

Background: LpxK is an essential membrane-bound kinase in the lipid A biosynthetic pathway.
Results: Structural and kinetic studies reveal the molecular basis of lipid binding.
Conclusion: The LpxK active site recognizes the lipid's glucosamine/phosphate headgroups and only accommodates disaccharides.
Significance: These studies provide a structural template for designing novel antibiotics and a glimpse of catalysis at the membrane interface.
Keywords: Crystal Structure, Enzyme, Lipid A, Lipid-binding Protein, Lipopolysaccharide (LPS), Membrane Protein, Antibiotic, Lipid Kinase
Abstract
The membrane-bound tetraacyldisaccharide-1-phosphate 4′-kinase, LpxK, catalyzes the sixth step of the lipid A (Raetz) biosynthetic pathway and is a viable antibiotic target against emerging Gram-negative pathogens. We report the crystal structure of lipid IVA, the LpxK product, bound to the enzyme, providing a rare glimpse into interfacial catalysis and the surface scanning strategy by which many poorly understood lipid modification enzymes operate. Unlike the few previously structurally characterized proteins that bind lipid A or its precursors, LpxK binds almost exclusively to the glucosamine/phosphate moieties of the lipid molecule. Steady-state kinetic analysis of multiple point mutants of the lipid-binding pocket pinpoints critical residues involved in substrate binding, and characterization of N-terminal helix truncation mutants uncovers the role of this substructure as a hydrophobic membrane anchor. These studies make critical contributions to the limited knowledge surrounding membrane-bound enzymes that act upon lipid substrates and provide a structural template for designing small molecule inhibitors targeting this essential kinase.
Introduction
Lipid molecules that reside in the membrane are acted upon by enzymes that either reside in the membrane themselves or are recruited there by other effectors. Despite the countless critical cellular processes that occur at the membrane interface, how enzymatic catalysis proceeds in such a heterogeneous environment remains a poorly understood phenomenon. In particular, due to the complex nature of biological membranes, the strategies with which these enzymes find and act upon their lipid substrates are yet to be elucidated. The bacterial lipid A (Raetz) biosynthetic pathway provides a useful template for understanding how cytosolic precursors become fully embedded membrane lipids and illustrates the various stages of membrane catalysis that accompany this transition (1).
In Gram-negative bacteria, lipid A is the hydrophobic anchor of lipopolysaccharide (LPS), which makes up the outer leaflet of the asymmetric outer membrane of these organisms. This acylated disaccharide of glucosamine plays an important role in eliciting an immunogenic response to bacterial pathogens and is essential to the survival of the vast majority of these microbes (2, 3). The sixth step of the lipid A biosynthetic pathway in Escherichia coli is the phosphorylation at the 4′-position of the tetraacyldisaccharide-1-phosphate intermediate (DSMP)2 by the cytosol-facing inner membrane kinase LpxK to produce lipid IVA (Fig. 1A) (4, 5). Crystal structures of Aquifex aeolicus LpxK in apo-, nucleotide-bound, and nucleotide analog-bound forms have been reported (6, 7), revealing LpxK as a distinct member of the P-loop-containing nucleoside triphosphate hydrolase superfamily that acts upon a lipid substrate at the membrane surface (8–10). The LpxK C-terminal domain, a substructure unique to this enzyme, connects to the N-terminal domain by a long two-stranded β-sheet linker and serves as the “lid” to trap the nucleotide substrate between the two domains, forming the “closed” conformation (Fig. 1B). In the apo-form, the C-terminal lid domain is rotated away from the active site by a ∼25° angle, resulting in an “open” conformation of the ATP-binding pocket (6).
FIGURE 1.
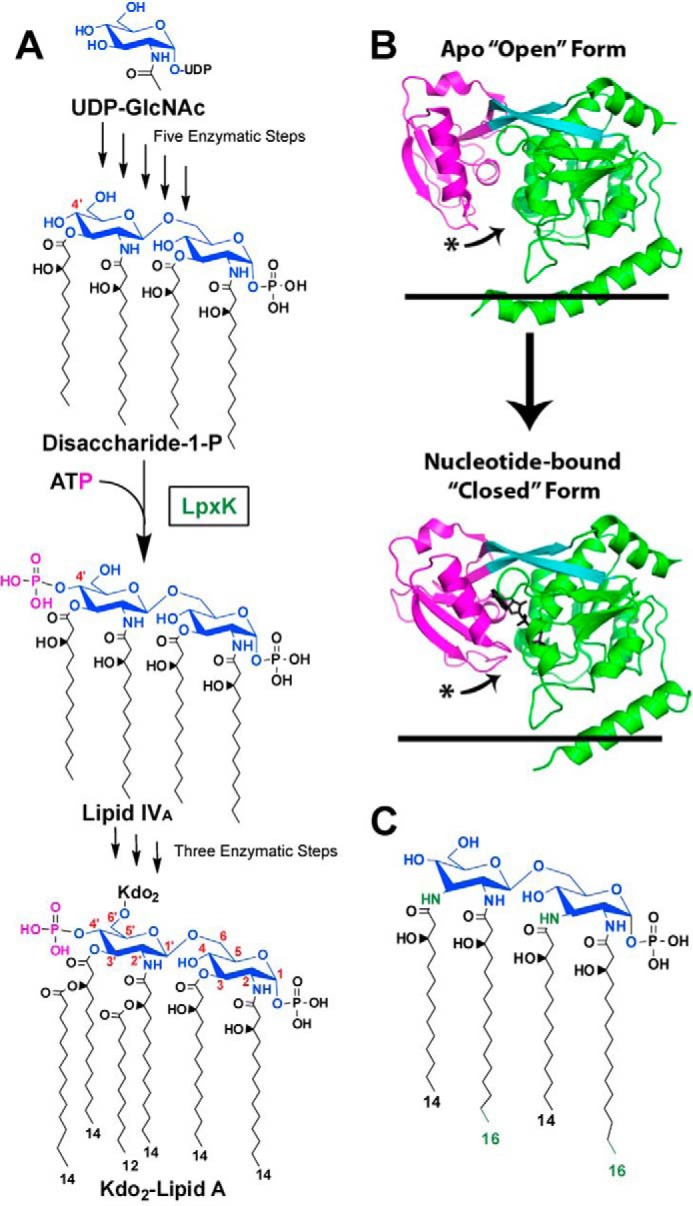
Role of LpxK in the Raetz pathway of lipid A biosynthesis. A, the biosynthesis of lipid A begins with uridine diphosphate-N-acetyl-d-glucosamine (UDP-GlcNAc). The pathway consists of nine steps, the sixth being the phosphorylation (magenta) of DSMP at the 4′-hydroxyl by LpxK, a membrane-bound kinase. Atoms of the glucosamine headgroups of lipid A are highlighted (blue), and positions on these sugars are indicated (red). Acyl chain length in Kdo2-lipid A is also indicated. Kdo, 3-deoxy-d-manno-oct-2-ulosonic acid. B, LpxK consists of an N-terminal domain (green), a linker region (cyan), and a C-terminal domain (magenta). The kinase undergoes a conformational transition upon catalysis in which its C-terminal “lid” domain closes around its nucleotide ligand (black sticks). The curved arrows indicate relative movement of C-terminal loops undergoing this hinge motion. Black lines represent the cytoplasmic inner membrane surface (Protein Data Bank entries 4EHX and 4ITL). C, predicted structure of A. aeolicus DSMP based on the structure of Aquifex lipid A (11, 12). A. aeolicus DSMP contains longer acyl chains at the 2- and 2′-positions on the glucosamine backbone as well as amide linkages for the 3- and 3′-acyl chains, which differentiate it from the E. coli DSMP. Differences are colored in green.
Much less is known regarding recognition by LpxK of its lipid ligands DSMP and lipid IVA. Kinetic studies have revealed that DSMP and ATP/Mg2+ must concurrently bind to the enzyme in order for catalysis to occur, and the kinase activity is a surface-dependent process (7). A. aeolicus LpxK has an apparent Km for Escherichia coli-derived DSMP of 7.0 ± 0.3 μm and shows activity in vitro despite molecular differences between E. coli DSMP and the predicted structure of A. aeolicus DSMP, which include longer acyl chains at the 2- and 2′-positions along with amide-linked acyl chains at the 3- and 3′-positions as opposed to ester linkages (Fig. 1C) (11, 12). Based on the directionality of the nucleotide phosphates in the AMP-PCP-bound LpxK structure, DSMP is predicted to bind to a shallow basic cleft on the N-terminal domain formed between the N-terminal helix and core of the domain (7). The size and location of this cleft suggest that LpxK binds a smaller portion of its lipid substrate when compared with the few other proteins that have been crystallized in complex with lipid A or its precursors and imply a distinct strategy for finding its lipid substrate.
Knowledge of how LpxK interacts with the membrane and its lipid substrate will directly facilitate the development of small molecule antibiotics targeting this essential lipid A enzyme in Gram-negative bacteria and provide molecular insight into the activity of membrane-bound lipid modification enzymes, about which there exist precious little structural or kinetic data to date.
EXPERIMENTAL PROCEDURES
Complementation of an E. coli lpxK Knockout with A. aeolicus LpxK
Methods for manipulating DNA were adapted from previously described techniques (13). The E. coli lpxK gene was amplified from the plasmid pJK2 (5) via PCR by primers designed to incorporate 5′ NdeI and 3′ XhoI restriction sites into the PCR product. PCR was performed using the KOD Hot Start kit (EMD Millipore, Billerica, MA) on a Mastercycler gradient thermocycler (Eppendorf, Hamburg, Germany). This oligonucleotide, along with the pET21b vector (EMD Millipore, Billerica, MA), was digested with NdeI and XhoI (New England Biolabs, Ipswich, MA), and the digested PCR product containing the E. coli lpxK gene was ligated into the expression vector by T4 ligase (Invitrogen), creating the plasmid pRPE10, and transformed into chemically competent DH5(α) cells (14) (Invitrogen). In order to transfer both the E. coli and A. aeolicus lpxK genes into the pBAD30 vector (15), the plasmids pRPE7 (A. aeolicus lpxK) and pRPE10 (E. coli lpxK) were digested with XbaI and XhoI in order to maintain the ribosome-binding site from pET21b. The inserts were gel-purified and ligated into the pBAD30 vector, which had been digested by XbaI and SalI (SalI and XhoI have compatible sticky ends). The pBAD30 vectors containing the pET21b ribosome binding site and E. coli and A. aeolicus lpxK were designated pRPE43 and pRPE44, respectively, and transformed into electrocompetent TG1/pTAG1 cells (16). The presence of the correct construction was confirmed by sequencing.
TG1/pTAG1 is an E. coli strain with a chromosomal knockout of lpxK replaced by a kanamycin cassette. The knockout is complemented by the lpxK gene on a pMAK705 plasmid, which is temperature-sensitive and stops being replicated at 42 °C (17). TG1/pTAG1 was transformed with the pRPE43, pRPE44, or empty pBAD30 vector and grown on LB-agar plates supplemented with 50 μg/ml chloramphenicol, 25 μg/ml kanamycin, and 50 μg/ml ampicillin at 30 °C. One colony from each plate was used to inoculate 6 ml of LB overnight supplemented with chloramphenicol, kanamycin, and ampicillin. The overnight cultures grew to an OD of ∼2, at which point they were diluted in LB to an OD of 1 and serially diluted 10-fold eight successive times. 3 μl of each dilution was spotted onto duplicate LB-agar plates supplemented with kanamycin and ampicillin. One plate was incubated at 30 °C and the other at 42 °C for 24 h, at which point relative colony growth between the three strains was assessed for the two temperatures.
In order to further confirm the ability of A. aeolicus LpxK to complement the E. coli enzyme in vivo, W3110 cells (18) that had been chemically transformed with the plasmids pBAD30 (vector control), pRPE43 (EcLpxK in pBAD30), or pRPE44 (AaLpxK in pBAD30) were transduced by P1vir bacterial lysates of the TG1/pTAG1 strain, transferring the chromosomal knockout of the lpxK gene. Upon outgrowth of the transductions onto fresh LB-agar plates supplemented with kanamycin/ampicillin, colonies were only observed for the strains of W3110 that harbored plasmid pRPE43 or pRPE44 but not the vector control. To confirm the correct placement of the kanamycin cassette disrupting the endogenous lpxK gene, colony PCR was performed on each of the surviving strains with a forward primer complementary to a sequence on the upstream msbA gene (prmsbaF) and reverse primers complementary to either the downstream ycaR gene (prycarR) or the transduced kanamycin cassette (prkanR). Results of colony PCR were analyzed on a 1% (w/v) agarose gel.
Crystal Structure of LpxK Bound to Lipid IVA
Wild type A. aeolicus LpxK was generated by growth of C41(DE3) (19) cultures expressing the construct pRPE7 and purified as described previously (6). Crystals were grown in 24-well sitting drop vapor diffusion trays from Hampton (Aliso Viejo, CA) at 20 °C. All conditions included a 700-μl well volume and 10-μl drop volume, and crystals were immediately flash-frozen in liquid nitrogen after being looped. Small cube-shaped crystals (0.03 × 0.03 × 0.03 mm) were obtained when LpxK was incubated with lipid IVA, ADP, and Mg2+. Lipid IVA was a gift from Dr. Hak Suk Chung, which was purified from the CMR300 E. coli strain, as has been previously described (20). The lipid IVA stock solution contained 0.1% (w/v) Triton X-100 and 25 mm Tris, pH 6.8, along with 2 mm lipid. The drop included seven parts reservoir solution containing 60% (v/v) 2-methyl-2,4-propanediol and 0.1 m HEPES, pH 7.5, and three parts protein solution containing 7.6 mg/ml LpxK, 1 mm lipid IVA, 12 mm Tris, pH 6.8, 0.05% (w/v) Triton X-100, 0.28% (w/v) dodecyl maltoside, 8.5% (v/v) glycerol, 21 mm HEPES, pH 8, 570 mm NaCl, 2.2 mm MgCl2, and 2.2 mm ADP. Initial crystals were harvested after 5 months of incubation and diffracted poorly. A microcrystalline seed stock was then made with three of these crystals homogenized in a 500 μl of 60% (v/v) 2-methyl-2,4-propanediol and 0.1 m HEPES, pH 7.5, solution.
Seed stock (see above) was used to nucleate a second batch of crystals. These crystals were grown in 24-well sitting drop trays as above with 0.5 μl of the seed stock per well. The drop consisted of seven parts reservoir solution containing 70% (v/v) 2-methyl-2,4-propanediol and 0.1 m HEPES, pH 7.5, and three parts protein solution containing 6.7 mg/ml LpxK, 1 mm lipid IVA, 12 mm Tris, pH 6.8, 0.05% (w/v) Triton X-100, 0.42% (w/v) dodecyl maltoside, 8.5% (v/v) glycerol, 21 mm HEPES, pH 8, 570 mm NaCl, 1 mm MgCl2, and 1 mm ADP. Cube-shaped crystals similar to the original seed stock were obtained after 1 month. Data were collected at a single wavelength (1.0 Å) at the SER-CAT (Southeast Regional Collaborative Access Team) 22-ID line at the Advanced Photon Source (Argonne National Laboratory) to a resolution of 3.5 Å in the C2221 space group with unit cell dimensions of a = 136.6 Å, b = 140.7 Å, c = 173.8 Å. Data were reduced and scaled using HKL-3000, and the structure was solved using the PHASER molecular replacement module within the PHENIX suite (21, 22). Apo-LpxK was used as the search model (Protein Data Bank code 4EHX). Iterative structure refinement was accomplished using the PHENIX software suite, included optimized atomic displacement parameters and stereochemistry weights, and used the search model as a restraint reference model. Riding hydrogen atoms were included throughout the refinement. There were four molecules in the asymmetric unit, and ligands (lipid IVA and HEPES) were added to the model using an electron density acceptance criterion of d ≥ 3 σ (d) in the Fo − Fc difference electron density map. The final model was validated via MOLPROBITY (23). Data collection and refinement statistics can be found in Table 1. A simulated annealing composite omit map for the lipid ligands was generated using CNS (24).
TABLE 1.
Data collection and refinement statistics for LpxK-lipid IVA complex
The data set was collected from two crystals.
| Parameters | Values |
|---|---|
| Data collection | |
| Space group | C2221 |
| Unit cell a, b, c (Å) | 136.6, 140.7, 173.8 |
| Wavelength (Å) | 1.0 |
| Resolution (Å) | 50.0–3.5 (3.56–3.50)a |
| Rmerge | 0.107 (0.687) |
| I/σ | 21.2 (2.0) |
| Completeness (%) | 94.2 (74.2) |
| Redundancy | 12.8 (5.7) |
| Reflections/unique | 257,205/20,053 |
| Refinement | |
| Rwork/Rfree (%) | 26.7/33.1 |
| No. of atoms (average B-factor, Å2) | |
| Protein | 10,272 (88.3) |
| Lipid IVA | 94 (74.5) |
| Ramachandran plot | |
| Favored/allowed/outlier (%) | 96.7/3.0/0.3 |
| Root mean square deviations | |
| Bond length (Å) | 0.01 |
| Bond angles (degrees) | 0.82 |
a Values in parenthesis are for the highest resolution shell.
Point Mutagenesis of Lipid-binding LpxK Residues
QuikChange mutagenesis (Stratagene, La Jolla, CA) was employed to generate point mutants N43A, R72A, R119A, Q142A, H143A, R171A, and E172A. Previously constructed point mutants Y74A, E100A, D138A, D138N, D139A, D139N, and H261A (6, 7) were also obtained. To generate partially purified LpxK point mutants, the plasmids were transformed into C41(DE3), expressed, and solubilized from membranes as described (7).
Solubilized membranes of each of the point mutants were assayed in the standard assay condition but with varied DSMP concentrations (1.56–100 μm) and a fixed concentration of ATP and MgCl2 of 5 mm. The other assay components were 50 mm Tris, pH 8.5, 0.5% (w/v) Triton X-100 (Thermo Scientific, Rockford, IL), 1 mg/ml BSA (Sigma-Aldrich), 0.1 m NaCl, and enzyme. The TLC-based assay was performed as described previously (6, 7), and the data were fit to the Michaelis-Menten equation using KaleidaGraph (Synergy Software, Reading, PA) in order to determine apparent Km and kcat with respect to DSMP. Enzyme concentration was varied to capture the linear range of activity for each mutant.
N-terminal Helix Truncation Studies
Two truncated forms of A. aeolicus LpxK were generated, one in which amino acids 2–12 were removed (referred to as Δ12LpxK) and another in which amino acids 2–29 were removed (referred to as Δ29LpxK). This corresponds to deleting roughly half of the N-terminal helix and the full N-terminal helix, respectively. Primers were used to amplify the Δ12LpxK or Δ29LpxK PCR product from pRPE7 and bestow 5′ NdeI and 3′ XhoI restriction sites. PCR was performed on a T3000 Thermocycler (Biometra, Göttingen, Germany) with the KOD Hot Start kit (EMD Millipore, Billerica, MA). The PCR products were digested with the respective enzymes, ligated with T4 ligase (Invitrogen) into the pET21b expression vector (EMD Millipore, Billerica, MA), and transformed into chemically competent DH5(α) cells (Invitrogen). Sequencing confirmed the correct construction of the Δ12LpxK (pRPE72) and Δ29LpxK (pRPE73) mutants. These plasmids were transformed into C41(DE3) cells, and membranes expressing these constructs were generated as with wild type LpxK (6, 19). All constructs expressed to comparable levels as judged by SDS-PAGE. Cell-free extract from cells expressing these constructs along with wild type LpxK and empty vector were assayed using the standard assay conditions (6, 7).
Membranes derived from C41(DE3) overexpressing the empty vector, full-length LpxK, Δ12LpxK, and Δ29LpxK were obtained and suspended in 50 mm HEPES, pH 8.0, to ∼9 mg/ml. Each of these membrane samples was diluted to 1.5 mg/ml into 0.5 ml of either 50 mm HEPES, pH 8.0, 1 m NaCl, 0.2 m sodium carbonate, pH 11.5, or the normal solubilization buffer consisting of 0.5 m NaCl, 1% (w/v) Triton X-100, and 20% (v/v) glycerol. Samples were incubated for 2 h at 4 °C in ultracentrifuge-safe microtubes (Beckman Coulter, Brea, CA) and centrifuged at 150,000 × g for 1 h at 4 °C. The supernatant was collected, and the pellet was homogenized in 0.5 ml of 50 mm HEPES, pH 8.0. Both soluble and insoluble fractions were analyzed using SDS-PAGE with 20 μl of each fraction per lane.
RESULTS
Complementation of an E. coli lpxK Knockout with A. aeolicus LpxK
To determine whether the LpxK ortholog from A. aeolicus could complement an E. coli knockout of the lpxK gene, pBAD30 vectors harboring the genes from both E. coli and A. aeolicus were transformed into the TG1/pTAG1 strain (16). When grown at 30 °C, TG1/pTAG1 transformed with E. coli lpxK, A. aeolicus lpxK, or the empty pBAD30 vector all grew robustly (15) (Fig. 2). In these cells, the temperature-sensitive replicon of the pMAK705 plasmid is able to function, and a sufficient amount of LpxK is expressed for the cells to survive (17). However, on identical plates that had been incubated at 42 °C, TG1/pTAG1 cells transformed with pBAD30 containing either E. coli lpxK or A. aeolicus lpxK were able to grow, whereas the empty vector could not (Fig. 2), indicating that A. aeolicus LpxK can perform the function of E. coli LpxK sufficiently well to complement a chromosomal knockout of the gene.
FIGURE 2.

Complementation of E. coli LpxK with A. aeolicus LpxK. The TG1/pTAG1 strain of E. coli, containing a chromosomal knockout of lpxK complemented with E. coli LpxK on a pMAK plasmid with a temperature-sensitive replicon, was transformed with pBAD vectors containing no insert, E. coli lpxK, or A. aeolicus lpxK. 3 μl of eight successive 10-fold serial dilutions (from left to right) of cultures starting at an A600 of 1 were spotted and grown at 30 or 42 °C. Cells containing the pBAD vectors with either LpxK ortholog, but not with an empty insert, were able to support growth at 42 °C upon loss of the pMAK vector, indicating that they could complement the chromosomal knockout of the lpxK gene.
In a second set of experiments designed to further confirm the complementation of E. coli LpxK with the A. aeolicus ortholog in vivo, a chromosomal knockout of the lpxK gene in E. coli (16) was transduced into W3110 strains (18) expressing E. coli or A. aeolicus LpxK on a pBAD30 vector. Both strains were able to survive upon transduction, whereas vector control was not. Colony PCR confirmed the presence of the lpxK::kan insertion at the correct location on the chromosome. These results indicate that the molecular differences between E. coli and A. aeolicus DSMP do not prevent A. aeolicus LpxK from utilizing the E. coli-derived substrate and validate our pursuit of structural and kinetic studies involving A. aeolicus LpxK with the more accessible E. coli-derived reaction product lipid IVA.
Crystal Structure of LpxK Bound to Its Lipid Product
In order to identify the specific molecular interactions between LpxK and its lipid ligands, A. aeolicus LpxK was crystallized in the presence of its product, lipid IVA. The resulting crystals diffracted to 3.5 Å with four molecules in the asymmetric unit (Table 1 and Fig. 3A). Each of the LpxK protomers adopted the “open” form of the enzyme with respect to nucleotide binding (6) (Figs. 1B and 3B). Interpretable density was found in two of the four putative lipid-binding pockets (chains A and C), and lipid IVA was modeled into both (Fig. 3A). The fact that the shape of the electron density at these two sites within the asymmetric unit is similar enough to allow for the placement of lipid IVA in the same orientation (Fig. 3B) further supports the inclusion of these ligands within the overall structure. Because the electron density is best defined in the pocket of chain A and the B-factors are considerably lower for these atoms (average B-factor of 59.4 Å2 for the chain A ligand versus 90.3 Å2 for the chain C ligand), further analysis regarding lipid IVA binding will be in reference to this protomer. A simulated annealing composite omit map for the lipid ligand of chain A was generated using CNS, and the corresponding difference density showed a strong signal for the lipid IVA headgroups (24) (Fig. 3C).
FIGURE 3.

Crystal structure of LpxK bound to lipid IVA. A, the asymmetric unit contains four LpxK molecules (green, cyan, magenta, and yellow for chains A, B, C, and D, respectively) and two molecules of lipid IVA (black sticks). B, alignment of polypeptide chains A–D illustrates that lipid IVA is bound to the “open” form of the enzyme in this crystal. C, stick representation of lipid IVA in the crystal structure (from chain A) with the corresponding simulated annealing composite omit electron density (blue mesh) calculated with coefficients 2Fo − Fc contoured at 1.5 σ. For comparison, the chemical structure of lipid IVA is included, with atoms omitted due to poor electron density shown in gray. The 4′-position is indicated in red for reference. D, the crystal structure of the LpxK-lipid IVA complex (chain A) reveals that the lipid is bound in a cleft at the base of the N-terminal domain between the N-terminal helix and core domain. Polypeptide backbone (purple diagram) is traced through the surface representation of LpxK (gray) with the lipid displayed as green sticks.
Lipid IVA is bound in the putative lipid-binding pocket on the underside of the N-terminal domain (Figs. 3D and 4A). Electron density for the glucosamine/phosphate headgroups of the lipid is strong, whereas density for the acyl chains is comparatively poor (Fig. 3C). Conserved residues of the L1 (purple), L4 (cyan), and L5 (orange) loops participate in lipid binding, as hinted at in previous studies (6) (Fig. 4). Loop L1, containing RGY from the highly conserved SRGY motif among LpxK orthologs, runs along the internal face of the lipid-binding pocket. The side chain of Arg72 is positioned to assist in binding the distal lipid IVA headgroup via a hydrogen bond with the 6′-oxygen and the 1-phosphate, whereas the Tyr74 side chain contributes a potential van der Waals interaction (Fig. 4, B and C). The putative hydrogen bond formed between the Arg72 guanidinium and the 6′-hydroxyl of the distal glucosamine may protect this oxygen from being erroneously phosphorylated.
FIGURE 4.

LpxK residues involved in lipid binding. A, surface representation of LpxK from the point of view of the inner membrane, with the N-terminal domain, N-terminal helix, and C-terminal domain substructures indicated. The surface created by residues of the conserved loops L1, L4, and L5 (magenta, cyan, and orange, respectively) makes up the left, back, and right sides of the lipid-binding pocket. B, schematic overview of lipid-binding residues of LpxK. Side chain atoms are highlighted in red. Coloring for the lipid is conserved from Fig. 3C. Straight blue dotted lines indicate putative hydrogen bonds, whereas curved blue dotted lines represent potential van der Waals contacts. C, orthogonal view of LpxK residues involved in lipid binding. Orientation of the lipid is conserved from Fig. 3D with the 4′-phosphate group oriented to the left (left) and front (right), respectively. Nearby side chains are represented as sticks with coloring conserved from A. Purple sticks indicate side chains that do not belong to the three highlighted motifs.
The L4 loop, which includes the Walker B motif, also appears to contain important residues for binding the lipid substrate. These residues make up the top of the lipid-binding pocket and are involved in the interaction with the glucosamine/phosphate moieties of lipid IVA (Fig. 4). Asp138, a residue implicated in magnesium binding according to previous structural data of both LpxK and other P-loop kinases (7, 9, 10), faces away from the lipid-binding site and points toward that of ATP/Mg2+. Asp139 of the Walker B motif appears to form a hydrogen bond with both the Arg171 and Arg72 guanidinium moieties, probably positioning these residues for substrate binding. Gln142 also interacts with Arg171 and may also contribute to van der Waals interactions with the lipid IVA 1-phosphate. His143, as hinted at upon analysis of HEPES sulfate binding to the “apo” structure (6), forms a hydrogen bond to the 1-position phosphate of the lipid.
The L5 loop (Leu165, Arg171, and Glu172) makes up a portion of the external wall of the lipid-binding pocket (Fig. 4). Leu165 is located in a favorable position to make hydrophobic contacts with the distal acyl chains of the lipid ligand, although a lack of acyl chain density prevents a more specific assignment. As previously mentioned, the Arg171 guanidinium group interacts with the Asp139 carboxylate and may also help LpxK bind to DSMP through extensive van der Waals contact with the lipid headgroups and C2′-acyl chain (Fig. 4). A slight shift in conformation of this residue could conceivably see this guanidinium side chain interacting with glucosamine oxygens or with the carboxamide oxygen of the 2′-acyl chain. Glu172, another highly conserved residue of the L5 loop, faces away from the lipid-binding site, forming a hydrogen bond with Gln142.
Other residues outside these three loops also appear to have potential roles in lipid binding. Asn43, a conserved residue of the N-terminal domain, may help position residues such as Arg171 and the catalytic Lys51 (Fig. 4). Val46, a residue that precedes the P-loop motif, is less conserved and may contribute van der Waals interactions to the distal glucosamine of the 4′-phosphate. Glu117, a less conserved N-terminal domain β-sheet residue, is hydrogen-bonded to the 4-position oxygen of the proximal glucosamine moiety (Fig. 4B). Tyr13 of the N-terminal helix is well positioned to interact with the acyl chains of lipid IVA, especially the 2′-hydroxymyristoyl group, given the extended electron density for this acyl chain compared with that of the other hydrocarbons (Figs. 3C and 4). Residues Glu100 and His261, previously implicated in ATP/Mg2+ binding and catalysis, respectively, appear to be uninvolved in lipid binding in this open conformation. Glu100 remains out of bonding range to the lipid (Fig. 4C), and His261 resides ∼15 Å away from the lipid-binding pocket on the C-terminal domain (Fig. 7A).
FIGURE 7.
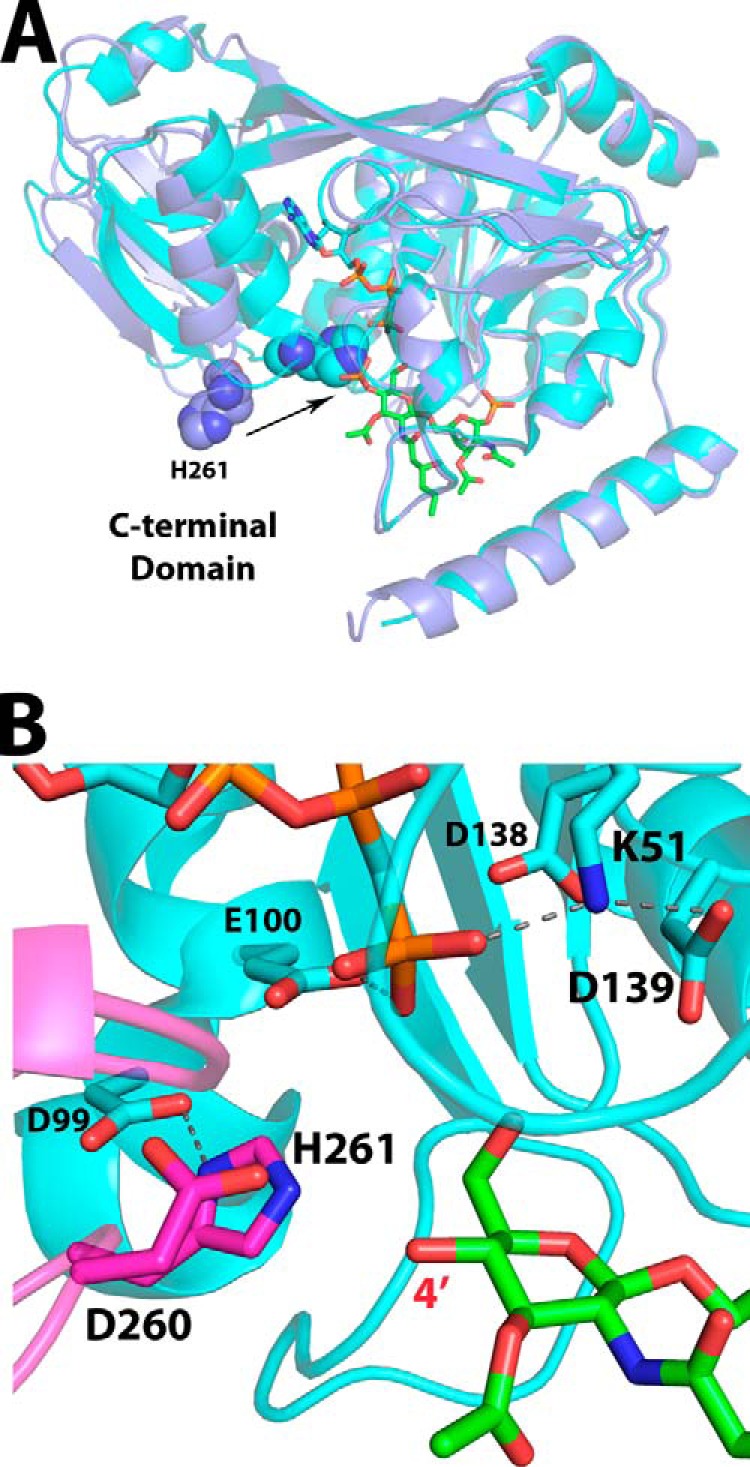
Overlay of AMP-PCP and lipid IVA-bound structures as a model for the catalytic ATP-DSMP-LpxK ternary complex. A, alignment of the LpxK-lipid IVA complex (purple schematic) and the nucleotide-bound complex in the “closed” form (Protein Data Bank code 4ITL) (cyan schematic) illustrates how the motion of the C-terminal domain closing around the nucleotide assembles the active site. The movement of the catalytic histidine His261 (spheres) upon closure of the C-terminal domain to assemble the active site is indicated by an arrow. B, visualization of the active site of the AMP-PCP bound LpxK structure (cyan schematic and sticks) with DSMP modeled (green sticks, with atoms for the 4′-phosphate removed) based on the alignment of the N-terminal domains from A. The C-terminal domain (magenta) closure moves the α-carbon of His261, the putative catalytic base, over 7 Å so that it may interact with Asp99 to be activated. Among the plausible catalytic bases Glu100, Asp139, and His261, the histidine resides nearest to the 4′-hydroxyl position (red) at 3.3 Å in this model, giving further credence to the hypothesis that this residue is the catalytic base. Hydrogen bonds within the AMP-PCP-bound LpxK structure are indicated with gray dashes. Font size is indicative of depth.
Point Mutagenesis of Lipid Binding Residues in LpxK
In order to further characterize lipid binding on a residue-by-residue basis, various point mutants of conserved residues of the DSMP binding pocket were generated and evaluated through steady-state kinetics. Apparent Km and kcat values with respect to DSMP are displayed in Table 2. The alanine mutant of Asn43, a residue that precedes the P-loop and appears to help position Arg171 for lipid binding, has little effect on Km for DSMP and decreases kcat a modest 6.7-fold (Table 2; Fig. 5 visually maps the Km data onto the structure). Alanine mutants of Glu100 and His261, conserved residues that do not directly bind the lipid according to the lipid IVA complex, only increase Km for DSMP 2–3-fold. The corresponding kcat decrease (2,000- and 770-fold, respectively) is much more significant, consistent with their primarily catalytic roles, as demonstrated previously (7).
TABLE 2.
Kinetic parameters of LpxK point mutants with respect to DSMP
| LpxK mutant | Apparent Km | Km increase | Apparent kcat | kcat decrease |
|---|---|---|---|---|
| μm | -fold | s−1 | -fold | |
| WT | 7 ± 1 | 1 | 2.4 ± 0.1 | 1 |
| N43A | 9 ± 2 | 1 | (3.6 ± 0.3) ×10−1 | 6.7 |
| R72A | 28 ± 3 | 4 | (3.9 ± 0.1) ×10−1 | 6.2 |
| Y74A | 48 ± 7 | 7 | (3.1 ± 0.2) ×10−2 | 77 |
| E100A | 17.4 ± 0.8 | 3 | (1.23 ± 0.02) ×10−3 | 2,000 |
| R119A | 9.7 ± 0.6 | 1 | (3.60 ± 0.07) ×10−2 | 67 |
| D138A | 47 ± 5 | 7 | (1.06 ± 0.05) ×10−3 | 2,300 |
| D138N | 7 ± 2 | 1 | (2.8 ± 0.2) ×10−2 | 87 |
| D139A | 46 ± 8 | 7 | (5.1 ± 0.4) ×10−4 | 4,700 |
| D139N | 14 ± 2 | 2 | (6.3 ± 0.2) ×10−3 | 380 |
| Q142A | 28 ± 3 | 4 | (3.5 ± 0.1) ×10−1 | 6.8 |
| H143A | 50 ± 10 | 7 | (1.6 ± 0.2) ×10−1 | 15 |
| R171A | 128 ± 9 | 20 | (1.38 ± 0.06) ×10−2 | 170 |
| E172A | 18 ± 3 | 3 | (7.0 ± 0.1) ×10−1 | 3.4 |
| H261A | 15 ± 1 | 2 | (3.1 ± 0.9) ×10−3 | 770 |
FIGURE 5.
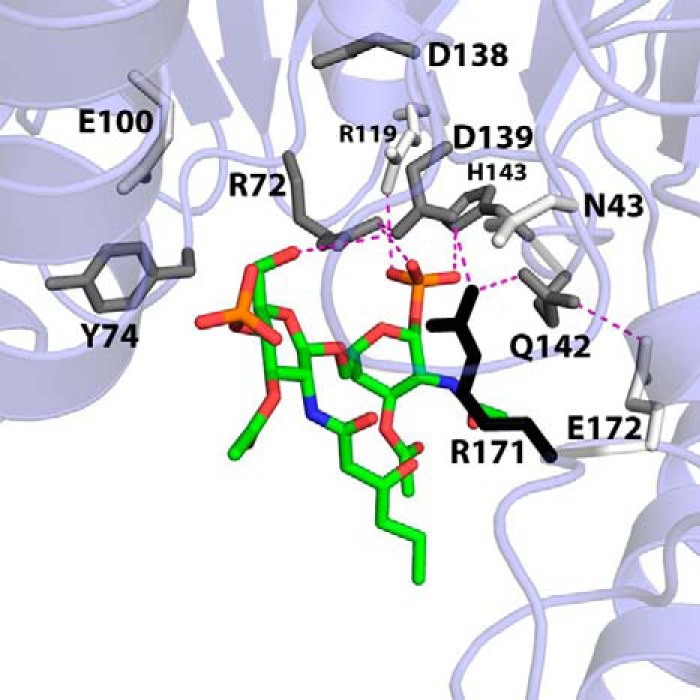
Apparent DSMP Km perturbations of lipid-binding pocket alanine mutants mapped onto the LpxK-lipid IVA coordinates. In order to visualize the effects of alanine mutants of conserved residues within the lipid-binding pocket on the apparent DSMP Km, these residues are shaded in the model based on how much this parameter is increased when mutated to alanine using the following criteria: 0–4-fold Km increase, white; 4–8-fold Km increase, gray; >8-fold Km increase, black. With the exception of Arg119, the alanine mutants of residues that appear to contact the lipid increase the Km at least 4-fold. Arg171 appears to be the most critical residue for lipid substrate binding, with an increased DSMP Km 20-fold higher than wild type. Potential hydrogen bonding interactions are shown as magenta dashes. Font size is indicative of depth.
Side chains of the conserved L1 loop SRGY motif (Arg72 and Tyr74) are positioned to extend into the putative lipid-binding pocket and interact with the lipid substrate. Alanine mutants of Arg72 and Tyr74 have a more significant effect on Km (4- and 7-fold increase, respectively) (Table 2), whereas their kcat values differ by an order of magnitude (6.2- and 77-fold decrease, respectively). Side chains of the L4 loop, which immediately follows the Walker B motif, also show a significant increase in DSMP Km. The Q142A and H143A mutants increase this parameter 4- and 7-fold, respectively (Table 2) while decreasing kcat a modest 6.8- and 15-fold. Another residue in the same locale, Arg119, has little effect on Km and decreases kcat by 67-fold when mutated to alanine, despite its proximity to the 1-phosphate of lipid IVA in the crystal structure (Fig. 4). Such a modest effect may reflect a lack of conservation of Arg119 among LpxK orthologs compared with other lipid-binding residues (6).
Another substructure thought to be involved in DSMP binding, based on residue conservation and the lipid IVA crystal structure, is the L5 loop (Fig. 4). Arg171, held in place through a strong interaction with Asp139, appears to provide extensive van der Waals contacts for the lipid. This residue, when mutated to alanine, results in a 20-fold increase in apparent Km for the DSMP substrate and a 170-fold decrease in apparent kcat (Table 2). Glu172, another residue of this loop, which appears to be indirectly involved in lipid binding through an interaction with Gln142, was also mutated to alanine, resulting in a 3-fold Km increase and a 3.4-fold kcat decrease.
Point mutants of the Walker B motif carboxylates (common to P-loop kinases) were also analyzed for their effect on the kinetic parameters of LpxK with respect to the lipid substrate. The D138A and D139A mutants both increase the apparent DSMP Km 7-fold (Table 2) while also having a significant impact on kcat (2,300- and 4,700-fold decrease, respectively). Asparagine mutants D138N and D139N result in rescued DSMP Km values and partially rescue the kcat value (87- and 380-fold, respectively) (Table 2). Surprisingly, Asp138, a residue that is hypothesized to bind to the catalytic Mg2+ ion, had a non-trivial effect on the DSMP Km. This residue, besides its catalytic role, may be critical for maintaining the correct orientation of the L4 loop. The mutation of Asp138 may directly affect the position of adjacent residue Asp139, which, according to the lipid-bound crystal structure and point mutagenesis, is involved in lipid binding (Fig. 4). The interaction of Asp139 with Arg171 appears to be highly relevant because effects on Km and kcat are much more drastic for the D139A mutant compared with the D139N mutant, which at least preserves some bonding characteristics. This result may indicate that the D139A point mutant alters the conformation of the crucial Arg171 side chain so that this moiety can no longer position the lipid substrate for efficient phosphate transfer.
Characterization of N-terminal Helix Truncation Mutants
According to the crystal structure of lipid IVA-bound LpxK, the only density-supported interaction between the lipid substrate and the N-terminal helix is a potential van der Waals interaction with Tyr13 (Fig. 4). Intrigued by the lack of visible interaction between the hydrophobic acyl chains and hydrophobic residues of the N-terminal helix, we endeavored to more concretely assign a role to this substructure. Based on surface charge and hydrophobicity analysis, we previously predicted that this helix is the primary mediator of membrane binding (6). To test this hypothesis, truncation mutants missing the first half of the N-terminal helix and the full helix were generated (designated Δ12LpxK and Δ29LpxK, respectively) and expressed on pET21b vectors in the C41(DE3) strain (19) (Fig. 6A). The cell-free extracts of Δ12LpxK, Δ29LpxK, and WT A. aeolicus LpxK were assayed for specific activity (7). The Δ12LpxK truncation mutant decreased activity 81-fold, whereas the Δ29LpxK truncation mutant decreased activity 16,000-fold to near background levels.
FIGURE 6.
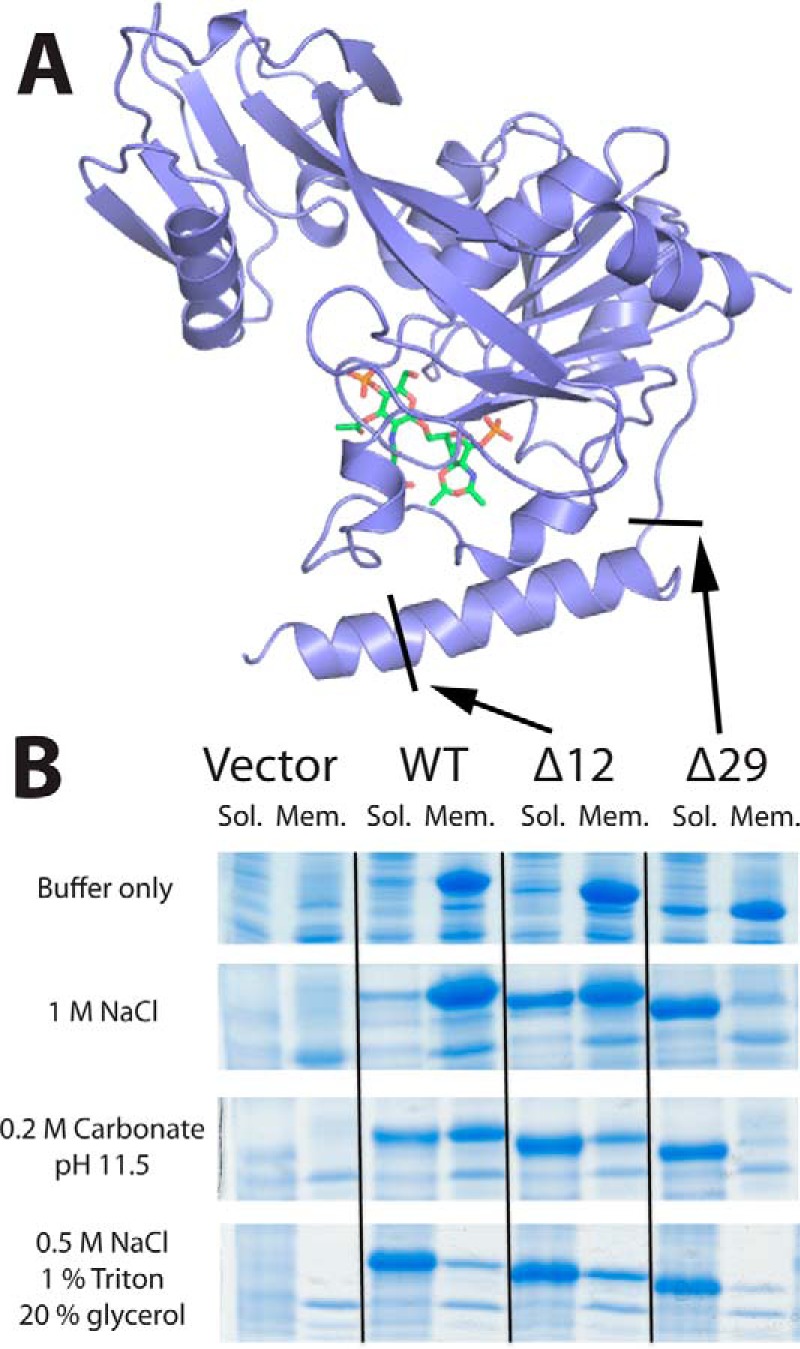
N-terminal truncation mutants reduce hydrophobic contact with the membrane. A, schematic representation of LpxK with arrows indicating truncation points for the Δ12 and Δ29 LpxK mutants. Coloring is conserved from Fig. 3D. B, SDS-polyacrylamide gels revealing the differential partitioning of the LpxK protein between insoluble membrane fractions and soluble fractions upon ultracentrifugation based on different solubilization buffer conditions. Truncation mutants are better solubilized than WT protein in conditions that disrupt the charge-charge interactions with the membrane, such as high salt or high pH. Band migration of the WT, Δ12, and Δ29 peptides was comparatively consistent with their predicted molecular masses of 36.5, 35.2, and 33.1 kDa, respectively.
In order to assess the nature of the interaction of the N-terminal helix with the membrane, membrane fractions of the C41(DE3) strain expressing empty vector, WT LpxK, Δ12LpxK, or Δ29LpxK were isolated from the cell-free extract by ultracentrifugation. Upon SDS-PAGE analysis, it was revealed that the vast majority of the protein for each construct continued to partition with the membrane. These membrane fractions were then treated with various buffer conditions, ultracentrifuged in order to separate solubilized protein from the remaining membrane-bound fraction, and analyzed by SDS-PAGE (Fig. 6B). In a simple HEPES buffer condition, all constructs remained bound to the membrane fraction. In a high salt condition designed to disrupt the charge-charge interactions between LpxK and the phospholipids of the membrane, wild type enzyme remained bound to the membrane. However, the Δ12 mutant displayed significant solubilization of the enzyme in this salt condition, whereas the Δ29 mutant showed nearly full solubilization. In another condition designed to disrupt charge interactions, which involved membrane fraction treatment with 0.2 m carbonate at pH 11.5, a similar trend emerged, in which elimination of half or all of the N-terminal helix resulted in partial or full solubilization, respectively. Carbonate was also able to solubilize a portion of the wild type enzyme. As a positive control, the solubilization condition of 0.5 m NaCl, 1% (w/v) Triton X-100, and 20% (v/v) glycerol successfully solubilized every construct, combining disruption of both charge and hydrophobic contacts with the membrane.
This result indicates that the primary function of the N-terminal helix is hydrophobic interaction with the membrane because when the N-terminal helix was partially or fully removed, the enzyme primarily relied on charge-charge interactions to remain membrane-bound, and conditions that disrupt these charge-charge interactions were sufficient to fully solubilize these constructs. This result also provides a plausible explanation for why the truncation mutants perform so poorly in the standard assay. Because the enzymes are performing their function on Triton X-100 micelles, which have uncharged (tetramethylbutyl)-phenyl headgroups, LpxK in the assay condition relies on the hydrophobicity of the N-terminal helix to remain bound to the micelles at the location of the DSMP substrate. Without this helix, LpxK probably disassociates from the membrane and is unable to access its lipid substrate, thus precluding efficient catalysis.
DISCUSSION
The crystal structure of lipid IVA-bound LpxK reveals almost exclusive interaction with the glucosamine/phosphate headgroups of the lipid, consistent with the notion that LpxK resides at the membrane interface and scans the surface of the inner membrane for its substrate DSMP (6). Such a lipid recognition mode is distinct from structurally characterized lipid A or lipid A precursor interactions with receptors and enzymes. For example, the β-sandwich fold of myeloid differentiation factors MD-1 and MD-2 form a hydrophobic pocket for the acyl chains of their lipid ligands both alone (25, 26) and in complex with Toll-like receptor 4 (TLR4) (27, 28), whereas the glucosamine/phosphate moieties are not extensively coordinated. LpxI of Caulobacter crescentus, the UPD-2,3-diacylglucosamine hydrolase that resides two steps upstream of LpxK in the lipid A biosynthetic pathway of this organism, also makes extensive van der Waals contacts with the acyl chain of its diacylated lipid ligands but comparatively few contacts with the glucosamine headgroup (29). Furthermore, structurally characterized ligand binding modes for lipid kinases, such as sphingosine kinase, display extensive acyl chain burial unlike LpxK (30).
In contrast to the aforementioned lipid-binding proteins, a headgroup-centric recognition mode, as seen in LpxK, has been noted in eukaryotic phosphatidylinositol-binding domains (31–33). Unlike LpxK, however, these effectors are transient peripheral membrane proteins and do not possess enzymatic activity on their own. The binding of soluble phosphoinositide analogs in complex with polyphosphate 5-phosphatase has been described recently (34), but this enzyme is not a bona fide membrane protein like LpxK, which possesses a hydrophobic binding component contributing to membrane affinity.
The crystal structure of LpxK bound to lipid is perhaps most comparable with the Staphylococcus aureus membrane transglycosylase structure with a lipid II analog, where lipid recognition also appears to occur primarily at the headgroup and involves two carbohydrate moieties (35). Although there are multiple arginine residues contributing to lipid binding as with LpxK, they are not nearly as critical for catalytic activity as Arg171 of LpxK. Furthermore, the transglycosylase is anchored in the membrane through a transmembrane helix in contrast to the amphiphilic helix of LpxK, and its lipid substrate contains a single polyprenol chain, unlike the tetraacylated lipid IVA product captured in the LpxK structure.
The complex presented in the work involves the “open” form of LpxK, in which the catalytic dyad of Asp99-His261 is not formed, and this histidine lies ∼12 Å away from the aspartate (6, 7) (Figs. 1B and 7A). Because the bound lipid is the phosphorylated LpxK product, this crystal structure most likely represents a postcatalytic complex in which ADP/Mg2+ has already left its binding site. Although the order of binding of the two substrates is not known, the fact that crystal complexes of LpxK bound to either the nucleotide or lipid alone can be attained may suggest a random order. Alignment of AMP-PCP-bound LpxK (7) and lipid IVA-bound LpxK provides a good model for the arrangement of the substrates in the putative LpxK-ATP/Mg2+-DSMP ternary complex (Fig. 7A). The N-terminal domains of the two structures are nearly superimposable, with the most noticeable structural differences being C-terminal domain “closure” around AMP-PCP, a motion that effectively assembles the catalytic residues in the active site (Fig. 7A). Among the possible catalytic bases outlined in previous studies (7), His261 appears closest to the 4′-hydroxyl of the DSMP substrate (3.3 Å) according to the model (Fig. 7B). Alanine mutants of residues Glu100 and His261, which are of catalytic importance according previous kinetic analysis (7), have a small effect on the apparent Km for the DSMP substrate while decreasing kcat significantly, confirming their primary role in catalysis (Table 2). These residues do not appear to participate in DSMP binding and are brought into play only upon domain closure with nucleotide-bound LpxK.
Several additional key residues were identified both structurally and kinetically as critical for LpxK lipid binding. His143 anchors the 1-phosphate in position for correct binding orientation (Fig. 4) and ensures that molecules like lipid X (2,3-diacylglucosamine-1-phosphate), whose headgroup consists of the 1-phosphate with only a single glucosamine, are not phosphorylated (4). As demonstrated previously, lipids that contain both glucosamine moieties but with altered acyl chain number are still substrates for the E. coli LpxK kinase activity (5), giving further support for the hypothesis that substrate recognition occurs primarily at the DSMP headgroup. Of all of the residues studied through point mutagenesis, Arg171 of the L5 loop reveals itself to be the most crucial to lipid binding, increasing the DSMP Km 20-fold when mutated to alanine (Table 2). Arg171 appears to provide a critical anchor point for residues of the lipid-binding pocket, may maintain van der Waals interactions with lipid IVA along its side chain, and may “lure” the negatively charged phosphates of the lipid substrate into this pocket using its guanidinium functionality.
The extensive characterization of the interaction between LpxK, its lipid ligands, and the cell membrane have provided the underpinnings for further study of membrane-embedded lipid modification enzymes. LpxK has differentiated itself from other P-loop kinases significantly through the development of a hydrophobic N terminus and surrounding basic residues that direct LpxK to the membrane and allow it to recognize the glucosamine/phosphate headgroups of its lipid substrate. Furthermore, this lipid-bound crystal structure makes LpxK a highly attractive target for the development of novel antimicrobials for three reasons. First, the dynamic conformational switch (Fig. 7A) that LpxK undergoes upon substrate binding makes for a specific high-affinity binding site which, if probed with an inhibitor, could trap LpxK in the closed state and prevent catalysis. Second, the large surface area of the lipid-binding site is naturally primed for a conjugated glucosamine sugar scaffold, providing an additional pathway toward the development of potent inhibitors. Finally, the existence of an LpxK crystal structure in complex with either nucleotide or lipid ligand of this bisubstrate enzyme presents the possibility of targeting both binding sites with a single drug. Such a strategy has been employed to engineer highly specific and highly potent inhibitory compounds for both protein kinases and other P-loop kinases, such as adenylate kinase (36, 37), providing a plausible template with which to combat the spread of Gram-negative bacteria with resistance to commercial antimicrobials.
Acknowledgments
We thank the late Dr. Christian R. H. Raetz for contributions to the conception of this work as well as supportive guidance. We thank Dr. Teresa A. Garrett for providing the TG1/pTAG1, Dr. Hak Suk Chung for providing the purified lipid IVA used in crystallization, and Dr. Charles Pemble for assistance with data collection. We also thank Dr. Seok-Yong Lee, Dr. K. V. Rajagopalan, Dr. Ken Kruezer, and members of the Raetz and Zhou laboratories for helpful discussion. Diffraction data were collected at the Southeast Regional Collaborative Access Team 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory, through the Duke University X-ray Crystallography Shared Resource.
This work was supported, in whole or in part, by National Institutes of Health Grant GM-51310 (to C. R. H. R. and P. Z.) and AI-055588 (to P. Z.). Use of the Advanced Photon Source was supported by the United States Department of Energy, Office of Science, and the Office of Basic Energy Sciences under Contract W-31-109-Eng-38.
The atomic coordinates and structure factors (code 4LKV) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- DSMP
- tetraacyldisaccharide-1-phosphate
- AMP-PCP
- 5′-adenylyl (β,γ-methylene)diphosphonate
- Walker B motif
- carboxylate-containing metal-binding motif of P-loop kinases.
REFERENCES
- 1. Raetz C. R., Guan Z., Ingram B. O., Six D. A., Song F., Wang X., Zhao J. (2009) Discovery of new biosynthetic pathways: the lipid A story. J. Lipid Res. 50, S103–S108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Raetz C. R., Whitfield C. (2002) Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Raetz C. R., Reynolds C. M., Trent M. S., Bishop R. E. (2007) Lipid A modification systems in gram-negative bacteria. Annu. Rev. Biochem. 76, 295–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ray B. L., Raetz C. R. (1987) The biosynthesis of gram-negative endotoxin. A novel kinase in Escherichia coli membranes that incorporates the 4′-phosphate of lipid A. J. Biol. Chem. 262, 1122–1128 [PubMed] [Google Scholar]
- 5. Garrett T. A., Kadrmas J. L., Raetz C. R. (1997) Identification of the gene encoding the Escherichia coli lipid A 4′-kinase: facile phosphorylation of endotoxin analogs with recombinant LpxK. J. Biol. Chem. 272, 21855–21864 [DOI] [PubMed] [Google Scholar]
- 6. Emptage R. P., Daughtry K. D., Pemble C. W., 4th, Raetz C. R. (2012) Crystal structure of LpxK, the 4′-kinase of lipid A biosynthesis and atypical P-loop kinase functioning at the membrane interface. Proc. Natl. Acad. Sci. U.S.A. 109, 12956–12961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Emptage R. P., Pemble C. W., 4th, York J. D., Raetz C. R., Zhou P. (2013) Mechanistic characterization of the tetraacyldisaccharide-1-phosphate 4′-kinase LpxK involved in lipid A biosynthesis. Biochemistry 52, 2280–2290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Punta M., Coggill P. C., Eberhardt R. Y., Mistry J., Tate J., Boursnell C., Pang N., Forslund K., Ceric G., Clements J., Heger A., Holm L., Sonnhammer E. L., Eddy S. R., Bateman A., Finn R. D. (2012) The Pfam protein families database. Nucleic Acids Res. 40, D290–D301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cheek S., Zhang H., Grishin N. V. (2002) Sequence and structure classification of kinases. J. Mol. Biol. 320, 855–881 [DOI] [PubMed] [Google Scholar]
- 10. Leipe D. D., Koonin E. V., Aravind L. (2003) Evolution and classification of P-loop kinases and related proteins. J. Mol. Biol. 333, 781–815 [DOI] [PubMed] [Google Scholar]
- 11. Plötz B. M., Lindner B., Stetter K. O., Holst O. (2000) Characterization of a novel lipid A containing d-galacturonic acid that replaces phosphate residues. The structure of the lipid a of the lipopolysaccharide from the hyperthermophilic bacterium Aquifex pyrophilus. J. Biol. Chem. 275, 11222–11228 [DOI] [PubMed] [Google Scholar]
- 12. Mamat U., Schmidt H., Munoz E., Lindner B., Fukase K., Hanuszkiewicz A., Wu J., Meredith T. C., Woodard R. W., Hilgenfeld R., Mesters J. R., Holst O. (2009) WaaA of the hyperthermophilic bacterium Aquifex aeolicus is a monofunctional 3-deoxy-d-manno-oct-2-ulosonic acid transferase involved in lipopolysaccharide biosynthesis. J. Biol. Chem. 284, 22248–22262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sambrook J. G., Russell D. W. (2001). Molecular Cloning: A Laboratory Manual, 3rd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 14. Inoue H., Nojima H., Okayama H. (1990) High efficiency transformation of Escherichia coli with plasmids. Gene 96, 23–28 [DOI] [PubMed] [Google Scholar]
- 15. Guzman L. M., Belin D., Carson M. J., Beckwith J. (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177, 4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garrett T. A., Que N. L., Raetz C. R. (1998) Accumulation of a lipid A precursor lacking the 4′-phosphate following inactivation of the Escherichia coli lpxK gene. J. Biol. Chem. 273, 12457–12465 [DOI] [PubMed] [Google Scholar]
- 17. Hamilton C. M., Aldea M., Washburn B. K., Babitzke P., Kushner S. R. (1989) New method for generating deletions and gene replacements in Escherichia coli. J. Bacteriol. 171, 4617–4622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bachmann B. J. (1972) Pedigrees of some mutant strains of Escherichia coli K-12. Bacteriol. Rev. 36, 525–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miroux B., Walker J. E. (1996) Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260, 289–298 [DOI] [PubMed] [Google Scholar]
- 20. Chung H. S., Raetz C. R. (2010) Interchangeable domains in the Kdo transferases of Escherichia coli and Haemophilus influenzae. Biochemistry 49, 4126–4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 22. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 25. Ohto U., Fukase K., Miyake K., Satow Y. (2007) Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa. Science 316, 1632–1634 [DOI] [PubMed] [Google Scholar]
- 26. Yoon S. I., Hong M., Han G. W., Wilson I. A. (2010) Crystal structure of soluble MD-1 and its interaction with lipid IVa. Proc. Natl. Acad. Sci. U.S.A. 107, 10990–10995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Park B. S., Song D. H., Kim H. M., Choi B. S., Lee H., Lee J. O. (2009) The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195 [DOI] [PubMed] [Google Scholar]
- 28. Ohto U., Fukase K., Miyake K., Shimizu T. (2012) Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2. Proc. Natl. Acad. Sci. U.S.A. 109, 7421–7426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Metzger L. E., 4th, Lee J. K., Finer-Moore J. S., Raetz C. R., Stroud R. M. (2012) LpxI structures reveal how a lipid A precursor is synthesized. Nat. Struct. Mol. Biol. 19, 1132–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang Z., Min X., Xiao S. H., Johnstone S., Romanow W., Meininger D., Xu H., Liu J., Dai J., An S., Thibault S., Walker N. (2013) Molecular basis of sphingosine kinase 1 substrate recognition and catalysis. Structure 21, 798–809 [DOI] [PubMed] [Google Scholar]
- 31. Lemmon M. A. (2008) Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9, 99–111 [DOI] [PubMed] [Google Scholar]
- 32. Stahelin R. V. (2009) Lipid binding domains: more than simple lipid effectors. J. Lipid Res. 50, S299–S304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moravcevic K., Oxley C. L., Lemmon M. A. (2012) Conditional peripheral membrane proteins: facing up to limited specificity. Structure 20, 15–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Trésaugues L., Silvander C., Flodin S., Welin M., Nyman T., Gräslund S., Hammarström M., Berglund H., Nordlund P. (2014) Structural basis for phosphoinositide substrate recognition, catalysis, and membrane interactions in human inositol polyphosphate 5-phosphatases. Structure 22, 744–755 [DOI] [PubMed] [Google Scholar]
- 35. Huang C. Y., Shih H. W., Lin L. Y., Tien Y. W., Cheng T. J., Cheng W. C., Wong C. H., Ma C. (2012) Crystal structure of Staphylococcus aureus transglycosylase in complex with a lipid II analog and elucidation of peptidoglycan synthesis mechanism. Proc. Natl. Acad. Sci. U.S.A. 109, 6496–6501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lavogina D., Enkvist E., Uri A. (2010) Bisubstrate inhibitors of protein kinases: from principle to practical applications. ChemMedChem 5, 23–34 [DOI] [PubMed] [Google Scholar]
- 37. Lienhard G. E., Secemski I. I. (1973) P1, P5-Di(adenosine-5′)pentaphosphate, a potent multisubstrate inhibitor of adenylate kinase. J. Biol. Chem. 248, 1121–1123 [PubMed] [Google Scholar]