414–425; discussion 426–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Semerano L., Clavel G., Assier E., Denys A., Boissier M. C. (2011) Blood vessels, a potential therapeutic target in rheumatoid arthritis? Joint Bone Spine 78, 118–123 [DOI] [PubMed] [Google Scholar]
- 32. Leong T. T., Fearon U., Veale D. J. (2005) Angiogenesis in psoriasis and psoriatic arthritis: clues to disease pathogenesis. Curr. Rheumatol. Rep. 7, 325–329 [DOI] [PubMed] [Google Scholar]
- 33. Folkman J. (1974) Tumor angiogensis: role in regulation of tumor growth. Symp. Soc. Dev. Biol. 30, 43–52 [PubMed] [Google Scholar]
- 34. Hanahan D., Folkman J. (1996) Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86, 353–364 [DOI] [PubMed] [Google Scholar]
- 35. Calabro A., Hascall V. C., Midura R. J. (2000) Adaptation of FACE methodology for microanalysis of total hyaluronan and chondroitin sulfate composition from cartilage. Glycobiology 10, 283–293 [DOI] [PubMed] [Google Scholar]
- 36. Lennon F. E., Mirzapoiazova T., Mambetsariev B., Poroyko V. A., Salgia R., Moss J., Singleton P. A. (2014) The μ-opioid receptor promotes opioid and growth factor-induced proliferation, migration, and epithelial mesenchymal transition (EMT) in human lung cancer. PLoS ONE 9, e91577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arnaoutova I., George J., Kleinman H. K., Benton G. (2009) The endothelial cell tube formation assay on basement membrane turns 20: state of the science and the art. Angiogenesis 12, 267–274 [DOI] [PubMed] [Google Scholar]
- 38. Singleton P. A., Mirzapoiazova T., Guo Y., Sammani S., Mambetsariev N., Lennon F. E., Moreno-Vinasco L., Garcia J. G. (2010) High molecular weight hyaluronan is a novel inhibitor of pulmonary vascular leakiness. Am. J. Physiol. Lung Cell. Mol. Physiol. 299, L639–L651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brandwijk R. J., Mulder W. J., Nicolay K., Mayo K. H., Thijssen V. L., Griffioen A. W. (2007) Anginex-conjugated liposomes for targeting of angiogenic endothelial cells. Bioconjug. Chem. 18, 785–790 [DOI] [PubMed] [Google Scholar]
- 40. Griffioen A. W., van der Schaft D. W., Barendsz-Janson A. F., Cox A., Struijker Boudier H. A., Hillen H. F., Mayo K. H. (2001) Anginex, a designed peptide that inhibits angiogenesis. Biochem. J. 354, 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. West D. C., Hampson I. N., Arnold F., Kumar S. (1985) Angiogenesis induced by degradation products of hyaluronic acid. Science 228, 1324–1326 [DOI] [PubMed] [Google Scholar]
- 42. Deed R., Rooney P., Kumar P., Norton J. D., Smith J., Freemont A. J., Kumar S. (1997) Early-response gene signalling is induced by angiogenic oligosaccharides of hyaluronan in endothelial cells. Inhibition by non-angiogenic, high molecular weight hyaluronan. Int. J. Cancer 71, 251–256 [DOI] [PubMed] [Google Scholar]
- 43. Otrock Z. K., Mahfouz R. A., Makarem J. A., Shamseddine A. I. (2007) Understanding the biology of angiogenesis: review of the most important molecular mechanisms. Blood Cells Mol. Dis. 39, 212–220 [DOI] [PubMed] [Google Scholar]
- 44. Toole B. P., Wight T. N., Tammi M. I. (2002) Hyaluronan-cell interactions in cancer and vascular disease. J. Biol. Chem. 277, 4593–4596 [DOI] [PubMed] [Google Scholar]
- 45. Slevin M., Kumar S., Gaffney J. (2002) Angiogenic oligosaccharides of hyaluronan induce multiple signaling pathways affecting vascular endothelial cell mitogenic and wound healing responses. J. Biol. Chem. 277, 41046–41059 [DOI] [PubMed] [Google Scholar]
- 46. Bourguignon L. Y., Wong G., Earle C., Krueger K., Spevak C. C. (2010) Hyaluronan-CD44 interaction promotes c-Src-mediated twist signaling, microRNA-10b expression, and RhoA/RhoC up-regulation, leading to Rho-kinase-associated cytoskeleton activation and breast tumor cell invasion, J. Biol. Chem. 285, 36721–36735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roskoski R., Jr. (2005) Src kinase regulation by phosphorylation and dephosphorylation. Biochem. Biophys. Res. Commun. 331, 1–14 [DOI] [PubMed] [Google Scholar]
- 48. Hsieh H. L., Lin C. C., Chan H. J., Yang C. M., Yang C. M. (2012) c-Src-dependent EGF receptor transactivation contributes to ET-1-induced COX-2 expression in brain microvascular endothelial cells. J. Neuroinflammation 9, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Carpenter T. C., Schroeder W., Stenmark K. R., Schmidt E. P. (2012) Eph-A2 promotes permeability and inflammatory responses to bleomycin-induced lung injury, Am. J. Respir. Cell Mol Biol. 46, 40–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fang W. B., Brantley-Sieders D. M., Hwang Y., Ham A. J., Chen J. (2008) Identification and functional analysis of phosphorylated tyrosine residues within EphA2 receptor-tyrosine kinase, J. Biol. Chem. 283, 16017–16026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Faoro L., Singleton P. A., Cervantes G. M., Lennon F. E., Choong N. W., Kanteti R., Ferguson B. D., Husain A. N., Tretiakova M. S., Ramnath N., Vokes E. E., Salgia R. (2010) EphA2 mutation in lung squamous cell carcinoma promotes increased cell survival, cell invasion, focal adhesions, and mammalian target of rapamycin activation. J. Biol. Chem. 285, 18575–18585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Krupinski J., Ethirajan P., Font M. A., Turu M. M., Gaffney J., Kumar P., Slevin M. (2008) Changes in hyaluronan metabolism and RHAMM receptor expression accompany formation of complicated carotid lesions and may be pro-angiogenic mediators of intimal neovessel growth. Biomark. Insights 2, 361–367 [PMC free article] [PubMed] [Google Scholar]
- 53. Sáinz-Jaspeado M., Huertas-Martinez J., Lagares-Tena L., Martin Liberal J., Mateo-Lozano S., de Alava E., de Torres C., Mora J., Del Muro X. G., Tirado O. M. (2013) EphA2-induced angiogenesis in ewing sarcoma cells works through bFGF production and is dependent on caveolin-1, PLoS ONE 8, e71449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ernkvist M., Luna Persson N., Audebert S., Lecine P., Sinha I., Liu M., Schlueter M., Horowitz A., Aase K., Weide T., Borg J. P., Majumdar A., Holmgren L. (2009) The Amot/Patj/Syx signaling complex spatially controls RhoA GTPase activity in migrating endothelial cells. Blood 113, 244–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dings R. P., Van Laar E. S., Loren M., Webber J., Zhang Y., Waters S. J., Macdonald J. R., Mayo K. H. (2010) Inhibiting tumor growth by targeting tumor vasculature with galectin-1 antagonist anginex conjugated to the cytotoxic acylfulvene, 6-hydroxylpropylacylfulvene. Bioconjug. Chem. 21, 20–27 [DOI] [PubMed] [Google Scholar]
- 56. Thijssen V. L, Hulsmans S., Griffioen A. W. (2008) The galectin profile of the endothelium: altered expression and localization in activated and tumor endothelial cells. Am. J. Pathol. 172, 545–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hughes C. S., Postovit L. M., Lajoie G. A. (2010) Matrigel: a complex protein mixture required for optimal growth of cell culture. Proteomics 10, 1886–1890 [DOI] [PubMed] [Google Scholar]
- 58. Beauchamp A., Debinski W. (2012) Ephs and ephrins in cancer: ephrin-A1 signalling. Semin. Cell Dev. Biol. 23, 109–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Singleton P. A., Salgia R., Moreno-Vinasco L., Moitra J., Sammani S., Mirzapoiazova T., Garcia J. G. (2007) CD44 regulates hepatocyte growth factor-mediated vascular integrity. Role of c-Met, Tiam1/Rac1, dynamin 2, and cortactin. J. Biol. Chem. 282, 30643–30657 [DOI] [PubMed] [Google Scholar]
- 60. Matou-Nasri S., Gaffney J., Kumar S., Slevin M. (2009) Oligosaccharides of hyaluronan induce angiogenesis through distinct CD44- and RHAMM-mediated signalling pathways involving Cdc2 and γ-adducin. Int. J. Oncol. 35, 761–773 [DOI] [PubMed] [Google Scholar]
- 61. Cheng N., Brantley D. M., Liu H., Lin Q., Enriquez M., Gale N., Yancopoulos G., Cerretti D. P., Daniel T. O., Chen J. (2002) Blockade of EphA receptor-tyrosine kinase activation inhibits vascular endothelial cell growth factor-induced angiogenesis. Mol. Cancer Res. 1, 2–11 [PubMed] [Google Scholar]
- 62. Zhou N., Zhao W. D., Liu D. X., Liang Y., Fang W. G., Li B., Chen Y. H. (2011) Inactivation of EphA2 promotes tight junction formation and impairs angiogenesis in brain endothelial cells. Microvasc. Res. 82, 113–121 [DOI] [PubMed] [Google Scholar]
- 63. Chan B., Sukhatme V. P. (2009) Receptor-tyrosine kinase EphA2 mediates thrombin-induced upregulation of ICAM-1 in endothelial cells in vitro. Thromb. Res. 123, 745–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shin K., Wang Q., Margolis B. (2007) PATJ regulates directional migration of mammalian epithelial cells. EMBO Rep. 8, 158–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Whitehead I., Kirk H., Kay R. (1995) Retroviral transduction and oncogenic selection of a cDNA encoding Dbs, a homolog of the Dbl guanine nucleotide exchange factor. Oncogene 10, 713–721 [PubMed] [Google Scholar]
- 66. Yamauchi J., Hirasawa A., Miyamoto Y., Kokubu H., Nishii H., Okamoto M., Sugawara Y., Tsujimoto G., Itoh H. (2002) Role of Dbl's big sister in the anti-mitogenic pathway from α1B-adrenergic receptor to c-Jun N-terminal kinase. Biochem. Biophys. Res. Commun. 296, 85–92 [DOI] [PubMed] [Google Scholar]
- 67. Kostenko E. V., Olabisi O. O., Sahay S., Rodriguez P. L., Whitehead I. P. (2006) Ccpg1, a novel scaffold protein that regulates the activity of the Rho guanine nucleotide exchange factor Dbs. Mol. Cell. Biol. 26, 8964–8975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Spindler V., Schlegel N., Waschke J. (2010) Role of GTPases in control of microvascular permeability. Cardiovasc. Res. 87, 243–253 [DOI] [PubMed] [Google Scholar]
- 69. Park D., Kim Y., Kim H., Kim K., Lee Y. S., Choe J., Hahn J. H., Lee H., Jeon J., Choi C., Kim Y. M., Jeoung D. (2012) Hyaluronic acid promotes angiogenesis by inducing RHAMM-TGFβ receptor interaction via CD44-PKCδ. Mol. Cells 33, 563–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang Y. Z., Cao M. L., Liu Y. W., He Y. Q., Yang C. X., Gao F. (2011) CD44 mediates oligosaccharides of hyaluronan-induced proliferation, tube formation, and signal transduction in endothelial cells. Exp. Biol. Med. (Maywood) 236, 84–90 [DOI] [PubMed] [Google Scholar]
- 71. Olofsson B., Porsch H., Heldin P. (2014) Knock-down of CD44 regulates endothelial cell differentiation via NFκB-mediated chemokine production. PLoS ONE 9, e90921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Taylor K. R., Yamasaki K., Radek K. A., Di Nardo A., Goodarzi H., Golenbock D., Beutler B., Gallo R. L. (2007) Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on Toll-like receptor 4, CD44, and MD-2. J. Biol. Chem. 282, 18265–18275 [DOI] [PubMed] [Google Scholar]
- 73. Taylor K. R., Trowbridge J. M., Rudisill J. A., Termeer C. C., Simon J. C., Gallo R. L. (2004) Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J. Biol. Chem. 279, 17079–17084 [DOI] [PubMed] [Google Scholar]
- 74. Savani R. C., Cao G., Pooler P. M., Zaman A., Zhou Z., DeLisser H. M. (2001) Differential involvement of the hyaluronan (HA) receptors CD44 and receptor for HA-mediated motility in endothelial cell function and angiogenesis. J. Biol. Chem. 276, 36770–36778 [DOI] [PubMed] [Google Scholar]
- 75. Gao F., Yang C. X., Mo W., Liu Y. W., He Y. Q. (2008) Hyaluronan oligosaccharides are potential stimulators to angiogenesis via RHAMM mediated signal pathway in wound healing. Clin. Invest. Med. 31, E106–E116 [DOI] [PubMed] [Google Scholar]