

Background: STIM1 activates store-operated Ca2+ entry (SOCE). The transcriptional regulation of STIM1 during sepsis is not known.
Results: STIM1 expression occurs downstream of TLR4 via activation of NF-κB and p38α-mediated c-Fos expression in endothelial cells.
Conclusion: Cooperative signaling of NF-κB and p38 MAPK downstream of TLR4 is essential for STIM1 expression during sepsis.
Significance: Selective p38α inhibitors may represent a potential therapeutic strategy to prevent sepsis-mediated lung vascular leaks.
Keywords: AP1 Transcription Factor (AP-1), Calcium, Endothelium, Gene Regulation, NF-κB Transcription Factor, p38 MAPK, Permeability, Sepsis, Thrombin
Abstract
Stromal interacting molecule 1 (STIM1) regulates store-operated Ca2+ entry (SOCE). Here we show that STIM1 expression in endothelial cells (ECs) is increased during sepsis and, therefore, contributes to hyperpermeability. LPS induced STIM1 mRNA and protein expression in human and mouse lung ECs. The induced STIM1 expression was associated with augmented SOCE as well as a permeability increase in both in vitro and in vivo models. Because activation of both the NF-κB and p38 MAPK signaling pathways downstream of TLR4 amplifies vascular inflammation, we studied the influence of these two pathways on LPS-induced STIM1 expression. Inhibition of either NF-κB or p38 MAPK activation by pharmacological agents prevented LPS-induced STIM1 expression. Silencing of the NF-κB proteins (p65/RelA or p50/NF-κB1) or the p38 MAPK isoform p38α prevented LPS-induced STIM1 expression and increased SOCE in ECs. In support of these findings, we found NF-κB and AP1 binding sites in the 5′-regulatory region of human and mouse STIM1 genes. Further, we demonstrated that LPS induced time-dependent binding of the transcription factors NF-κB (p65/RelA) and AP1 (c-Fos/c-Jun) to the STIM1 promoter. Interestingly, silencing of c-Fos, but not c-Jun, markedly reduced LPS-induced STIM1 expression in ECs. We also observed that silencing of p38α prevented c-Fos expression in response to LPS in ECs, suggesting that p38α signaling mediates the expression of c-Fos. These results support the proposal that cooperative signaling of both NF-κB and AP1 (via p38α) amplifies STIM1 expression in ECs and, thereby, contributes to the lung vascular hyperpermeability response during sepsis.
Introduction
The lung endothelium is interfaced strategically between the blood and airspace to tightly regulate the exchange of fluid from the circulation to the underlying tissue. Endothelial barrier dysfunction results in uncontrolled fluid extravasation and lung edema (1). In septic patients and in rodent models of sepsis, the lungs are generally the first organ to fail (2) because of rapid loss of endothelial barrier function (1). Sepsis, a systemic inflammatory response to bacterial infection, results in high mortality in hospitalized patients (1–5). The basis of the severe lung injury that often occurs in the setting of sepsis remains a mystery. In this study, we considered the possibility that excess intracellular Ca2+ in endothelial cells is involved in severe septic lung injury and, therefore, set out to investigate whether key components of the calcium entry system in endothelial cells become up-regulated during sepsis, resulting in calcium overload.
We have shown previously that Ca2+ overload in endothelial cells (ECs)2 results in markedly increased endothelial permeability (6–10). Bacterial endotoxin (LPS) can directly induce Ca2+ overload in ECs to cause endothelial barrier dysfunction and vascular injury (11, 12). Bacterial cell wall component LPS ligates TLR4 to induce the expression of cytokines, chemokines, adhesion molecules, apoptotic factors, and several other mediators through NF-κB and p38 MAPK activation (13–15). Experimental animal model studies have demonstrated that inhibition of NF-κB activation suppresses inflammatory responses (16, 17). This transcription factor may also be involved in signaling increased Ca2+ entry in ECs (9, 18). The role of TLR4 signaling in the endothelium during sepsis has been investigated using mice that express TLR4 exclusively in ECs (19). The results of this study showed that endothelial TLR4 signaling is sufficient to detect and clear intraperitoneal Escherichia coli infection. Further, studies using a mouse model in which a degradation-resistant form of IκBα, the inhibitor of NF-κB, is selectively expressed in ECs showed protection against LPS- or E. coli-induced lung injury (20). These findings suggest that TLR4 signaling in ECs is critical to the inflammatory responses.
Inflammatory mediators generated and released during sepsis, such as thrombin and oxidants, are also known to activate Ca2+ entry through store-operated Ca2+ channels (SOCs) in ECs (6). We have reported previously that transient receptor potential canonical (TRPC) 1 and 4 channels function as SOCs in ECs (6–10, 21). Other studies have shown that current (ICRAC) through the Ca2+ selective channel Orai1 also contributes to SOCE in ECs (22, 23). Importantly, studies have elucidated the mechanism of the endoplasmic reticulum (ER)-localized Ca2+ sensor protein STIM1 in activating SOCE through TRPC and Orai1 channels (24, 25). Agonist-induced ER store Ca2+ depletion causes clustering of STIM1 at the ER/plasma membrane interface, which, in turn, binds to and activates SOCs in the plasma membrane (24, 25). Furthermore, studies have shown that reactive oxygen species can induce STIM1-mediated Ca2+ entry through Orai1 channels in ECs by activating STIM1 proteins via S-glutathionylation of the N-terminal cysteine residue in the STIM1 protein (26). We have shown that thrombin-induced endothelial barrier disruption in lung ECs is mediated by STIM1 activation of SOCE (10, 21). In the in vivo setting, an LPS-induced lung vascular permeability increase was abrogated in EC-restricted STIM1 knockout (Stim1EC−/−) mice (27). Therefore, in this study, we hypothesized that increased STIM1 expression during sepsis signals abnormally high intracellular Ca2+ in ECs, resulting in loss of endothelial barrier function and a rise in vascular permeability. This hypothesis predicts that TLR4 signaling in ECs activates STIM1 transcription and, thereby, contributes to lung vascular hyperpermeability.
A large body of evidence suggests that the blood coagulation system, which is known to modulate the inflammatory response of the endothelium (5, 28–31), is dysregulated during sepsis. Thrombin, the agonist for the protease-activated receptor 1 (PAR-1), is generated in excessive quantities at the site of infection during sepsis (28, 30, 32). Activation of PAR-1 expressed on the endothelial cell surface promotes Ca2+ entry, vascular inflammation, and injury (7, 9, 33, 34). Recent studies using mouse models have shown that blocking PAR-1 activation in the early stages of sepsis improves vascular barrier function (35). We showed that thrombin activation of PAR-1 on ECs increases vascular permeability through SOCE (6–10). Therefore, in this study, we utilized both in vitro and in vivo approaches to test whether LPS-induced STIM1 expression in ECs is indeed responsible for the hyperpermeability response observed in sepsis. We observed that LPS induced STIM1 transcription in ECs via the transcription factors NF-κB and AP1. LPS also increased the expression of the SOC components TRPC1, TRPC4, and Orai1 in ECs. The increased expression of STIM1 and SOC components was associated with augmented PAR-1-mediated SOCE and elevated vascular permeability.
EXPERIMENTAL PROCEDURES
Materials
Human lung microvessel endothelial cells (HLMVECs) and endothelial growth medium 2 were from Lonza Walkersville, Inc. (Walkersville, MD). FBS was from Hyclone (Logan, UT). Hanks' balanced salt solution, l-glutamine, trypsin, TRIzol reagent, TaqDNA polymerase, and Fura-2/AM were from Invitrogen. Human α-thrombin was obtained from Enzyme Research Laboratories (South Bend, IN). LPS (ultrapure E. coli 0111:B4) was obtained from InvivoGen (San Diego, CA). Actinomycin D, thapsigargin, SB203580, PD98059, SP600125, and 6-amino-4-(4-phenoxyphenylamino)quinzoline (an NF-κB inhibitor) were from Calbiochem (La Jolla, CA). Quantitative PCR primers were custom-synthesized by IDT (Coralville, IA). Human (h)-specific siRNA to target STIM1, p65/RelA, p38α, p38β, c-Fos, and scrambled siRNA (Sc-siRNA) were obtained from Dharmacon (Lafayette, CO). Anti-STIM1 mAb was purchased from BD Transduction Laboratories. Anti-STIM1 pAb was from Proteintech Group (Chicago, IL). Antibodies specifically reacting with TRPC1, Orai1, and siRNAs specific to p38γ, p50/NF-κB1, c-Jun, and siRNA transfection reagent were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-p65/RelA pAb was purchased from Millipore Corp. (Billerica, MA). Anti-p38α pAb, anti-p38β mAb, anti-p38γ pAb, c-Fos pAb, anti-c-Jun pAb, anti-phospho-ERK1/2 pAb, anti-ERK1/2 pAb, anti-phospho-JNK mAb, and anti-JNK mAb were from Cell Signaling Technology (Danvers, MA). Anti-β-actin mAb was from Sigma. Phospho (Thr-325)-c-Fos pAb was from Abcam (Cambridge, MA). Anti-TRPC4 pAb was purchased from Everest Biotech Ltd. (Ramona, CA). PAR-1-activating peptide (TFLLRNPNDK-NH2) was custom-synthesized by GenScript (Piscataway, NJ). Fast SYBR Green master mix was purchased from Applied Biosystems (Grand Island, NY).
Cell Culture
HLMVECs grown in endothelial growth medium 2 microvascular supplemented with 10% FBS were used between passages 2 and 4. Lung endothelial cells from C57BL6J mice were isolated and cultured as we described previously (7). Cells between passages 3 and 4 were used for experiments. Mouse lung endothelial cells (MLECs) were characterized by their cobblestone morphology, PECAM-1 (platelet/endothelial cell adhesion molecule 1 or CD31) expression, and Dil-Ac-LDL uptake (7). To study the LPS effect, ECs were incubated with 1% FBS-containing medium overnight at 37 °C and then exposed to LPS in 1% FBS-containing medium.
Quantitative Real-time-PCR
HLMVECs were incubated with either vehicle (dimethyl sulfoxide) or actinomycin D (1 μm) for 1 h, SB203580 (10 μm) for 30 min, or NF-κB inhibitor (5 μm) for 12 h and then exposed to LPS (1 μg/ml) for the indicated times. Total RNA, extracted according to the recommendations of the manufacturer with an RNeasy kit (Qiagen, CA) was used to generate first-strand complementary DNA by reverse transcriptase (Invitrogen). cDNA (10 ng) mixed with SYBR Green PCR master mix was used for real-time quantitative PCR. STIM1 mRNA was normalized to β-actin mRNA. Human STIM1 and β-actin were amplified using the following primer sets: STIM1, 5′-ACCAGCATGAAGTCCTTGAG-3′ (sense) and 5′-TGAAGATGACAGACCGGAGT-3′ (antisense); β-actin, 5′ TTGCTGACAGGATGCAGAAGGAGA-3′ (sense) and 5′ACTCCTGCTTGCTGATCCACATCT-3′ (antisense).
Immunoblotting
Endothelial cell lysates or total lung tissue homogenates from C57BL6J mice were resolved by SDS-polyacrylamide gel electrophoresis on a 10% separating gel under reducing conditions and transferred to a Duralose membrane. Membranes were blocked with 5% dry milk in TBST (10 mm Tris-HCl (pH 7.5), 150 mm NaCl, and 0.05% Tween 20) for 1 h at 22 °C. Membranes were then incubated with the indicated primary antibody (diluted in blocking buffer) overnight. After three washes, membranes were incubated with the appropriate horseradish peroxidase-conjugated secondary antibody. Protein bands were detected by enhanced chemiluminescence.
Cytosolic Ca2+ Measurement
The cytoplasmic Ca2+ concentration ([Ca2+]i) in ECs was measured using the Ca2+-sensitive fluorescent dye Fura-2/AM (10, 21). Cells were grown to confluence on gelatin-coated glass coverslips and then washed two times and incubated for 12 h at 37 °C in medium containing 1% FBS. Cells exposed to LPS for the indicated time intervals were washed and loaded with 3 μm Fura-2/AM for 30 min. After loading, cells were washed with Hanks' balanced salt solution, and the coverslips were transferred to a perfusion chamber at 37 °C and imaged using a semimotorized microscope (Axio Observer D1, Carl Zeiss GmbH, Jena, Germany) equipped with an AxioCam HSm camera (Carl Zeiss) and a Fluar ×40 oil immersion objective. Light was provided by a DG-4 wavelength switcher (Princeton Scientific Instruments, Monmouth Junction, NJ). Dual excitation at 340 and 380 nm was used, and emission was collected at 520 nm. The AxioVision physiology software module was used to acquire the images at 1-s intervals, and the data were analyzed offline. In each experiment, 20–30 cells were selected to measure the change in ([Ca2+]i).
siRNA Transfection
ECs grown to ∼70% confluence on gelatin-coated culture dishes were transfected with target siRNA or sc-siRNA as described previously (10). 72 h after transfection, cells were challenged with LPS (1 μg/ml) and then used for Ca2+ measurements or harvested for Western blot analysis.
Transendothelial Electrical Resistance Measurement
The real-time change in transendothelial monolayer electrical resistance (TER) was measured as described by us (36). The confluent endothelial monolayer was incubated with 1% FBS-containing medium overnight and challenged with LPS. Four hours after LPS exposure, the thrombin-induced real-time change in TER was measured. Data are presented as resistance normalized to its value at time 0 (36).
Mouse Lung Capillary Filtration Coefficient Measurement
C57BL6J mice obtained from Charles River Laboratories (Wilmington, MA) were housed in the University of Illinois Animal Care Facility and used according to approved animal protocols. Pulmonary capillary filtration coefficient (Kf,c) was measured to determine the microvascular permeability to liquid, as described previously (7, 34). To test the effects of LPS, the mice were given a dose of LPS (5 mg/kg intraperitoneally), and, at 4 h after the LPS challenge, the isolated lungs were used for Kf,c measurements (7, 34). In experiments testing the effects of PAR-1 agonist peptide alone on Kf,c, PAR-1 agonist peptide (30 μm) containing perfusion buffer was infused via a side port at a rate of 0.2 ml/min. Kf,c measurements were made at baseline and after a 20-min exposure to PAR-1 agonist peptide. The values are expressed as the ratio of experimental to basal Kf,c values in the same lung preparation. To study the in vivo relevance of p38 MAPK inhibition, mice were anesthetized with ketamine/xylazine (100/5 mg kg intraperitoneally), and then SB203580 (1.0 mg/kg) or vehicle (dimethyl sulfoxide) was injected through the retro-orbital vein 60 min prior to LPS (5 mg/kg intraperitoneally) injection. Kf,c was then evaluated in isolated lung preparations as described.
Assessment of Mouse Lung Microvessel Permeability in Vivo
C57BL6J mice were anesthetized (2.5% sevoflurane in room air) for insertion of an indwelling jugular catheter and were then allowed to recover for 30 min. Next, mice were injected with LPS (5 mg/kg intraperitoneally) or saline. 195 min after LPS or saline administration, mice received either saline (100 μl) or a 100-μl bolus of PAR-1-activating peptide (1 mg/kg) through the jugular vein. Four hours later, the mice were sacrificed, and the lungs were harvested. Thirty minutes before sacrifice, mice received a 100-μl bolus of Evans blue dye conjugated with albumin (EBA, 20 mg/kg) through the jugular vein. The EBA present in lung tissue was measured as described previously (10, 20).
ChIP Assays
ChIP assays were performed as described previously (16) using a ChIP assay kit from Upstate Biotechnology. HLMVECs exposed to LPS (1 μg/ml) for the indicated time periods were used for ChIP. The primers used for quantitative PCR after Chip were as follows: NF-κB sites 1–3, 5′-GAG GCT AAC GTC GTG TCC TG-3′ (forward) and 5′-AGC TGG ATC CCG GAA TAA CC-3′ (reverse); AP1 site, 5′-GTG TCC TGG GCC TCT GTT TA-3′ (forward) and 5′-GTG AAG ACC TCC CCG GAA TC-3′ (reverse). DNA-protein interaction was calculated with the following formula: 2ΔCtx − 2ΔCtb, where ΔCtx is the cycling threshold of input DNA minus the cycling threshold of sample DNA, and ΔCtb is the cycling threshold of input DNA minus the cycling threshold of control antibodies.
Statistical Analysis
Data were analyzed by unpaired two-tailed Student's t test. Difference in mean values were considered significant at p ≥ 0.05.
RESULTS
LPS Induces STIM1 Expression and Augments PAR-1-induced SOCE in HLMVECs
To determine whether LPS activation of TLR4 increases STIM1 expression, we first measured STIM1 mRNA expression in response to LPS in HLMVECs. LPS induced STIM1 transcript expression in HLMVECs, and the expression level was maximal at 4 h (Fig. 1A). Pretreatment with the transcriptional inhibitor actinomycin D blocked LPS-induced STIM1 transcript expression (Fig. 1A). We also observed increased STIM1 protein expression in response to LPS challenge (Fig. 1B). STIM1 protein expression was increased more than 6-fold within 6 h after LPS exposure (Fig. 1B). However, another STIM1 homolog, STIM2, localized in the ER, was not increased in response to LPS in HLMVECs (Fig. 1B). We and others have shown that STIM1 activates TRPC1 and TRPC4 (SOC channels) and Orai1 (Ca2+ release-activated Ca2+ channel (CRAC)) (10, 21–23) to induce Ca2+ entry in ECs. Increased STIM1 expression alone is not sufficient to activate SOCE, but STIM1 interaction with plasma membrane-localized TRPC or Orai channels is essential to activate SOCE (24, 25). Therefore, we determined the expression of TRPC1, TRPC4 and Orai1 proteins in HLMVECs in response to LPS challenge. LPS challenge substantially increased the expression of TRPC1, TRPC4, and Orai1 in HLMVECs (Fig. 1B). These results suggest that TLR4 signaling not only induces STIM1 expression but also induces the expression of SOC channel components in HLMVECs.
FIGURE 1.
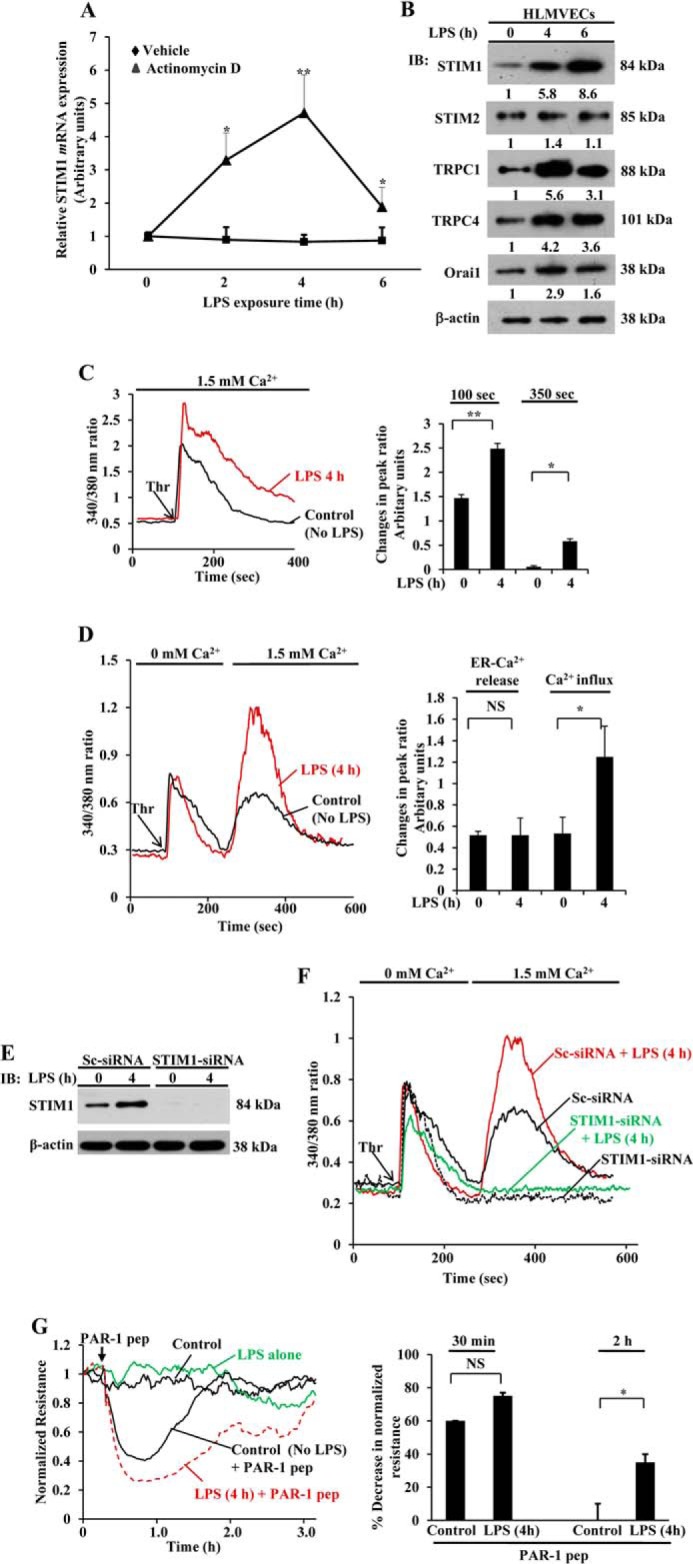
LPS induces the expression of STIM1 and SOCE channel components in HLMVECs. A, HLMVECs were treated with LPS (1 μg/ml) in the presence and absence of actinomycin D (0.5 μm) for 0, 2, 4, and 6 h. After this treatment, total RNA was isolated, and quantitative real-time PCR was performed to determine STIM1 mRNA expression. STIM1 mRNA induction fold was calculated by measuring the ratio of STIM1 to β-actin. Results are mean ± S.E. of four experiments. *, p < 0.01; **, p < 0.001 compared with cells treated with actinomycin D. B, HLMVECs grown to confluence were exposed to LPS (1 μg/ml) for 0, 4, and 6 h. After this treatment, total cell lysates were used for immunoblot (IB) analysis to determine STIM1, STIM2, TRPC1, TRPC4, Orai1, and β-actin. Blots were quantified by densitometry, and fold increase over basal relative to β-actin is shown. Results are mean of two experiments. C, HLMVECs were exposed to LPS (1 μg/ml) for 0 and 4 h, loaded with 3 μm FURA-2/AM, and used to measure the thrombin-induced increase in [Ca2+]i in the presence of extracellular Ca2+ (left panel). The arrow indicates the time point at which thrombin (Thr) (50 nm) was added. Results are representative of four experiments. The bar graph (right panel) shows the quantification of change in fluorescence ratio at peak (100 s) and at 350 s. *, p < 0.01; **, p < 0.001 compared with control cells. D, HLMVECs pretreated with LPS as above were used to measure thrombin-induced (50 nm) ER-stored Ca2+ release and Ca2+ release-activated Ca2+ entry (left panel). Results are representative of three experiments. The bar graph (right panel) shows the quantification of three experiments. NS, not significant; *, p < 0.01 compared with control cells. E, HLMVECs were transfected with control siRNA (Sc-siRNA) or STIM1-siRNA (100 nm), and, 48 h after transfection, cells were treated with LPS (1 μg/ml) for 0 and 4 h. After LPS treatment, cells were used to determine STIM1 protein expression by immunoblot analysis. Data are representative of two experiments. F, HLMVECs transfected with Sc-siRNA or STIM1-siRNA were exposed to LPS as above and used to measure thrombin-induced ER-stored Ca2+ release and Ca2+ release-activated Ca2+ entry. Data are representative of two experiments. G, HLMVECs were grown to confluence on gold electrodes (see details under “Experimental Procedures”). Cells were washed and incubated in 1% FBS medium for 12 h, and then the cells were treated with LPS (1 μg/ml) for 4 h or left untreated. After this treatment, the cells were challenged with PAR-1 agonist peptide (pep, TFLLRNPNDK, 25 μm). Decreases in resistance quantified from four independent experiments are mean ± S.E. (bottom panel). NS = not significant; *, p < 0.001 compared with control cells.
To address the functional relevance of increased expression of STIM1, TRPC1, TRPC4, and Orai1 proteins, we measured the thrombin-induced increase in [Ca2+]i. LPS pretreatment significantly increased the thrombin-induced increase in [Ca2+]i in HLMVECs (Fig. 1C). Next we measured thrombin-induced, ER-stored Ca2+ release and Ca2+ release-activated Ca2+ entry in control and LPS-pretreated HLMVECs. In both control and LPS-primed cells, thrombin-induced ER store Ca2+ release (initial peak) was similar. However, when adding back Ca2+, Ca2+ entry was more than 2-fold higher in the LPS-primed cells compared with unprimed cells (Fig. 1D).
To study the basis of augmented SOCE in LPS primed cells, we silenced STIM1 expression by transfecting HLMVECs with siRNA specific to STIM1 and then studied the LPS effect. In STIM1 siRNA-transfected cells, STIM1 protein expression was blocked compared with control siRNA (Sc-siRNA) (Fig. 1E). Also, thrombin-induced Ca2+ entry was blocked in STIM1 siRNA-transfected cells with or without LPS pretreatment (Fig. 1F), therefore indicating the crucial role of STIM1 in activating SOCE in ECs.
We have shown previously that Ca2+ overload in ECs signals an increase in vascular permeability (6–10). To address the functional relevance of LPS priming-mediated SOCE augmentation in response to PAR-1 agonists, we studied the effect of LPS priming on TER, a measure of endothelial monolayer permeability (36). After 4 h of LPS priming, cells were challenged with PAR-1-activating peptide to assess real-time changes in TER. In control cells (not pretreated with LPS), challenge with PAR-1-activating peptide produced an ∼55% decrease in TER (Fig. 1G), and TER returned to baseline within ∼2 h (Fig. 1G). In the LPS-primed cells, the PAR-1 agonist produced an ∼75% decrease in TER, and there was a prolonged recovery time to the baseline lasting ∼3 h (Fig. 1G), indicating that increased SOCE contributes to the potentiated EC monolayer permeability following LPS priming.
LPS Induces STIM1 Expression in Mouse Lung Endothelial Cells
To highlight the relevance of mouse studies to human disease, we studied the effects of LPS on STIM1 protein expression in MLECs. MLECs exposed to LPS for various time periods were used for Western blot analysis to determine STIM1 protein expression. LPS induced STIM1 protein expression in a time-dependent manner (Fig. 2A). STIM1 expression was increased ∼10-fold over basal levels after 6 h of LPS challenge (Fig. 2A). To address the functional relevance of LPS-induced STIM1 expression in MLECs, we measured thrombin-induced as well as pharmacologically induced SOCE using thapsigargin (TG) in control and LPS-primed MLECs. Similar to HLMVECs, LPS priming enhanced SOCE in response to thrombin or TG challenge without altering ER store Ca2+ release in MLECs (Fig. 2, B and C).
FIGURE 2.
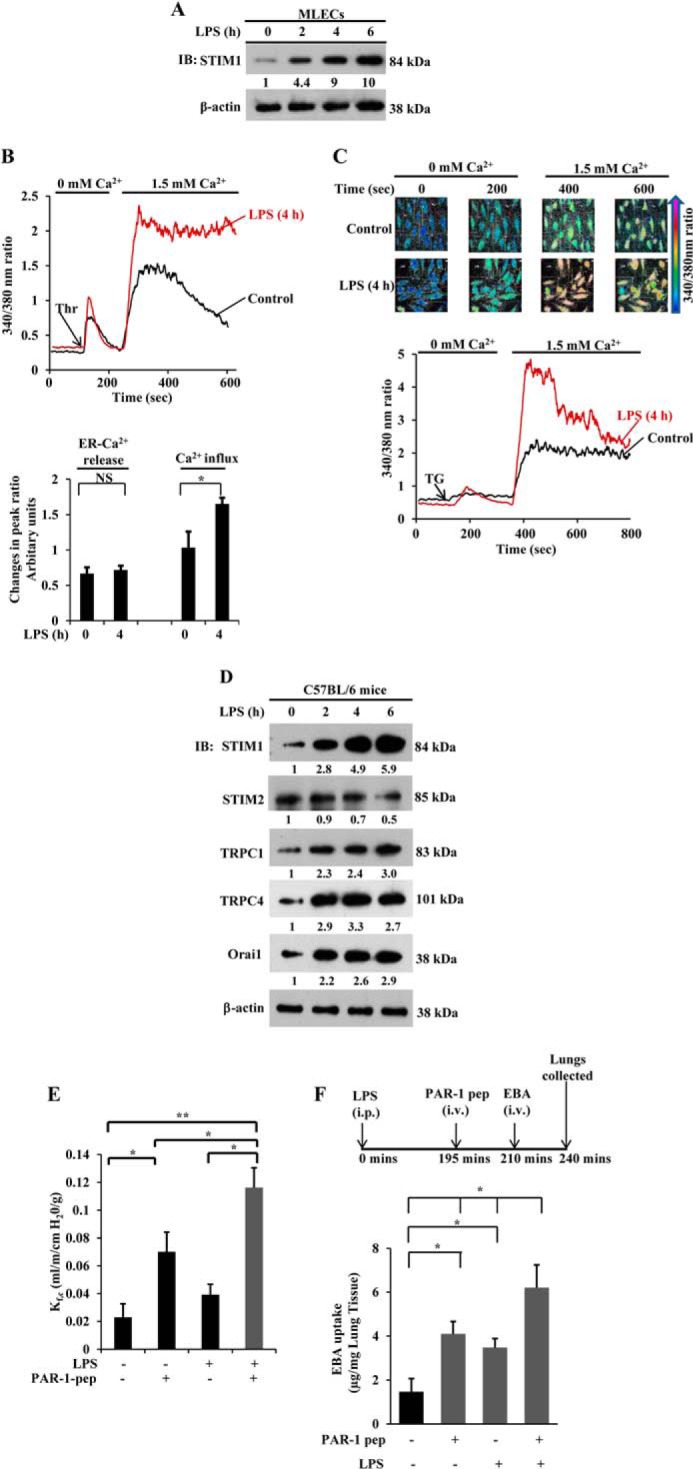
LPS induces STIM1 expression and augments lung vascular permeability. A, MLECs grown to confluence were exposed to LPS (1 μg/ml) for the indicated time points. Total cell lysates were used for immunoblotting (IB) as described in Fig. 1B. Results are representative of two experiments. Blots were quantified, and the fold increase over basal levels relative to β-actin is shown. B, MLECs were pretreated with or without LPS (1 μg/ml) for 4 h. Cells loaded with Fura-2/AM were used to measure thrombin-induced (50 nm) ER-stored Ca2+ release and Ca2+ release-activated Ca2+ entry (top panel). The change in peak fluorescence ratio (340/380) for ER-stored Ca2+ release and Ca2+ entry over basal levels was calculated. The bar graph (bottom panel) shows the quantification of three experiments. NS, not significant; *, p < 0.001 compared with control cells. C, MLECs pretreated treated with LPS as above or left untreated were used to measure TG-induced (1 μm) ER-stored Ca2+ release and Ca2+ release-activated Ca2+ entry. TG-induced changes in 340/380 nm ratio color images are shown for control and LPS-pretreated cells (top row). The change in [Ca2+]i is depicted by the colors indicated in the arrow. Bottom row, TG-induced fluorescence ratio (340/380) change over basal for ER-stored Ca2+ release and Ca2+ release-activated Ca2+ entry in control and LPS-pretreated cells. Results are representative of two experiments. D, C57BL/6 mice injected with LPS (5.0 mg/kg intraperitoneally) or saline. After LPS or saline injection, at the indicated time periods, lungs harvested were used for immunoblot analysis to determine STIM1, STIM2, TRPC1, TRPC4, and Orai1. The blots were stripped and reprobed for β-actin antibody as a loading control. Results are representative of three experiments. Blots were quantified, and the fold increase over basal levels relative to β-actin is shown. E, C57BL/6 mice injected with LPS or saline as above. 4 h after LPS or saline injection, lungs harvested were used for isolated lung preparation to determine lung vascular permeability (see details under “Experimental Procedures”). PAR-1 agonist peptide (pep, 30 μm) was included in the perfusion buffer to assess the PAR-1-induced liquid filtration coefficient (Kf,c) (n = 5). *, p < 0.05; **, p < 0.001; control group versus PAR-1 peptide, LPS treated group versus LPS + PAR-1 peptide group, or PAR-1 peptide group versus LPS + PAR-1 peptide group. F, C57BL/6 mice either injected with LPS or saline as above were used to measure PAR-1-induced EBA uptake in lungs. Top panel, experimental design. Results are mean ± S.E. of changes in lung EBA uptake (n = 5/group). *, p < 0.01; control (saline injected) versus PAR-1 peptide treated, control versus LPS treated, or PAR-1 peptide treated versus LPS + PAR-1 peptide-treated. i.p., intraperitoneal; i.v., intravenous.
LPS Priming in Vivo Potentiates PAR-1-induced Increase in Lung Microvessel Permeability
To validate the in vivo pathophysiologic relevance of increased STIM1 expression in ECs, we injected mice (C57BL6J) with LPS intraperitoneally, and lungs harvested at different time intervals after LPS injection were used for Western blot analysis. We observed substantially increased protein expression for STIM1, TRPC1, TRPC4, and Orai1, but not STIM2, in LPS-treated mice compared with control mice injected with saline (Fig. 2D). Furthermore, to address whether the increased expression of STIM1, TRPC1, TRPC4, and Orai1 contributes to lung vascular hyperpermeability, we measured microvessel liquid permeability in isolated intact lung preparations. Here mice received intraperitoneal saline (control injection) or LPS for 4 h, followed by lung isolation for determination of the lung vascular liquid filtration coefficient Kf,c, a measure of intact lung vascular permeability (7, 34). We observed that PAR-1-activating peptide induced an ∼3-fold increase in lung Kf,c when compared with basal conditions (Fig. 2E). Mice receiving the indicated dose of LPS alone also showed a significant increase in Kf,c (Fig. 2E). Interestingly, lung preparations from LPS-treated mice showed an ∼5-fold increase in Kf,c, over basal levels when challenged by PAR-1-activating peptide (Fig. 2E). To further support this observation, we determined the PAR-1-induced lung vascular leak in vivo by measuring EBA uptake into the lung in control and LPS-primed mice (20). The PAR1 agonist caused a 6-fold increase in EBA uptake with LPS priming compared with a 3-fold increase without priming (Fig. 2F). These in vivo results further support the hypothesis that LPS-induced expression of STIM1 and SOC components in intact lung microvessels may contribute to the hyperpermeability response during sepsis.
LPS Promotes NF-κB and p38 MAPK Activation to Induce STIM1 Expression in Endothelial Cells
Next we focused on the signaling pathways activated downstream of TLR4 that mediate STIM1 expression because STIM1 is crucial for activating SOCE in ECs to induce a vascular permeability increase. It is now well known that signaling via both the NF-κB and p38 MAPK pathways contributes to the vascular inflammatory responses seen in sepsis (13–16). To determine the role of the NF-κB and p38 MAPK pathways in mediating LPS-induced STIM1 expression, we inhibited the LPS-induced activation of NF-κB and p38 MAPK with specific pharmacological inhibitors. The NF-κB inhibitor 6-amino-4-(4-phenoxyphenylamino) quinzoline, used in this study, has been shown previously to prevent LPS-induced TNF-α production in murine splenocytes and also reduced carrageenin-induced edema formation in the rat hind paw (37). We observed that the NF-κB inhibitor prevented LPS-induced STIM1 mRNA expression in HLMVECs (Fig. 3A). We also pretreated HLMVECs with the p38 MAPK inhibitor SB203580, which inhibits both p38α and p38β isoforms (38, 39), and observed that SB203580 prevented LPS-induced STIM1 mRNA expression in HLMVECs (Fig. 3A).
FIGURE 3.

Inhibition of NF-κB or p38 MAPK activation prevents LPS-induced STIM1 expression in endothelial cells. A, HLMVECs were preincubated with p38 MAPK inhibitor (10 μm) for 30 min or NF-κB inhibitor (5 μm) for 12 h, and then cells were exposed to LPS for the indicated time periods. After LPS exposure, cells were used for quantitative real-time PCR to determine mRNA expression for STIM1 and β-actin. STIM1 mRNA induction fold was calculated by measuring the ratio of STIM1 to β-actin. Results are mean ± S.E. of four experiments. *, p < 0.01 compared with cells treated with either p38 MAPK or NF-κB inhibitor. B and C, HLMVECs transfected with control siRNA (Sc-siRNA, 100 nm), p65/RelA siRNA (100 nm), or p50/NF-κB1 siRNA (100 nm). Left panels, cell lysates were used for immunoblotting (IB) to determine p65/RelA (B) or p50/NF-κB1 (C) expression. Right panels, cells exposed to LPS for 0 and 6 h were used for immunoblotting to determine STIM1 or β-actin expression. Results are representative of at least two experiments. SE, short exposure; LE, long exposure. D, HLMVECs transfected with Sc-siRNA or p65/RelA-siRNA (100 nm). After LPS treatment, cells were used to measure thrombin-induced ER-stored Ca2+ release and Ca2+ release-activated Ca2+ entry. Results are representative of at least two experiments. E, HLMVECs were preincubated with SB203580 (10 μm) for 30 min, and then cells were exposed to LPS for the indicated time periods. After LPS treatments, cells were used for immunoblotting to determine STIM1 protein expression. Results are representative of three experiments. F, HLMVECs pretreated with vehicle (dimethyl sulfoxide) or SB203580 (10 μm) for 30 min and then challenged with or without LPS for 4 h. After LPS treatment, cells were used to measure thrombin (Thr)-induced ER-stored Ca2+ release and Ca2+ release-activated Ca2+ entry. Results are representative of two experiments. G, HLMVECs were preincubated with the ERK inhibitor PD98059 (40 μm) for 30 min, and then cells were exposed to LPS for the indicated time periods. After LPS treatment, cell lysates were used for immunoblotting to determine ERK1/2 phosphorylation (top panel) or STIM1 expression (bottom panel). H, HLMVECs were preincubated with the JNK inhibitor SP600125 (20 μm) for 30 min, and then cells were exposed to LPS for the indicated time periods. After LPS treatment, cell lysates were used for immunoblotting to determine JNK phosphorylation (top panel) or STIM1 expression (bottom panel). G and H, the numbers below the lanes indicate quantification of band intensity from two separate experiments presented as the ratio of phosphorylated ERK1/2 to total ERK1/2 or the ratio of phosphorylated JNK to total JNK. P/T, phospho/total. I, C57BL/6 mice pretreated with vehicle or the p38 inhibitor SB203580 were injected with LPS or saline as in Fig. 2E. Four hours after LPS or saline injection, lungs harvested were used for isolated lung preparation to determine lung vascular permeability (see details under “Experimental Procedures”). PAR-1 agonist peptide (pep, 30 μm) was included in the perfusion buffer to assess the PAR-1-induced Kf,c (n = 5 mice/group). **, p < 0.001; control group versus LPS + PAR-1 peptide group or dimethyl sulfoxide + LPS + PAR-1 peptide group versus SB203580 + LPS + PAR-1 peptide group. DMSO, dimethyl sulfoxide.
LPS signaling usually triggers the translocation of the NF-κB dimer p65/p50 into the nucleus to induce target gene expression (40). Therefore, to strengthen the role of NF-κB signaling in the mechanism of LPS-induced STIM1 expression, we first silenced the NF-κB protein p65/RelA in HLMVECs by transfecting HLMVECs with siRNA specific to p65/RelA. In this experiment, we observed that STIM1 protein expression in response to LPS was blocked in p65/RelA-siRNA-transfected cells compared with control cells or Sc-siRNA- transfected cells (Fig. 3B). Next we observed that silencing of the NF-κB protein p50/NF-κB1 also prevented LPS-induced STIM1 expression in HLMVECs (Fig. 3C). Consistent with these results, LPS potentiation of the thrombin-induced increase in SOCE was suppressed in HLMVECs after p65/RelA knockdown (Fig. 3D), indicating that NF-κB signaling is vital for the induction of STIM1 in HLMVECs in response to LPS.
Next we investigated the effect of p38 MAPK inhibition on STIM1 protein expression in HLMVECs. We pretreated HLMVECs with the p38 MAPK inhibitor SB203580 and challenged control (vehicle-treated) and SB203580-treated cells with LPS for different time intervals. Then the cells were used to determine STIM1 protein expression. LPS-induced STIM1 protein expression was reduced markedly in cells pretreated with SB203580 compared with vehicle-treated control cells (Fig. 3E). Next we examined the effect of inhibition of p38 MAPK on LPS-induced STIM1 expression and subsequent potentiation of thrombin-induced SOCE. In this experiment, we pretreated HLMVECs with LPS for 4 h in the presence and absence of SB203580, and then cells were used to measure thrombin-induced ER store Ca2+ release and SOCE. We observed that SB203580 blocked LPS potentiation of SOCE in response to thrombin (Fig. 3F).
LPS is known to activate the ERK and JNK signaling pathways (41). Therefore, we investigated the possibility that ERK and JNK activation also contribute to LPS-induced STIM1 expression in HLMVECs. Here we used a pharmacological approach to prevent the activation of ERK and JNK in HLMVECs (42). We observed that the ERK inhibitor PD98059 prevented LPS-induced ERK phosphorylation (Fig. 3G, top panel) but failed to inhibit LPS-induced STIM1 expression in HLMVECs (Fig. 3G, bottom panel). Next, we observed that the JNK inhibitor SP600125 blocked LPS-induced JNK phosphorylation (Fig. 3H, top panel) but had no significant effect on LPS-induced STIM1 expression in HLMVECs (Fig. 3H, bottom panel). These results suggest that the ERK and JNK signaling pathways are not involved in the mechanism of LPS-induced STIM1 expression in HLMVECs.
Because we observed that only the p38 inhibitor, but not ERK or JNK inhibitors, suppressed LPS-induced STIM1 expression in HLMVECs, we determined the effect of inhibition of p38 on LPS potentiation of the PAR-1-mediated increase in lung vascular permeability. In this experiment, we injected mice with SB203580 or left them untreated, followed by LPS challenge for 4 h, and then lung vascular liquid permeability (Kf,c,) was measured as above (Fig. 2E). We observed that LPS-induced STIM1 expression was reduced markedly in lungs from SB203580-injected mice compared with lungs from vehicle-injected mice (Fig. 4I, top panel). Interestingly, SB203580 pretreatment prevented LPS potentiation of the PAR-1-mediated increase in Kf,c, (Fig. 4I). These results support the key role for p38 signaling in the mechanism of LPS-induced lung vascular leaks.
FIGURE 4.

p38α signaling mediates LPS-induced STIM1 expression in HLMVECs. A, HLMVECs were transfected with control siRNA (Sc-siRNA) or p38α-siRNA (100 nm). Left panel, after transfection, cells were used for immunoblotting (IB) to determine p38 isoform (p38α, p38β, and p38γ) expression. Right panel, after transfection, cells exposed to LPS for different time intervals were used for immunoblotting to determine the expression of STIM1, p38α, and β-actin. B, HLMVECs transfected with the indicated concentrations of Sc-siRNA or p38β-siRNA. Left panel, after transfection, cells were for immunoblotting to determine the expression of p38β and β-actin. Right panel, after transfection, cells exposed to LPS were used for immunoblotting to determine the expression of STIM1 and β-actin. C) HLMVECs transfected with the indicated concentrations of Sc-siRNA or p38γ-siRNA. Left panel, after transfection, cells were used for immunoblotting to determine the expression of p38γ and β-actin. Right panel, after transfection, cells exposed to LPS were used for immunoblotting to determine the expression of STIM1 and β-actin. A–C, results are representative of at least two experiments.
Using a pharmacological approach, we showed that p38 signaling is required for triggering LPS responses in endothelial cells. We have recently shown that the p38 MAPK isoforms p38α, p38β, and p38γ are expressed in HLMVECs (10). We also showed that silencing of p38α suppresses constitutive STIM1 expression in HLMVECs (10). To determine which isoform of p38 is involved in LPS-induced STIM1 expression in HLMVECs, we first silenced p38α in HLMVECs by transfecting HLMVECs with siRNA specific to p38α. In p38α-siRNA-transfected cells, expression of p38α, but not the other p38 isoforms under study (p38β and p38γ), was blocked (Fig. 4A, left panel). Interestingly, we observed that p38α-siRNA transfection prevented LPS-induced STIM1 expression (Fig. 4A, right panel). Next we silenced p38β expression in HLMVECs (Fig. 4B, left panel) and observed that p38β silencing had no significant effect on LPS-induced STIM1 expression (Fig. 4B, right panel). Similarly, we observed that silencing of p38γ had no effect on LPS-induced STIM1 expression in HLMVECs (Fig. 4C). Collectively, these results support the notion that p38α signaling plays a critical role in the mechanism of LPS-induced STIM1 expression in HLMVECs.
LPS Induces Binding of Transcription Factors NF-κB and AP1 to the STIM1 Promoter
To gain insight into the transcriptional mechanism of LPS-induced STIM1 expression, we analyzed the 5′-regulatory region of the human (h) STIM1 gene utilizing Genomatix software. We observed the presence of three putative NF-κB binding sites at −172, −198, and −216 (NF-κB1, 2, and 3, respectively) and one AP1 site (−347) upstream of the transcription start site in the hSTIM1 promoter (Fig. 5A). Interestingly, we also observed the presence of binding sites for the transcription factors NF-κB and AP1 in the 5′ regulatory region of the mouse (m) STIM1 gene (Fig. 5B). To address whether LPS induces the binding of transcription factors NF-κB and AP1 to the hSTIM1 promoter, we performed ChIP assays. HLMVECs exposed to LPS for different time intervals were used for ChIP assays. LPS induced time-dependent binding of the NF-κB protein p65/RelA to the hSTIM1 promoter in HLMVECs (Fig. 5C). The binding of p65/RelA to the hSTIM1 promoter was ∼6-fold higher than basal levels 60 min after LPS challenge and returned to basal levels 120 min after LPS challenge (Fig. 5C). LPS also induced the binding of the AP1 components c-Fos and c-Jun to the hSTIM1 promoter in HLMVECs (Fig. 5, D and E), and the binding was maximal 30 min after LPS stimulation (Fig. 5, D and E). These results suggest that LPS activates transcription factors NF-κB and AP1 to induce STIM1 transcription in ECs.
FIGURE 5.

LPS induces p65/RelA, c-Fos, and c-Jun binding to the STIM1 promoter. A and B, the putative nucleotide sequence identified in the 5′-regulatory region of human (A) and mouse (B) STIM1 genes is shown. Nucleotides are numbered relative to the transcription start site as +1. Potential consensus sequences for the transcription factors NF-κB and AP1 are indicated in boldface and are underlined. C, D, and E, ChIP assay of the interaction of p65/RelA (C), c-Fos (D), and c-Jun (E) with the hSTIM1 promoter. Primers specific to the NF-κB binding sites 1–3 and AP1 binding sites were used to perform quantitative PCR (see details under “Experimental Procedures”). HLMVECs exposed to LPS (1 μg/ml) for 0, 30, 60, and 120 min were used for the ChIP assay. Results are mean ± S.E. of four experiments. Results were normalized to those of input DNA and are presented relative to basal values. C, *, p < 0.01, 0 min versus 30 min; **, p < 0.001, 0 min versus 60 min. D, *, p < 0.01, 0 min versus 60 or 120 min; **, p < 0.001, 0 min versus 30 min. E, **, p < 0.001, 0 min versus 30 min.
p38α Signaling Downstream of TLR4 Mediates c-Fos Expression to Induce STIM1 Expression in Endothelial Cells
We showed above that silencing of NF-κB proteins (p65/RelA or p50) or p38α prevented LPS-induced STIM1 expression in HLMVECs. Furthermore, we showed that LPS induced the binding of the NF-κB and the immediate early genes c-Fos and c-Jun, the components of activator protein 1 (AP1), to the STIM1 promoter in HLMVECs. To determine whether c-Fos and c-Jun signaling is essential for STIM1 expression in response to LPS, we first silenced c-Fos expression in HLMVECs. In c-Fos siRNA-transfected cells, basal as well as LPS-induced c-Fos expression was reduced markedly in HLMVECs (Fig. 6A) compared with control or Sc-siRNA-transfected cells (Fig. 6A). Interestingly, we observed that LPS-induced STIM1 expression was also prevented in c-Fos-siRNA-transfected cells (Fig. 6A). Next we silenced c-Jun expression and verified that c-Jun expression was nearly eliminated compared with control cells or cells transfected with Sc-siRNA (Fig. 6B). Surprisingly, we observed that LPS-induced STIM1 expression was not reduced significantly in c-Jun-siRNA-treated cells compared with controls (Fig. 6B). These observations suggest that c-Fos, but not c-Jun signaling plays a dominant role in LPS-induced STIM1 transcription in HLMVECs.
FIGURE 6.

p38α-mediated c-Fos expression is required for LPS-induced STIM1 expression. A, HLMVECs were transfected with control siRNA (Sc-siRNA) or c-Fos-siRNA (100 nm). After transfection, cells were exposed to LPS (1 μg/ml) for 0, 2, 4, and 6 h. After LPS treatment, cells were used for immunoblotting to determine STIM1, c-Fos, and β-actin expression. The experiment was repeated three times, and the results are mean ± S.E. (center and bottom panels). *, p < 0.05; **, p < 0.001; significantly different from control cells not stimulated with LPS. B, HLMVECs transfected with control siRNA or c-Jun-siRNA (100 nm). After siRNA transfection, cells exposed to LPS as above were used for immunoblotting to determine STIM1, c-Jun, and β-actin expression. The experiment was repeated three times, and results are mean ± S.E. (center and bottom panels). N.S., not significant; *, p < 0.05; **, p < 0.001; significantly different from control cells not stimulated with LPS. C, HLMVECs transfected with control siRNA or p38α-siRNA (100 nm). After transfection, cells exposed to LPS were used for immunoblotting to determine c-Fos, c-Jun, and β-actin expression. The experiment was repeated three times, and results are mean ± S.E. (center and bottom panels). *, p < 0.05; **, p < 0.001; significantly different from control cells not stimulated with LPS. D, HLMVECs transfected with control siRNA or p65/RelA-siRNA (100 nm). After transfection, cells exposed to LPS as above were used for immunoblotting to determine c-Fos and β-actin expression. Results are representative of at least two experiments. E, HLMVECs transfected with control siRNA or p38α-siRNA (100 nm). After transfection, cells were exposed to LPS (1 μg/ml) for 0, 15, 30, and 60 min. After LPS exposure, cells were used for immunoblotting with antibodies specific to phospho-c-Fos or total c-Fos. Experiments were repeated three times, and results are mean ± S.E. (center and bottom panels). *, p < 0.01, significantly different from control cells not stimulated with LPS. F, HLMVECs were pretreated with the p38 inhibitor SB203580 (10 μm) for 5 min or left untreated, and then cells exposed to LPS as above were used for immunoblotting with antibodies specific to phospho-c-Fos or total c-Fos. The numbers below the lanes indicate quantification of band intensity from two separate experiments presented as the ratio of phosphorylated c-Fos to total c-Fos.
p38 MAPK signaling activates the AP1 transcription factor (41, 43–45). Because we observed that suppression of p38α expression in HLMVECs prevented LPS-induced STIM1 expression, we investigated the possible role of LPS-induced p38α activation in mediating the induction of AP1 components in HLMVECs. In this experiment, we silenced p38α expression in HLMVECs and then measured the expression of c-Fos and c-Jun. Interestingly, silencing of p38α inhibited LPS-induced c-Fos but not c-Jun expression (Fig. 6C). In fact, c-Jun expression was increased in p38α knockdown cells (Fig. 6C). This observation is in agreement with previous reports suggesting that p38α negatively regulates c-Jun (46, 47). Because we observed that NF-κB signaling is required for LPS-induced STIM1 expression, we determined whether NF-κB signaling is required for LPS-induced c-Fos expression in HLMVECs. We observed that silencing of p65/RelA had no effect on c-Fos expression in HLMVECs (Fig. 6D), indicating that p38α, but not NF-κB, signaling controls c-Fos expression in HLMVECs. Next we investigated whether p38α signaling is required for the activation of c-Fos. We observed that LPS induced time-dependent phosphorylation of c-Fos at Thr-325 in control and Sc-siRNA-transfected cells (Fig. 6E), whereas, in p38α knockdown cells, this response was blocked (Fig. 6E). To investigate the possible loss of c-Fos expression, we pretreated HLMVECs with the p38 inhibitor SB203580 for 5 min and then stimulated the cells with LPS. We observed that LPS-induced c-Fos phosphorylation was largely eliminated in SB203580-treated cells without loss of c-Fos expression (Fig. 6F). Collectively, these results indicate that p38-mediated c-Fos expression and activation is required for STIM1 expression in endothelial cells.
DISCUSSION
Sepsis associated with acute lung injury is a common cause of death in hospitalized patients (1–5). Acute lung injury is, in large part, the result of lung vascular leaks and protein-rich pulmonary edema (1, 48). However, the signaling switch that mediates lung vascular leaks during sepsis is defined poorly. We have shown previously that Ca2+ entry through SOCs regulates lung vascular barrier function (6, 7). We have also shown that the edema-inducing factor thrombin, which is generated during sepsis at abnormal levels in the vascular system (28–32), can induce Ca2+ entry through activation of TRPC1 and TRPC4 in the endothelial cells to cause vascular barrier dysfunction (7–10, 21). In addition, we showed that inflammatory mediators induce the expression of TRPC1 in human vascular ECs via NF-κB activation (9, 18). Increased TRPC1 expression has been associated with augmented SOCE and permeability increase in human ECs. Recent studies from our laboratory have shown that the ER-localized Ca2+-sensing protein STIM1 is crucial for activating SOCE in ECs (10, 21). Importantly, it has been shown recently that endotoxin-induced lung vascular leaks and injury responses were suppressed in endothelial cell-restricted STIM1 knockout mice (27), suggesting that endothelial STIM1-mediated Ca2+ entry (i.e. SOCE) plays a vital role in the mechanism of sepsis-induced lung vascular leaks. However, whether STIM1 expression is increased during sepsis and whether the increased STIM1 expression in ECs contributes to the abnormal lung vascular leaks was not known. To address these questions, we utilized in vitro and in vivo experimental models and showed that endotoxin induced the expression of STIM1 in both human and murine lung ECs. We also showed that endotoxin induced the expression of the SOC components TRPC1, TRPC4, and Orai1 channels in ECs. In addition, we observed that endotoxin-induced expression of STIM1 and SOC components was associated with augmented PAR-1-mediated endothelial permeability both in vitro and in vivo, indicating that increased STIM1 expression-mediated SOCE may signal the lung vascular hyperpermeability associated with sepsis-induced acute lung injury.
The current paradigm is that inflammatory mediators such as endotoxin and TNF-α trigger inflammatory responses through the up-regulation of inflammatory genes by activating the NF-κB and p38 MAPK signaling pathways (13–16, 49). Because STIM1 is crucial for activating SOCE in ECs, we investigated whether activation of both the NF-κB and p38 MAPK pathways downstream of TLR4 is required for STIM1 expression in ECs. We observed that pharmacological inhibition of either NF-κB or p38 MAPK prevented LPS-induced STIM1 expression and subsequent PAR-1-mediated SOCE. We also observed that pharmacological inhibition of p38 in vivo blocked LPS potentiation of the PAR-1-mediated increase in lung vascular permeability. Further, we showed that knockdown of either NF-κB (p65/RelA or p50/NF-κB1) or the p38 MAPK isoform p38α prevented LPS-induced STIM1 expression in ECs. These findings support the postulation that cooperative signaling of both NF-κB and p38α is required for the expression of STIM1 in response to LPS challenge in ECs.
It has been shown recently that the serum and glucocorticoid-inducible kinase SGK1 controls constitutive STIM1 transcription via NF-κB signaling in bone marrow-derived mast cells (50). However, the transcriptional mechanism of STIM1 expression in response to inflammatory stimuli in ECs was not studied. Therefore, we analyzed the STIM1 gene sequence and found consensus binding sites for the transcription factors NF-κB and AP1 in the 5′ regulatory regions of both human and mouse STIM1 genes. Because LPS can activate both transcription factors (NF-κB and AP1), we performed ChIP assays to determine whether these transcription factors bind to the STIM1 promoter in response to LPS. LPS induced the binding of the NF-κB protein p65/RelA to the hSTIM1 promoter in a time-dependent manner. Maximal binding was observed 60 min after LPS stimulation in HLMVECs. This observation is in agreement with our finding that silencing of either the NF-κB protein p65/RelA or p50/NF-κB1 prevents LPS-induced STIM1 expression in HLMVECs. LPS also induced the binding of the AP1 components c-Fos and c-Jun to the hSTIM1 promoter in HLMVECs. Both c-Fos and c-Jun binding to the STIM1 promoter reached peak levels within 30 min of LPS challenge, indicating that the early binding of AP1 to the STIM1 promoter may recruit or facilitate NF-κB binding to NF-κB sites on the STIM1 promoter to initiate transcription. We also confirmed the role of AP1 in mediating STIM1 transcription utilizing knockdown experiments. Here we observed that loss of c-Fos expression prevented LPS-induced STIM1 expression in HLMVECs, whereas the loss of c-Jun expression had no significant effect on LPS-induced STIM1 expression in HLMVECs, indicating that c-Fos could replace c-Jun in inducing STIM1 expression in ECs. Importantly, silencing of either NF-κB or AP1 prevented LPS-induced STIM1 expression in HLMVECs. Collectively, these results support the proposal that binding of transcription factors NF-κB and AP1 to the STIM1 promoter is essential for STIM1 transcription in response to LPS in ECs.
Expression of four different p38 MAPK isoforms (MAPK14 (p38α), MAPK11 (p38β), MAPK12 (p38γ), and MAPK13 (p38δ)) have been identified in mammalian cells (51). In recent studies, we reported the expression of the p38α, p38β, and p38γ isoforms in HLMVECs (10). In this study, we also report that SOCE-mediated p38β activation induced STIM1 phosphorylation, which, in turn, inhibited SOCE in HLMVECs (10). Furthermore, we reported that silencing of the p38 MAPK isoform p38α markedly reduced constitutive STIM1 expression in HLMVECs (10). Here we observed that endotoxin-induced STIM1 expression was also prevented in p38α knockdown HLMVECs, suggesting that p38α signaling is required for endotoxin-induced STIM1 transcription. Mounting evidence suggests that p38 MAPK signaling may control inflammatory gene transcription through chromatin remodeling by inducing histone 3 (H3) phosphorylation at Ser-10 (14). This event could increase the accessibility of transcription factors to their binding sites in the promoter regions of a subset of genes, including c-Fos (14, 45). Therefore, to delineate the role of p38α signaling in the mechanism of LPS-induced STIM1 expression, we silenced p38α expression and measured c-Fos expression in response to LPS in HLMVECs. Interestingly, we observed that p38α silencing prevented c-Fos expression under basal conditions and in response to LPS challenge. In contrast to c-Fos expression, c-Jun expression was increased in p38α-silenced HLMVECs. Collectively, these results support the concept that p38α signaling induces STIM1 expression through the AP1 component c-Fos in HLMVECs.
In summary, we showed that bacterial endotoxin induced the expression of STIM1 and its interacting channel proteins TRPC1, TRPC4, and Orai1 in both human and mouse ECs. The increased expression of these of proteins was associated with augmented PAR-1-mediated permeability responses. Importantly, we showed that NF-κB activation and p38α-mediated c-Fos expression in response to endotoxin contribute to STIM1 transcription in lung vascular ECs, setting the stage for endothelial Ca2+ overload and resulting vascular leaks induced by mediators such as thrombin.
This work was supported, in whole or in part, by National Institutes of Health Grant P01HL077806 (Project 3).
- EC
- endothelial cell
- SOC
- store-operated Ca2+ channel
- TRPC
- transient receptor potential canonical
- SOCE
- store-operated Ca2+ entry
- ER
- endoplasmic reticulum
- HLMVEC
- human lung microvascular endothelial cell
- Sc-siRNA
- scrambled siRNA
- pAb
- polyclonal antibody
- MLEC
- mouse lung endothelial cell
- TER
- transendothelial electrical resistance
- EBA
- Evans blue dye conjugated with albumin
- TG
- thapsigargin
- Dil-Ac
- 1,1′-dioctadecyl-3,3,3′-tetramethylindocarbocyanine perchlorate-labeled acetylated low-density lipoprotein.
REFERENCES
- 1. Goldenberg N. M., Steinberg B. E., Slutsky A. S., Lee W. L. (2011) Broken barriers: a new take on sepsis pathogenesis. Sci. Transl. Med. 3, 88ps25. [DOI] [PubMed] [Google Scholar]
- 2. Baghai-Ravary R., Bellingan G. J. (2006) Pulmonary dysfunction in sepsis. Adv. Sepsis 5, 42–51 [Google Scholar]
- 3. Dombrovskiy V. Y., Martin A. A., Sunderram J., Paz H. L. (2007) Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: a trend analysis from 1993 to 2003. Crit. Care. Med. 35, 1244–1250 [DOI] [PubMed] [Google Scholar]
- 4. Matthay M. A., Zemans R. L. (2011) The acute respiratory distress syndrome: pathogenesis and treatment. Annu. Rev. Pathol. 6, 147–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aird W. C. (2003) The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 101, 3765–3777 [DOI] [PubMed] [Google Scholar]
- 6. Tiruppathi C., Ahmmed G. U., Vogel S. M., Malik A. B. (2006) Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation 13, 693–708 [DOI] [PubMed] [Google Scholar]
- 7. Tiruppathi C., Freichel M., Vogel S. M., Paria B. C., Mehta D., Flockerzi V., Malik A. B. (2002) Impairment of store-operated Ca2+ entry in TRPC4−/− mice interferes with increase in lung microvascular permeability. Circ. Res. 91, 7919–7923 [DOI] [PubMed] [Google Scholar]
- 8. Paria B. C., Vogel S. M., Ahmmed G. U., Alamgir S., Shroff J., Malik A. B., Tiruppathi C. (2004) Tumor necrosis factor-α-induced TRPC1 expression amplifies store-operated Ca2+ influx and endothelial permeability. Am. J. Physiol. Lung Cell Mol. Physiol. 287, L1303-L1313 [DOI] [PubMed] [Google Scholar]
- 9. Paria B. C., Bair A. M., Xue J., Yu Y., Malik A. B., Tiruppathi C. (2006) Ca2+ influx-induced by PAR-1 activates a feed-forward mechanism of TRPC1 expression via NF-κB activation in endothelial cells. J. Biol. Chem. 281, 20715–20727 [DOI] [PubMed] [Google Scholar]
- 10. Sundivakkam P. C., Natarajan V., Malik A. B., Tiruppathi C. (2013) Store-operated Ca2+ entry (SOCE) induced by protease-activated receptor-1 mediates STIM1 phosphorylation to inhibit SOCE in endothelial cells through AMP-activated protein kinase and p38β mitogen-activated protein kinase. J. Biol. Chem. 288, 17030–17041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tauseef M., Knezevic N., Chava K. R., Smith M., Sukriti S., Gianaris N., Obukhov A. G., Vogel S. M., Schraufnagel D. E., Dietrich A., Birnbaumer L., Malik A. B., Mehta D. (2012) TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J. Exp. Med. 209, 1953–1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kandasamy K., Bezavada L., Escue R. B., Parthasarathi K. (2013) Lipopolysaccharide induces endoplasmic store Ca2+-dependent inflammatory responses in lung microvessels. PLoS ONE 8, e63465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Banerjee A., Gerondakis S. (2007) Coordinating TLR-activated signaling pathways in cells of the immune system. Immunol. Cell Biol. 85, 420–424 [DOI] [PubMed] [Google Scholar]
- 14. Saccani S., Pantano S., Natoli G. (2002) p38-dependent marking of inflammatory genes for increased NF-κB recruitment. Nat. Immunol. 3, 69–75 [DOI] [PubMed] [Google Scholar]
- 15. Anderson P. (2008) Post-transcriptional control of cytokine production. Nat. Immunol. 8, 353–359 [DOI] [PubMed] [Google Scholar]
- 16. Tiruppathi C., Soni D., Wang D. M., Xue J., Singh V., Thippegowda P. B., Cheppudira B. P., Mishra R. K., Debroy A., Qian Z., Bachmaier K., Zhao Y. Y., Christman J. W., Vogel S. M., Ma A., Malik A. B. (2014) The transcription factor DREAM represses the deubiquitinase A20 and mediates inflammation. Nat. Immunol. 15, 239–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sehnert B., Burkhardt H., Wessels J. T., Schröder A., May M. J., Vestweber D., Zwerina J., Warnatz K., Nimmerjahn F., Schett G., Dübel S., Voll R. E. (2013) NF-κB inhibitor targeted to activated endothelium demonstrates a critical role of endothelial NF-κB in immune-mediated diseases. Proc. Natl. Acad. Sci. U.S.A. 110, 16556–16561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paria B. C., Malik A. B., Kwiatek A. M., Rahman A., May M. J., Ghosh S., Tiruppathi C. (2003) Tumor necrosis factor-α induces nuclear factor-κB-dependent TRPC1 expression in endothelial cells. J. Biol. Chem. 278, 37195–37203 [DOI] [PubMed] [Google Scholar]
- 19. Andonegui G., Zhou H., Bullard D., Kelly M. M., Mullaly S. C., McDonald B., Long E. M., Robbins S. M., Kubes P. (2009) Mice that exclusively express TLR4 on endothelial cells can efficiently clear a lethal systemic Gram-negative bacterial infection. J. Clin. Invest. 119, 1921–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ye X., Ding J., Zhou X., Chen G., Liu S. F. (2008) Divergent roles of endothelial NF-κB in multiple organ injury and bacterial clearance in mouse models of sepsis. J. Exp. Med. 205, 1303–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sundivakkam P. C., Freichel M., Singh V., Yuan J. P., Vogel S. M., Flockerzi V., Malik A. B., Tiruppathi C. (2012) Ca2+ Sensor STIM1 is necessary and sufficient for the store-operated Ca2+ entry function of transient receptor potential canonical (TRPC) 1 and 4 channels in endothelial cells. Mol. Pharmacol. 81, 510–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Abdullaev I. F., Bisaillon J. M., Potier M., Gonzalez J. C., Motiani R. K., Trebak M. (2008) Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 23, 1289–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shinde A. V., Motiani R. K., Zhang X., Abdullaev I. F., Adam A. P., González-Cobos J. C., Zhang W., Matrougui K., Vincent P. A., Trebak M. (2013) STIM1 controls endothelial barrier function independently of Orai1 and Ca2+ entry. Sci. Signal. 6, ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee K. P., Yuan J. P., Hong J. H., So I., Worley P. F., Muallem S. (2010) An endoplasmic reticulum/plasma membrane junction: STIM1/Orai1/TRPCs. FEBS Lett. 584, 471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zeng W., Yuan J. P., Kim M. S., Choi Y. J., Huang G. N., Worley P. F., Muallem S. (2008) STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol. Cell 32, 439–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hawkins B. J., Irrinki K. M., Mallilankaraman K., Lien Y. C., Wang Y., Bhanumathy C. D., Subbiah R., Ritchie M. F., Soboloff J., Baba Y., Kurosaki T., Joseph S. K., Gill D. L., Madesh M. (2010) S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J. Cell Biol. 190, 391–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gandhirajan R. K., Meng S., Chandramoorthy H. C., Mallilankaraman K., Mancarella S., Gao H., Razmpour R., Yang X. F., Houser S. R., Chen J., Koch W. J., Wang H., Soboloff J., Gill D. L., Madesh M. (2013) Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J. Clin. Invest. 123, 887–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Esmon C. T. (2005) The interactions between inflammation and coagulation. Br. J. Haematol. 131, 417–430 [DOI] [PubMed] [Google Scholar]
- 29. Levi M., van der Poll T. (2005) Two-way interactions between inflammation and coagulation. Trends Cardiovasc. Med. 15, 254–259 [DOI] [PubMed] [Google Scholar]
- 30. Minami T., Sugiyama A., Wu S. Q., Abid R., Kodama T., Aird W. C. (2004) Thrombin and phenotypic modulation of the endothelium. Arterioscler. Thromb. Vasc. Biol. 24, 41–53 [DOI] [PubMed] [Google Scholar]
- 31. Kataoka H., Hamilton J. R., McKemy D. D., Camerer E., Zheng Y. W., Cheng A., Griffin C., Coughlin S. R. (2003) Protease-activated receptors 1 and 4 mediate thrombin signaling in endothelial cells. Blood 102, 3224–3231 [DOI] [PubMed] [Google Scholar]
- 32. Gando S., Nanzaki S., Morimoto Y., Kobayashi S., Kemmotsu O. (1999) Systemic activation of tissue-factor dependent coagulation pathway in evolving acute respiratory distress syndrome in patients with trauma and sepsis. J. Trauma 47, 719–723 [DOI] [PubMed] [Google Scholar]
- 33. Fazal F., Bijli K. M., Murrill M., Leonard A., Minhajuddin M., Anwar K. N., Finkelstein J. N., Watterson D. M., Rahman A. (2013) Critical role of non-muscle myosin light chain kinase in thrombin-induced endothelial cell inflammation and lung PMN infiltration. PLoS ONE 8, e59965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vogel S. M., Gao X., Mehta D., Ye R. D., John T. A., Andrade-Gordon P., Tiruppathi C., Malik A. B. (2000) Abrogation of thrombin-induced increase in pulmonary microvascular permeability in PAR-1 knockout mice. Physiol. Genomics. 4, 137–145 [DOI] [PubMed] [Google Scholar]
- 35. Kaneider N. C., Leger A. J., Agarwal A., Nguyen N., Perides G., Derian C., Covic L., Kuliopulos A. (2007) “Role reversal” for the receptor PAR1 in sepsis-induced vascular damage. Nat. Immunol. 8, 1303–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tiruppathi C., Malik A. B., Del Vecchio P. J., Keese C. R., Giaever I. (1992) Electrical method for detection of endothelial cell shape change in real time: assessment of endothelial barrier function. Proc. Natl. Acad. Sci. U.S.A. 89, 7919–7923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tobe M., Isobe Y., Tomizawa H., Nagasaki T., Takahashi H., Fukazawa T., Hayashi H. (2003) Discovery of quinazolines as a novel structural class of potent inhibitors of NF-κB activation. Bioorg. Med. Chem. 11, 383–391 [DOI] [PubMed] [Google Scholar]
- 38. Kumar S., Boehm J., Lee J. C. (2003) p38 MAP kinases: key signaling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discov. 2, 717–726 [DOI] [PubMed] [Google Scholar]
- 39. Mayer R. J., Callahan J. F. (2006) p38 MAP kinase inhibitors: a future therapy for inflammatory diseases. Drug Discov. Today Ther. Strateg. 3, 49–54 [Google Scholar]
- 40. Shembade N., Harhaj E. W. (2012) Regulation of NF-κB signaling by the A20 deubiquitinase. Cell. Mol. Immunol. 2, 123–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sakurai H. (2012) Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol. Sci. 33, 522–530 [DOI] [PubMed] [Google Scholar]
- 42. Lowry J. L., Brovkovych V., Zhang Y., Skidgel R. A. (2013) Endothelial nitric-oxide synthase activation generates an inducible nitric-oxide synthase-like output of nitric oxide in inflamed endothelium. J. Biol. Chem. 288, 4174–4193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huang Q., Lan F., Wang X., Yu Y., Ouyang X., Zheng F., Han J., Lin Y., Xie Y., Xie F., Liu W., Yang X., Wang H., Dong L., Wang L., Tan J. (2014) IL-1β-induced activation of p38 promotes metastasis in gastric adenocarcinoma via upregulation of AP-1/c-fos, MMP2 and MMP9. Mol. Cancer 13, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ferreiro I., Barragan M., Gubern A., Ballestar E., Joaquin M., Posas F. (2010) The p38 SAPK is recruited to chromatin via its interaction with transcription factors. J. Biol. Chem. 285, 31819–31828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. O'Donnell A., Odrowaz Z., Sharrocks A. D. (2012) Immediate-early gene activation by the MAPK pathways: what do and don't we know? Biochem. Soc. Trans. 40, 58–66 [DOI] [PubMed] [Google Scholar]
- 46. Ventura J. J., Tenbaum S., Perdiguero E., Huth M., Guerra C., Barbacid M., Pasparakis M., Nebreda A. R. (2007) p38α MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nat. Genet. 39, 750–758 [DOI] [PubMed] [Google Scholar]
- 47. Hui L., Bakiri L., Stepniak E., Wagner E. F. (2007) A suppressor of cell proliferation and tumorigenesis. Cell Cycle 6, 2429–2433 [DOI] [PubMed] [Google Scholar]
- 48. Aman J., van Bezu J., Damanafshan A., Huveneers S., Eringa E. C., Vogel S. M., Groeneveld A. B., Vonk Noordegraaf A., van Hinsbergh V. W., van Nieuw Amerongen G. P. (2012) Effective treatment of edema and endothelial barrier dysfunction with imatinib. Circulation 126, 2728–2738 [DOI] [PubMed] [Google Scholar]
- 49. Guma M., Stepniak D., Shaked H., Spehlmann M. E., Shenouda S., Cheroutre H., Vicente-Suarez I., Eckmann L., Kagnoff M. F., Karin M. (2011) Constitutive intestinal NF-κB does not trigger destructive inflammation unless accompanied by MAPK activation. J. Exp. Med. 208, 1889–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Eylenstein A., Schmidt S., Gu S., Yang W., Schmid E., Schmidt E. M., Alesutan I., Szteyn K., Regel I., Shumilina E., Lang F. (2012) Transcription factor NF-κB regulates expression of pore-forming Ca2+ channel unit, Orai1, and its activator, STIM1, to control Ca2+ entry and affect cellular functions. J. Biol. Chem. 287, 2719–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wagner E. F., Nebreda A. R. (2009) Signaling integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 9, 537–549 [DOI] [PubMed] [Google Scholar]