

Background: In embryonic development, PTK7 regulates orientation of cells in a tissue plane.
Results: PTK7 controls cellular protrusions and, as a result, directional cell motility and metastasis in fibrosarcoma HT1080 cells.
Conclusion: Both PTK7 expression and proteolysis contribute to efficient cell motility and metastasis.
Significance: PTK7 is a potential diagnostic biomarker with predictive value and a promising drug target in cancer.
Keywords: Cancer, Cell Polarity, Matrix Metalloproteinase (MMP), Metastasis, Migration, Protein Kinase
Abstract
It is well established that widely expressed PTK7 is essential for vertebrate tissue morphogenesis. In cancer, the functionality of PTK7 is selectively regulated by membrane type-1 matrix metalloproteinase (MT1-MMP), ADAMs (a disintegrin domain and metalloproteinases), and γ-secretase proteolysis. Here, we established that the full-length membrane PTK7, its Chuzhoi mutant with the two functional MT1-MMP cleavage sites, and its L622D mutant with the single inactivated MT1-MMP cleavage site differentially regulate cell motility in a two-dimensional versus three-dimensional environment. We also demonstrated that in polarized cancer cells, the levels of PTK7 expression and proteolysis were directly linked to the structure and kinetics of cell protrusions, including lamellipodia and invadopodia. In the functionally relevant and widely accepted animal models of metastasis, mouse and chick embryo models, both the overexpression and knock-out of PTK7 in HT1080 cells abrogated metastatic dissemination. Our analysis of human tissue specimens confirmed intensive proteolysis of PTK7 in colorectal cancer tumors, but not in matching normal tissue. Our results provide convincing evidence that both PTK7 expression and proteolysis, rather than the level of the cellular full-length PTK7 alone, contribute to efficient directional cell motility and metastasis in cancer.
Introduction
Motile processes are essential for a plethora of physiological and pathological situations ranging from embryo development to tumor dissemination and metastasis. Migrating cells need to arrange both leading edge-rear end polarity consisting of cytoskeleton-driven extension and trafficking of necessary membrane extension and adhesion components (1–5). Widely expressed protein-tyrosine kinase 7 (PTK7)3 (also known as colon carcinoma kinase-4, CCK-4) is a member of the receptor protein tyrosine kinase family. In this family of kinases, the kinase-like domain lacks at least one of the usually conserved catalytic residues (6–8). In vertebrate tissue morphogenesis, PTK7 regulates the canonical and non-canonical Wnt pathways and orientation of cells in a tissue plane (planar cell polarity) (7–11). The full-length membrane PTK7 consists of seven extracellular immunoglobulin-like (Ig) domains, the transmembrane and juxtamembrane regions, and a catalytically inert cytoplasmic tyrosine kinase domain (12). Depending on the cell type, PTK7 was observed to bind to multiple proteins, including plexins and semaphorins (13, 14), RACK1 and PKCδ1 (15, 16), β-catenin (9), Wnt3a and Wnt8 (11, 17), LRP6 (6), and Src (18).
Recently, we and others (19–22) established that PTK7 is a subject of proteolysis in species ranging from zebrafish to humans. In humans, stepwise proteolysis involves the activities of MT1-MMP, the members of the ADAM/ADAMTS family, and γ-secretase. Proteolysis of the full-length PTK7 results in the soluble N-terminal fragment and the respective C-terminal, membrane-associated, and intracellular proteolytic fragments (21, 22). These proteolytic events reverse the anti-invasive effect of the full-length membrane PTK7.
Because of extensive proteolysis and fragmentation of PTK7 leading to its multiple proteolytic fragments with contrasting functions, the current measurements of PTK7 in human cancer tissue specimens remain controversial. These measurements are predominantly based on semi-quantitative immunohistochemistry using a single PTK7 antibody and on measurements of PTK7 mRNA levels in tumor samples. As a result, the literature suggests that the expression of PTK7 is up-regulated in colon (23, 24), gastric (25) and lung cancers, and acute myeloid leukemia (26, 27) and down-regulated in melanomas (28, 29) and breast carcinomas (30, 31), some of which exhibit deletions of the human chromosome 6p containing the genomic PTK7 gene. The most recent observations by others suggest the fundamental function and the high prognostic value of PTK7 in the most aggressive, metastatic cancer types, including breast, lung, and colorectal cancer and intrahepatic cholangiocarcinoma (32–35).
To shed more light on the function of PTK7 in cancer, we performed a series of cell migration, invasion, and metastasis assays in cultured cells and mouse and chick embryo tumor models. To corroborate our observations, we also analyzed the status of MT1-MMP and PTK7 in clinical cancer samples. Our results established that PTK7 plays a highly significant role in metastasis. We also suggest that the MT1-MMP/PTK7 axis, rather than PTK7 alone, is a potential biomarker and a promising drug target in malignancies in which the MT1-MMP activity and PTK7 are co-expressed.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
General reagents were purchased from Sigma-Aldrich unless indicated otherwise. A goat polyclonal AF4499 antibody against the N-terminal 31–199 PTK7 portion, a mouse monoclonal 3G4 antibody to the MT1-MMP catalytic domain, a rabbit polyclonal α-tubulin antibody, a rabbit polyclonal antibody to the trans-Golgi network integral membrane protein TGN46, and phalloidin conjugated with Alexa Fluor 594 were from R&D Systems, EMD Millipore, Cell Signaling Technology, Abcam, and Invitrogen, respectively. Alexa Fluor 488-conjugated species-specific secondary antibodies were from Invitrogen. A rabbit polyclonal antibody against the C-terminal portion of PTK7 was from Cell Signaling Technology.
Cells, Cloning, and Mutagenesis
Human fibrosarcoma HT1080 cells (HT1080 cells) were from ATCC. HT1080 cells transfected with the full-length 1–1070 PTK7 construct tagged with the C-terminal FLAG tag (PTK7 cells), the MT1-MMP-resistant L622D mutant in which the MT1-MMP cleavage site is inactivated by mutation, and the Chz mutant with the insertion of Ala-Asn-Pro509 (Chz cells) were described earlier (19–21, 36). The Ala-Asn-Pro509 insertion generated an additional MT1-MMP cleavage site (Pro-Glu-Lys↓Leu512) in the Chz mutant (19). Cells with the transcriptional silencing of MT1-MMP were obtained using the shRNA construct (shMT1 cells) (21, 37). shMT1 cells were transfected with the Chz construct using Lipofectamine LTX (Invitrogen), and the clones were then selected in the presence of G418 (200 μg/ml) to generate the shMT1-Chz cells. To facilitate their identification in vivo, HT1080 and PTK7 cells were transfected with the DsRed2 retroviral red fluorescent protein (RFP) (Origene; pRFP-C-RS; catalogue no. TF200451) (38) and selected using a cell sorter to isolate HT1080-RFP and PTK7-RFP cells, respectively. The 29-mer shRNA construct was used to silence the endogenous PTK7 in HT1080 cells. Following transfection of the cells using the visual reporter vector, the RFP-positive cells were selected using a cell sorter. Selected cells were then grown in the presence of puromycin (2 μg/ml). Western blotting with the goat polyclonal PTK7 antibody was used to select the silenced clones. Three individual clones with the silenced PTK7 were pooled together to generate shPTK7 cells.
Cell and Tissue Lysates
Cells were grown in a 10-cm tissue culture dish (BD Biosciences) to reach ∼80% confluence and then lysed, using 0.5 ml of 20 mm Tris-HCl, pH 7.4, containing 1% deoxycholate, 1% octylphenoxypolyethoxyethanol, 150 mm NaCl, a protease inhibitor mixture III, 1 mm phenylmethylsulfonyl fluoride, and 10 mm EDTA. Following centrifugation, the protein concentration in the supernatants was adjusted to 1.5 mg/ml. Human colon normal and tumor tissue lysates were prepared by Protein Biotechnologies. To assure the high quality of resected samples, tumor and matching normal tissue specimens were flash frozen to −120 °C in 5–10 min post-resection. Tissue samples were lysed in radioimmune precipitation assay buffer (PBS, pH 7.4, 1 mm EDTA, 0.25% sodium deoxycholate, 1 mm Na3VO4, 1 μg/ml of aprotinin, pepstatin-A, and leupeptin each, 1 mm NaF, 0.1% SDS, and 1 mm phenylmethylsulfonyl fluoride). Following centrifugation, protein concentration in the supernatant fractions was adjusted to 1 mg/ml. The extract aliquots (30 μg total protein each) were analyzed by Western blotting with the specific primary antibodies followed by horseradish peroxidase-conjugated species-specific secondary antibody (Fitzgerald Industries) and the SuperSignal West Dura extended duration substrate (Thermo Fisher Scientific). The chemiluminescent signal was acquired using the HyBlot CL autoradiography film (Denville).
Cell Surface Biotinylation
Cells were surface-biotinylated using EZ-Link sulfosuccinimidyl 2-(biotinamido)-ethyl-1,3-dithiopropionate (0.1 mg/ml in PBS; 1 h; 4 °C). Cells were lysed as above, and biotinylated proteins were precipitated from the supernatant samples using streptavidin-agarose beads. The captured material was eluted using 2× SDS sample loading buffer (125 mm Tris-HCl, pH 6.8, 4% SDS, 0.005% bromphenol blue, 20% glycerol, and 20 mm DTT).
Detection of the Soluble PTK7 Ectodomain in the Medium Samples
Cells were grown to reach a 60–90% confluence in wells of a 12-well tissue culture plate (Becton Dickinson). Cells were washed with and then transferred for 16 h to fresh serum-free DMEM (0.5 ml/well). The medium aliquots were then withdrawn and centrifuged to remove cell debris. The supernatant was concentrated until dryness using a SpeedVac. To remove salt, the pellet was washed using acetone, dissolved in 1% SDS and centrifuged to remove insoluble material (14,000 rpm; 10 min). The supernatant aliquots were analyzed by Western blotting using the PTK7 antibody to the N-terminal PTK7 sequence.
Scratch (Wound Healing) Assay
Confluent cells in wells of a 24-well culture plates (BD Biosciences) were scratched using a 0.2-ml pipette tip and incubated 8 h at 37 °C in a humidified chamber. Cells were then fixed using 4% paraformaldehyde and observed using a microscope (4× objective), and three images/well in three wells were recorded and digitized for each condition. The wound area was quantified using ImageJ software and presented as percentage relative to HT1080 cells (= 100%).
Invasion Assay in Three-dimensional Collagen
Cells (1 × 105/well) in serum-free DMEM (80 μl) were mixed with type I collagen (20 μl, 3 mg/ml, BD Biosciences). Aliquots (20 μl each) were then placed in wells of a 12-well plate. The plates were incubated for 30 min at 37 °C to allow the collagen gel to polymerize. Next, type I collagen (50 μl) was mixed with DMEM, containing 10% FBS as chemoattactant, and the mixture (200 μl) was added to each well. Following polymerization of the FBS-containing collagen layer (∼60 min), serum-free DMEM (0.5 ml) was added to each well, and incubation was continued for an additional 72 h. The cells were stained using 0.2% crystal violet/20% methanol (0.5 ml; 1 h). In the next 24 h, the excess dye was removed by water washes. Cell images were recorded using a digital camera. The invasion area was measured using ImageJ software and presented as percentage relative to HT1080 cells (= 100%). Each sample was analyzed in triplicate.
Transwell Invasion Assay
The assay was performed in wells of a 24-well Transwell plate with an 8-μm pore membrane (21). The insert membranes were coated with type I collagen (30 μg/well, 3 mg/ml, BD Biosciences). Cells (1 × 105/well) in serum-free DMEM (0.1 ml) were placed into the upper chamber. DMEM-10% FBS (chemoattractant, 0.6 ml) was placed in the lower chamber. Serum-free DMEM (0.6 ml) was used as a control. Cells were allowed to invade for 3.5 h. The cells were stained for 10 min using 0.2% crystal violet/20% methanol (0.3 ml). The cells on the upper membrane surface were removed with a cotton swab. The dye from the cells that migrated onto the lower surface of the membrane was extracted with 1% SDS (0.25 ml). Dye absorbance was measured at A570 nm using a plate reader. Cell invasion is presented as percentage relative to HT1080 cells (= 100%). Each sample was analyzed in triplicate.
Live Cell Imaging
Cells (1 × 103 cells/well) were grown in wells of a 96-well glass-bottomed plate (PerkinElmer Life Science). Cell images were acquired at 5-min intervals for 8 h using an inverted fully automated, incubated Nikon TE2000 Perfect Focus System epi-fluorescence microscope (10 × 0.3 numerical aperture objective) and an Orca II charge-coupled device camera. The cell tracking was performed using MetaMorph software (Molecular Devices), and the velocity/directionality data were analyzed by Excel. At least 100 cells were quantified for each condition. To measure lamellipodia dynamics and directionality, the images were acquired at 1-min intervals using a 20 × 0.75 numerical aperture objective. The images were analyzed using MetaMorph software.
Immunofluorescence
Cells grown on a microscope coverglass (Fisher) were fixed using 4% paraformaldehyde, permeabilized using 0.1% Triton X-100, and blocked in 1% casein. Cells were stained using the TGN46 primary antibody (1:1,000 dilution) for 16 h at 4 °C followed by Alexa Fluor 488-conjugated anti-rabbit secondary antibody (1:500 dilution). The Alexa Fluor 594-conjugated phalloidin (1:500 dilution) was used to visualize the actin cytoskeleton. The specimens were mounted in the Vectashield mounting medium with DAPI (Vector Laboratories). Images were acquired using an Olympus BX51 fluorescence microscope equipped with a MagnaFire digital camera and MagnaFire software (version 2.1C, Olympus).
In Situ Gelatin Zymography Using FITC-gelatin
Wells of a 96-well plate were coated with 50 μl of FITC-gelatin (100 μg/ml) for 2 h at 37 °C. The excess gelatin was removed by washes using prewarmed DMEM. Cells (1 × 103/well) in DMEM-10% FBS were added to the wells and incubated for 72 h at 37 °C. The cells were fixed with 4% paraformaldehyde for 10 min. The Alexa Fluor 594-conjugated phalloidin (Molecular Probes; 1:500 dilution) was used to visualize the actin cytoskeleton. The specimens were then mounted, and the images were acquired as above. The degraded FITC-gelatin area (10 images per condition) was quantified using ImageJ software (39).
Tumor Growth and Metastasis Model in Mice
Athymic nu/nu mice were kept in a barrier facility under HEPA filtration and fed with an autoclaved laboratory rodent diet. All animal studies were conducted in accordance with the procedures outlined in the NIH Guide for the Care and Use of Laboratory Animals under assurance number A3873-1. Four-week-old mice (five mice per group) were used. For the tumor growth experiments, the HT1080, PTK7, and L622D cells (1 × 105/animal) were injected subcutaneously. Three weeks after cell injection, the mice were anesthetized using a ketamine mixture (10 μl of ketamine-HCl, 7.6 μl of xylazine, 2.4 μl of acepromazine maleate, and 10 μl of H2O) via s.c. injection, and the tumors were snap-frozen immediately after extraction. Tissue lysates were prepared using radioimmune precipitation assay buffer (PBS, pH 7.4, 1 mm EDTA, 0.25% sodium deoxycholate, 1 mm Na3VO4, 1 μg/ml of aprotinin, pepstatin-A, and leupeptin each, 1 mm NaF, 0.1% SDS, and 1 mm phenylmethylsulfonyl fluoride).
For the metastasis model, the RFP-labeled HT1080, PTK7, and shPTK7 cells (1 × 105/animal, five mice per group, two independent experiments) were injected via the tail vein. The mice were examined daily for the signs of sickness, weight loss, and unusual behavior. At 11 weeks, if there were signs of sickness, mice were anesthetized using a ketamine mixture (10 μl of ketamine-HCl, 7.6 μl of xylazine, 2.4 μl of acepromazine maleate, and 10 μl of H2O) via s.c. injection and then subjected post-autopsy to the external fluorescent imaging of the lung from the ventral view. The OV100 Small Animal Imaging System (Olympus) was used to acquire fluorescence images of RFP-labeled metastases (38). The lungs were then surgically removed and weighted using analytical balances (Sartorius). Kaplan-Meier analysis was used to determine mouse survival.
Metastasis Model in Chick Embryos
Chick embryos (University of Alberta Farms) were placed into sterile weigh boats with plastic lids at day 4 post-fertilization. To model metastatic lesion formation, on day 10 post-fertilization, cancer cells (50 μl of 2 × 106 cells/ml in PBS) were injected into a feeding arteriole in the chorioallantoic membrane (CAM) using a disposable micropipette syringe. To model the growth of primary tumor, the cells (1 × 103) were implanted into the CAM of ex ovo embryos using the glass microcapillary (40). To label the vasculature, lectin-FITC conjugate (50 μl; Vector Biolabs) was injected using a disposable micropipette syringe into a feeding arteriole of the CAM. Cancer cells were allowed to form primary tumors and metastatic lesions for 5 days after which embryos were used for intravital imaging. A 200–300-μm image stack was acquired every 5 min in 5-μm step size increments for 2 h. Zeiss upright microscope (Carl Zeiss) fitted with a temperature regulated enclosure (Plastics), a Ludl-XY stage controller (Ludl), a 405/491/561/646/750 nm diode laser switcher (Quorum Technologies), a Hamamatsu 512 × 512 EMCCD camera (Hamamatsu), and a full range of Zeiss microscope objectives were used for image acquisition. Volocity software (PerkinElmer) was used to control the microscope, field movement correction, and single cell tracking.
RESULTS
Proteolytic Processing of PTK7 in Vitro and in Vivo
We specifically employed highly invasive fibrosarcoma HT1080 cells in our experiments. These cells express low levels of endogenous PTK7 and high levels of both active MT1-MMP and ADAMs (21). These parameters were favorable to establishing the PTK7 effects in the cells, which overexpressed the wild-type or mutant PTK7 or the PTK7-silencing constructs (PTK7, L622D, Chuzhoi (Chz), and shPTK7 cells, respectively). In our study, we also used HT1080 and Chz cells with the transcriptionally silenced MT1-MMP (shMT1 and shMT1-Chz cells, respectively). We have determined that the PTK7 ectodomain was processed at two distinct cleavage sites (21). The first cleavage caused the release of the soluble N-terminal PTK7–65 ectodomain fragment and the generation of the matching cell-associated C-terminal PTK7–50 species was the result of MT1-MMP proteolysis at the Pro-Lys-Pro↓Leu622 site. The MT1-MMP-resistent L622D PTK7 mutant was generated (20). The second cleavage in the C-terminal portion of the PTK7 ectodomain was performed by ADAMs. This cleavage led to the release of the soluble N-terminal PTK7–70 fragment and generation of the matching cell-associated C-terminal PTK7–45 form. Our experiments also suggested that MT1-MMP and ADAM proteolysis of PTK7 was a prerequisite for the follow-on intramembrane γ-secretase cleavage of the C-terminal membrane portion of PTK7 (21). Chz mutation, an Ala-Asn-Pro tripeptide insertion in the junction region between the fifth and the sixth Ig-like domains of PTK7 (36), causes an inclusion of an additional MT1-MMP cleavage site (Pro-Glu-Lys↓Leu512) and aberrant proteolysis of Chz relative to PTK7 (19). Total and cell surface levels of PTK7 and MT1-MMP were assessed by Western blotting of the total cell lysate and the biotinylated membrane samples, respectively (Fig. 1A). We also analyzed the levels of the soluble PTK7 ectodomain in the medium. To measure the levels of the full-length PTK7 and its N-terminal soluble and C-terminal membrane-tethered proteolytic fragments, we used the antibodies raised against the N-terminal and C-terminal PTK7 portions.
FIGURE 1.

PTK7 expression in cultured cells, tumor xenografts in mice, and colorectal cancer clinical samples. A, structure of the full-length PTK7 and the Chz mutant. MT1-MMP, ADAM, and γ-secretase cleavage sites are shown by arrows. The original MT1-MMP cleavage site (Pro-Lys-Pro↓Leu622) is localized in the seventh Ig-like domain of PTK7. The Chz mutation (Ala-Asn-Pro, underlined) results in an additional MT1-MMP cleavage site (Pro-Glu-Lys↓Leu512; boxed) in the junction region between the fifth and the sixth Ig-like domains of PTK7. S, signal peptide; Ig, immunoglobulin-like domain; TM, transmembrane domain; JM, juxtamembrane region; KINASE, the inert kinase domain. Residue numbering is shown below the structure. B, shed, soluble (Shed), biotinylated cell surface (Cell surface), and total cell lysate (Total Cell lysate) samples were analyzed using Western blotting with PTK7, MT1-MMP, and α-tubulin (loading control) antibodies. C, the status of PTK7 in tumor xenografts. Protein lysates were prepared from HT1080, PTK7, and L622D tumor xenografts developed in immunodeficient nu/nu mice. The lysate aliquots (30 μg/lane total protein each) were then analyzed using Western blotting with PTK7 antibodies directed to the N-terminal ectodomain (N-end) and to the C-terminal cytoplasmic tail (C-end). D, the status of PTK7 and MT1-MMP in human colon cancer tissue specimens. Tissue lysates (Protein Biotechnologies) prepared from colorectal tumors, and matching normal colonic tissues were analyzed by Western blotting with the PTK7, MT1-MMP, E-cadherin, and α-tubulin (loading control) antibodies. The two PTK7 antibodies we used were directed to the N-terminal ectodomain (N-end) and the C-terminal cytoplasmic tail (C-end). Black and open arrowheads indicate the full-length PTK7 protein and its proteolytic fragments, respectively. The enzyme and the C-terminal self-proteolytic fragment of membrane-tethered MT1-MMP are shown by the connected arrows. WB, Western blotting.
According to our assays, MT1-MMP was expressed on the cell surface of all cell types we tested, except shMT1 cells in which MT1-MMP was silenced. The solubilized, released PTK7 ectodomain fragments were readily detected in the medium. N-terminal PTK7–65 was the main ectodomain fragment in PTK7 cells. In turn, the N-terminal PTK7–70 fragment alone was recorded in L622D cells, thus confirming the resistance of the L622D PTK7 mutant to MT1-MMP proteolysis. Because of the presence of the additional MT1-MMP cleavage site (Pro-Glu-Lys↓Leu512) in the Chz construct, the soluble, released Chz mutant ectodomain was additionally degraded in the medium and, as a result, only very minor levels of Chz were detected in the medium. MT1-MMP silencing inactivated MT1-MMP proteolysis of Chz and led to the release of the N-terminal PTK7–70 Chz fragment into the shMT1-Chz cell medium. The ectodomain fragments of PTK7 were not detected in the HT1080, shPTK7, and shMT1 medium samples (Fig. 1B).
Full-length membrane PTK7 was detected in the surface-biotinylated and total cell lysate samples. Our analysis confirmed the efficient PTK7 silencing in shPTK7 cells. Cell surface levels of Chz were low compared with the full-length PTK7. This observation is consistent with the efficient proteolysis of Chz by MT1-MMP (19). Because of MT1-MMP silencing, cell surface levels of Chz increased in shMT1-Chz cells. The C-terminal PTK7–50 fragment was identified in PTK7 and Chz, but not in both the L622D and shMT1-Chz cell lysate samples. In contrast with the C-terminal PTK7–50 fragment, the levels of the C-terminal PTK7–45 fragment were negligible. The levels of the endogenous full-length PTK7 that we detected in total cell lysate samples of shMT1 and shMT1-Chz cells were increased relative to HT1080 cells. Overall, our data suggest that MT1-MMP is a major sheddase of PTK7 in our HT1080 cell system.
Furthermore, we analyzed the PTK7 levels in tissue samples derived from tumor xenografts (Fig. 1C). HT1080, PTK7, and L622D cells were inoculated subcutaneously in immune-deficient nude mice. All injected animals developed tumors. We did not observe a significant difference in tumor growth rate in the HT1080, PTK7, and L622D animal groups (n = 5; data not shown). In 3 weeks post-cell injection, animals were euthanized, and tumor xenografts were excised and analyzed to determine the status of PTK7. In agreement with the results of our cell-based tests, the MT1-MMP cleavage-dependent C-terminal PTK7–50 fragment was detected in the PTK7 tumors but not in the L622D samples. In turn, the ADAM cleavage-dependent C-terminal PTK7–45 fragment was present in both the PTK7 and L622D xenografts. An additional ∼35-kDa fragment that was the result of the further proteolysis of the C-terminal PTK7 portion was also detected in the PTK7 and L622D samples.
To further support our results, we evaluated the status of PTK7 and MT1-MMP in colorectal cancer biopsy specimens and matching normal tissues (Fig. 1D). In our Western blotting assays, the N-terminal 65–75-kDa proteolytic fragments of PTK7 were readily detected in tumor samples. In turn, these fragments were absent in normal tissue. The N-terminal 65-kDa and 75-kDa fragments directly correlated with those which we observed in our cell-based systems and which were generated by MT1-MMP- and ADAM-dependent cleavage of the PTK7 ectodomain, respectively (21). The levels of the N-terminal, soluble fragments of PTK7 in the tumor samples correlated well with the respective C-terminal PTK7 membraneous fragments. Several additional low molecular weight fragments, which were caused by the further fragmentation of the C-terminal portion of PTK7, were also observed in tumor samples. Notably, the ratio of the cleaved PTK7 fragments to the full-length PTK7 was high in colorectal cancer samples compared with the xenografts.
As additional controls, we determined the levels of E-cadherin and MT1-MMP in cancer and normal tissue specimens. Three tumors were E-cadherin-positive, whereas one tumor expressed low levels of E-cadherin. The MT1-MMP enzyme levels were similar in tumor samples relative to normal tissue. However, tumors, but not normal tissue, exhibited significant levels of the ∼45-kDa autolytic MT1-MMP fragment. This MT1-MMP fragment is the indicative of the presence of the active, TIMP-2-free species of MT1-MMP in the cancer samples alone (41). Our results indicate that PTK7 is proteolytically degraded in colon cancer, rather than in normal tissue, and that the proteolytic fragments we detected in colorectal tumors are similar to those we observed in our cell-based tests and animal models.
PTK7 Differentially Regulates Cell Motility in the Two-dimensional versus Three-dimensional Environment
To determine the effect of PTK7 on the directional cell locomotion, we next used three assays: the wound healing or scratch assay (Fig. 2A), the invasion assay in a three-dimensional collagen matrix (Fig. 2B), and the invasion assay in transwells (Fig. 2C). In the wound healing assay, the silencing of PTK7 in shPTK7 cells increased by 20% (p < 0.05), rather than suppressed, their efficiency of locomotion relative to HT1080 cells. In turn, in the invasion assays in the three-dimensional collagen matrix and in transwells, shPTK7 cells were ∼50% less motile compared with HT1080 cells (p < 0.05).
FIGURE 2.

PTK7 in cell locomotion. A, wound healing scratch assay. Following scratching of an artificial gap in a confluent cell monolayer, cells were allowed to migrate for 8 h to close the gap. The borders of the original scratch (time = 0) are shown by dotted lines. The yellow lines highlight cell borders after 8 h. B, cell invasion in three-dimensional collagen. Cells were embedded in collagen spheres in serum-free medium. Collagen solution that contained 10% FBS as a chemoattractant was then overlaid on the spheres and then also allowed to form a gel layer. Cells were allowed to migrate from the spheres and invade the collagen layer for 72 h. The original spheres (time = 0) are encircled. Cell images are shown at the bottom. C, relative locomotion of HT1080 (= 100%), PTK7, Chz, shPTK7, shMT1, and shMT1-Chz cells in the wound healing scratch assay and the invasion and invasion assays in the three-dimensional (3D) collagen matrix and transwells. The assays were performed in triplicate. ± S.D. (error bars) did not exceed 10%; p < 0.05.
According to the wound healing assay, locomotion of PTK7 cells, which overexpressed the membrane full-length PTK7, was incapacitated. Similarly, relative to HT1080 cells PTK7 cells were 2–3-fold less motile in the invasion assays in both the three-dimensional collagen matrix and transwells (p < 0.05). The expression of the Chz mutant, however, repressed motility by ∼50% (p < 0.05) in the wound healing assay alone but increased the efficiency of cell locomotion in both three-dimensional collagen and transwell assays (by ∼30 and 45%, respectively; p < 0.05) compared with HT1080 cells. MT1-MMP silencing in shMT1-Chz cells completely blocked cell locomotion in all three motility assays, whereas MT1-MMP silencing alone in shMT1 cells had no effect on cell motility in the wound healing assay but repressed cell locomotion by >50% (p < 0.05) in the three-dimensional collagen matrix and in transwells.
PTK7 Regulates Lamellipodia Protrusions
To establish the effect of PTK7 on directional cell motility in more detail, we employed live imaging of migrating cells (Fig. 3). For these purposes, we used a fully automated epi-fluorescence microscope to acquire images of HT1080, PTK7, Chz, shPTK7, and shMT1-Chz cells. Images were acquired every 5 min for 8 h. These time-lapse videos were used to track cells (Fig. 3A) and reconstruct their migration behavior. To assess directionality and persistence of migration, we plotted cell track coordinates (Fig. 3E) and quantified the number of cells that traveled significant distances from their original position. We determined that the number of motile cells that traveled >40 microns from their original position (time = 0) was 4.6% in HT1080 cells, whereas only 1.5% and 2.3% of PTK7 and shPTK7 cells were migratory, respectively (Fig. 3). On the contrary, 22.3% of Chz cells migrated over a 40-micron distance.
FIGURE 3.

Tracking and morphometric analysis of the cells. A, images were taken every 5 min for 8 h (10 × 0.3 numerical aperture objective) and analyzed with Metamorph software to reconstruct the tracks of the individual migrating cells (red lines). Cell positions at time = 0 and the reconstructed tracks are shown. B, lamellipodial dynamics in the cells. Images were acquired every 1 min for 1 h (20 × 0.75 numerical aperture objective), and the whole time lapse was then projected onto a single plane. Membrane areas with high lamellipodial activity appear dark in the merged images. Red shading outlines the areas with high lamellipodial activity. Note that in the polarized HT1080 and Chz cells lamellipodia represent a limited fraction of the cell perimeter, whereas in the non-polarized PTK7 and shMT1-Chz cells, lamellipodia occupy a significant portion of the cell perimeter. C, directionality of lamellipodial protrusion. Images were acquired as described in B and processed to show the outlines of the cells at time = 0 and at times = 20, 40, and 60 min. Red arrows point to the direction of migration. The arrow length and the numbers at the bottom panel indicate the migrated distance (based on nuclear position). D, labeling of HT1080, shPTK7, PTK7, Chz, and MT1-Chz cells using phalloidin (red, actin), DAPI (blue, nuclei), and the trans-Golgi network (green, TGN46 (TGN)). The nucleus-TGN46 axis is shown by the white lines drawn through the cell body. E, overlaid migration track coordinates (in microns) of HT1080, PTK7, shPTK7, and Chz cells. Images described in A were imported from Metamorph (100 cell tracks each). F, relative motility of HT1080, PTK7, shPTK7, and Chz cells. Migration tracks of individual HT1080, PTK7, shPTK7, and Chz cells were digitized and analyzed to calculate the percentage of cells with a migration distance >40 microns (measured from the cell origin; time = 0) relative to the total number of cells.
To analyze the reasons for different motility among these cells, we next focused on the lamellipodia, which behavior largely defines how cells migrate. The lamellipodium is a dynamic flattened cell protrusion formed at the leading edge of migrating cells that contains a branched dendritic network of actin filaments (42). To visualize lamellipodia dynamics in migrating cells, 1-min intervals were used between the images (Fig. 3, B and C). The polarized lamellipodia in HT1080 cells represented a small fraction of the cell circumference and were productive and directionally sustained in the course of our 1-h observation period, resulting in efficient cell locomotion. On the contrary, lamellipodia in shPTK7 cells exhibited a high frequency of protrusions and retractions and were less able to support cell migration. Both PTK7 and shMT1-Chz cells remained stationary and readily formed cell-cell contacts and multicellular clusters. These cells displayed actively ruffling non-polarized lamellipodia that occupied a significant portion of the cell perimeter and, as a result, were unsupportive of directional migration. According to our observations, Chz cells alone exhibited the polarized and persistent lamellipodia, which were confined to a small segment of the cell membrane.
Immunostaining of the cells using the TGN46 antibody (a trans-Golgi compartment marker) and phalloidin that binds the cellular actin cytoskeleton supported our above observations. Thus, the 4′,6-diamidino-2-phenylindole (DAPI)-stained nucleus, the trans-Golgi, and the lamellipodial actin immunoreactivity were well aligned in polarized HT1080 and Chz cells (Fig. 3). In turn, there was an evident misalignment among the nucleus, the trans-Golgi, and the lamellipodial actin in non-migratory PTK7 and shMT1-Chz cells.
PTK7 Regulates Invadopodia
Invadopodia are protrusions of the cell membrane that extend into the extracellular matrix and accumulate MMPs for matrix degradation (42). To visualize the proteolytic potential of invadopodia, we used the FITC-gelatin in situ degradation assay. After plating the cells on FITC-gelatin followed by a 72-h incubation, the actin cytoskeleton and nuclei were stained using phalloidin and DAPI, respectively (Fig. 4). Migratory polarized HT1080 and Chz cells efficiently degraded FITC-gelatin and, in addition, generated the well defined degraded gelatin tracks. On the contrary, PTK7 and L622D cells degraded gelatin less efficiently when compared with HT1080 and Chz cells and appeared as the non-motile multicellular clusters lacking the degraded gelatin tracks. The shMT1-Chz control cells did not degrade gelatin and appeared immobile.
FIGURE 4.
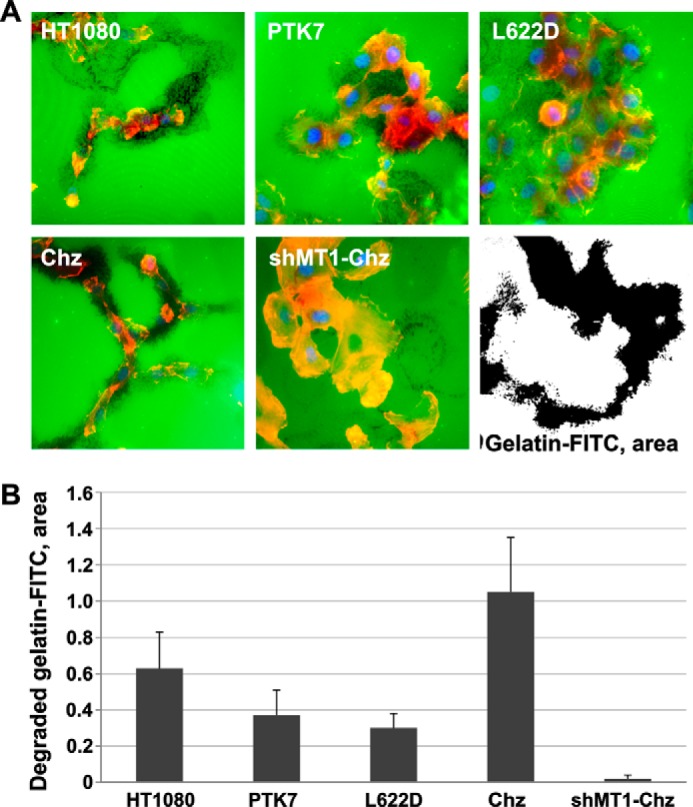
FITC-gelatin degradation assay. A, representative fluorescence images of cells grown on FITC-gelatin (green). Cells were plated on FITC-gelatin. At 72 h, cells were fixed and stained using phalloidin (red, actin) and DAPI (blue, nuclei). Black dots show the areas of the degraded gelatin. A representative black and white image shows the approach we used to highlight the degraded (black) FITC-gelatin area and quantify our data, which are presented in B. B, the degraded FITC-gelatin area was quantified with ImageJ (39).
PTK7 Affects Metastasis to the Lung in Mice
According to our current findings, PTK7 plays a significant role in cell migration and invasion. Because both efficient migration and invasion through the basement membrane and tissue are required for metastatic cancer, we next hypothesize that PTK7 affects metastasis of HT1080 cells to the lung. To test our hypothesis, we injected RFP-labeled HT1080, PTK7, and shPTK7 cells (1 × 105/animal; n = 5) in the tail vein of nude mice. When the symptoms of respiratory distress were observed, indicating the progression of the lung metastatic disease, fluorescence images of metastasis to the lung were obtained post-autopsy using the OV100 Small Animal Imaging System (Olympus). The RFP fluorescence was readily detected in metastatic lesions in mice injected with HT1080 cells, but not in mice, which received PTK7 or shPTK7 cells (Fig. 5). At 9 weeks post-cell injection, four mice of the HT1080 group were euthanized. All mice in the HT1080 group exhibited multiple metastases to the lung. In turn, five mice of the PTK7 and four mice of shPTK7 groups were alive at week 11 (Fig. 5B). One mouse from the shPTK7 group died on week 8, metastases-free. At the end of experiment at week 11, all surviving mice were euthanized, and their lungs were surgically removed, thoroughly analyzed, and weighed (Fig. 5C). There were no detectable metastases to the lungs of mice in the PTK7 and shPTK7 groups.
FIGURE 5.

PTK7 affects metastases to the lung in mice. A, representative fluorescence images of metastases to the lung. RFP-labeled HT1080, PTK7, and shPTK7 cells were injected via the tail vein of nude mice (n = 5). Animals were sacrificed and metastatic lesions (red-yellow) were recorded post-autopsy using fluorescence imaging. Metastases were observed only in the mice injected with HT1080 cells but not in mice injected with PTK7 and shPTK7 cells. The dashed line delineates the chest area. B, Kaplan-Maier survival curve. Mice received injections with the RFP-labeled HT1080, PTK7, and shPTK7 cells. C, lung weight post-autopsy.
PTK7 Affects Cell Invasion and Metastasis in the Chick Embryo Model
To analyze the role of PTK7 in cell invasion and metastasis in more detail, we used the chick embryo model (40). For these purposes, RFP-expressing HT1080, shPTK7, and L622D cells were injected into a feeding arteriole in the CAM. The intravital images of metastatic lesions were acquired at day 5 post-injection, every 5 min for 2 h using 5-μm step increments (Fig. 6A and supplemental Movie S1). HT1080 cells efficiently extravasated the blood vessels and then acquired the spindle-like, polarized morphology, exhibited an invasive behavior within the CAM tissue, and disseminated along the vasculature (Fig. 6, B and C), thus demonstrating the characteristics of metastatic cancer cells (Fig. 6, B and C). On the contrary, both shPTK7 and L622D cells were largely immotile, exhibited the flat, rounded morphology, and lacked membrane protrusions. These cells failed to disseminate and formed the small, compact colonies immediately outside the blood vessels.
FIGURE 6.

PTK7 regulates HT1080 cell invasion in the chick embryo model in vivo. A, representative images of the RFP-labeled HT1080, PTK7, and shPTK7 metastatic lesions on day 5 post-injection. RFP channel alone is shown for simplicity. Insets show the merged RFP and FITC images, in which FITC labels the vasculature. B, quantification of tumor cell tracks velocity in HT1080, shPTK7, and L622D lesions. C, quantification of cell displacement rate (migration productivity) in HT1080, shPTK7, and L622D lesions. D, representative images of HT1080, PTK7, and shPTK7-induced primary tumors at day 5 post-injection. The dashed line delineates invasive tumor front. Note the polarized, invasive HT1080 cells outside the primary tumor (white arrows). Scale bars, 30 μm.
To corroborate our results, we also implanted HT1080, shPTK7, and L622D cells into the CAM of ex ovo embryos. The resulting primary tumors were analyzed on day 5 post-implantation. The intravital images of these tumors suggested that HT1080 tumor cells disseminated into the stroma and were readily detectable outside the primary tumor explant. In contrast, no invasive cells were identified outside the shPTK7 and L622D primary tumor borders.
DISCUSSION
Our study is focused on understanding of how the pericellular proteolysis and cell polarity mechanisms converge in cancer cell migration. At its most basic level, polarity may be considered as the generation of asymmetry within a single cell, whether the result of the asymmetry is a directed movement that leads to cellular migration or to the relocalization of specific multicellular structures. Polarized cell locomotion correlates directly with the pericellular proteolysis and extracellular matrix remodeling both of which involve the action of MMPs and especially proinvasive MT1-MMP (43, 44). Our earlier results established that PTK7 is a major cleavage target of MT1-MMP in the plasma membrane (20). MT1-MMP directly cleaves the PKP621↓LI sequence localized in the exposed region of the seventh Ig-like domain of PTK7 generating, as a result, the N-terminal, soluble PTK7 species, which are likely to interact with membrane PTK7 in a dominant-negative fashion. Thus, soluble PTK7 generated by MT1-MMP activity induces RhoA signaling and induces actin cytoskeleton contractility. On the contrary, the full-length PTK7 down-regulates myosin light chain phosphorylation and, as a result, reduces actin cytoskeleton contractility (20).
Based on our previous results, we have hypothesized that MT1-MMP deactivates the anti-invasive function of the full-length PTK7 (19–21). However, in-depth understanding of these effects was lacking in the earlier studies by us and others (9, 17, 20, 22). We now demonstrate that the MT1-MMP/PTK7 axis is essential for the polarized cell motility through the regulation of cell protrusions, including lamellipodia and invadopodia. Lamellipodia dysfunction and, consequently, inefficient migration were observed in cells with both decreased and increased levels of PTK7. Thus, cells with PTK7 silencing displayed random non-productive lamellipodial activity and, as a result, they were unable to migrate efficiently. In the presence of excess of the full-length PTK7, abnormally active, but non-polarized, non-productive lamellipodia of large size occupied a significant portion of cells. The full-length PTK7 also suppressed invadopodia activity as evidenced by reduced gelatin degradation. In agreement, PTK7 cells were poorly migratory in the three-dimensional collagen matrix and transwell assays.
Additional evidence was obtained in our studies employing cells expressing the Chz PTK7 mutant alone or in combination with an MT1-MMP knock-out. These results suggested a primary role of MT1-MMP proteolysis of PTK7 in the regulation of cell protrusion dynamics and polarity. Thus, PTK7 proteolysis was essential for the sustained, productive, and well oriented lamellipodia and cell polarity. Notably, MT1-MMP is also localized in lamellipodia of migrating cells and in invadopodia of invading cells (45–47). MT1-MMP cleaves ECM proteins, initiates the activation pathway of soluble MMPs, and controls the functionality of cell adhesion and signaling receptors (48–53). The importance of receptor shedding by MT1-MMP, including CD44, integrins, and transglutaminase is well documented (54–56).
Consistent with our findings in cultured cells, both the PTK7 silencing and overexpression fully blocked the ability of HT1080 cells to metastasize to the lung in nude mice and diminished productive cell invasion in the chick embryo model. Although the shPTK7 and L622D cells were able to form metastatic lesions these cells exhibited non-oriented round cell morphology and failed to invade and disseminate the stroma. Correspondingly, shPTK7 and L622D also failed to invade out of the primary tumors. It is likely that the PTK7 expression levels and a certain ratio of cleaved to full-length PTK7 promote the directed extension of invasive membrane protrusions that control invasive cell behavior and metastasis.
Furthermore, the proteolysis of PTK7 was also readily observed in the human tissue specimens from colon cancer patients. The analysis of clinical samples suggested that the ratio of the full-length PTK7 to the PTK7 proteolytic fragments, rather than the total levels of cellular PTK7 alone, is a promising diagnostic biomarker with predictive value. While our manuscript was under preparation and review, several additional studies directly link the PTK7 functionality with cancer progression and metastasis in prostate, breast, esophageal, lung, and hepatic cancer types and, potentially, in many other cancers (32, 34, 35, 57–59). As a result, it is now highly likely that the PTK7 plays a global fundamental role in malignancy. Overall, our results and the most recent findings by others imply that the PTK7 function is a potential drug target and a novel approach to control metastasis in patients who co-express tumor PTK7 and metalloprotease activity.
Together, our results indicate that a tightly controlled limited proteolysis of PTK7 regulates the directionality and dynamics of cell membrane protrusions in a way that is critical for directed cell motility, invasion, and metastasis.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants R01CA83017, R01CA157328, and R01DE022757 (to A. Y. S.) and R03DA033979 (to V. S. G.).

This article contains supplemental Movie S1.
- PTK7
- protein-tyrosine kinase 7
- CAM
- chorioallantoic membrane
- ADAM
- a disintegrin domain and metalloproteinase
- Chz
- Chuzhoi mutant of PTK7
- MT1-MMP
- membrane type-1 matrix metalloproteinase
- RFP
- red fluorescent protein.
REFERENCES
- 1. Bisi S., Disanza A., Malinverno C., Frittoli E., Palamidessi A., Scita G. (2013) Membrane and actin dynamics interplay at lamellipodia leading edge. Curr. Opin. Cell Biol. 25, 565–573 [DOI] [PubMed] [Google Scholar]
- 2. Rauzi M., Lenne P. F., Lecuit T. (2010) Planar polarized actomyosin contractile flows control epithelial junction remodelling. Nature 468, 1110–1114 [DOI] [PubMed] [Google Scholar]
- 3. Muñoz-Soriano V., Belacortu Y., Paricio N. (2012) Planar cell polarity signaling in collective cell movements during morphogenesis and disease. Curr. Genomics 13, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang Y. (2009) Wnt/Planar cell polarity signaling: a new paradigm for cancer therapy. Mol. Cancer Ther. 8, 2103–2109 [DOI] [PubMed] [Google Scholar]
- 5. Jessen J. R. (2009) Noncanonical Wnt signaling in tumor progression and metastasis. Zebrafish 6, 21–28 [DOI] [PubMed] [Google Scholar]
- 6. Bin-Nun N., Lichtig H., Malyarova A., Levy M., Elias S., Frank D. (2014) PTK7 modulates Wnt signaling activity via LRP6. Development 141, 410–421 [DOI] [PubMed] [Google Scholar]
- 7. Lu X., Borchers A. G., Jolicoeur C., Rayburn H., Baker J. C., Tessier-Lavigne M. (2004) PTK7/CCK-4 is a novel regulator of planar cell polarity in vertebrates. Nature 430, 93–98 [DOI] [PubMed] [Google Scholar]
- 8. Yen W. W., Williams M., Periasamy A., Conaway M., Burdsal C., Keller R., Lu X., Sutherland A. (2009) PTK7 is essential for polarized cell motility and convergent extension during mouse gastrulation. Development 136, 2039–2048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Puppo F., Thomé V., Lhoumeau A. C., Cibois M., Gangar A., Lembo F., Belotti E., Marchetto S., Lécine P., Prébet T., Sebbagh M., Shin W. S., Lee S. T., Kodjabachian L., Borg J. P. (2011) Protein tyrosine kinase 7 has a conserved role in Wnt/β-catenin canonical signalling. EMBO Rep. 12, 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Winberg M. L., Tamagnone L., Bai J., Comoglio P. M., Montell D., Goodman C. S. (2001) The transmembrane protein Off-track associates with Plexins and functions downstream of Semaphorin signaling during axon guidance. Neuron 32, 53–62 [DOI] [PubMed] [Google Scholar]
- 11. Peradziryi H., Kaplan N. A., Podleschny M., Liu X., Wehner P., Borchers A., Tolwinski N. S. (2011) PTK7/Otk interacts with Wnts and inhibits canonical Wnt signalling. EMBO J. 30, 3729–3740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jung J. W., Ji A. R., Lee J., Kim U. J., Lee S. T. (2002) Organization of the human PTK7 gene encoding a receptor protein tyrosine kinase-like molecule and alternative splicing of its mRNA. Biochim. Biophys. Acta 1579, 153–163 [DOI] [PubMed] [Google Scholar]
- 13. Whitford K. L., Ghosh A. (2001) Plexin signaling via off-track and rho family GTPases. Neuron 32, 1–3 [DOI] [PubMed] [Google Scholar]
- 14. Toyofuku T., Zhang H., Kumanogoh A., Takegahara N., Suto F., Kamei J., Aoki K., Yabuki M., Hori M., Fujisawa H., Kikutani H. (2004) Dual roles of Sema6D in cardiac morphogenesis through region-specific association of its receptor, Plexin-A1, with off-track and vascular endothelial growth factor receptor type 2. Genes Dev. 18, 435–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shnitsar I., Borchers A. (2008) PTK7 recruits dsh to regulate neural crest migration. Development 135, 4015–4024 [DOI] [PubMed] [Google Scholar]
- 16. Wehner P., Shnitsar I., Urlaub H., Borchers A. (2011) RACK1 is a novel interaction partner of PTK7 that is required for neural tube closure. Development 138, 1321–1327 [DOI] [PubMed] [Google Scholar]
- 17. Lhoumeau A. C., Puppo F., Prébet T., Kodjabachian L., Borg J. P. (2011) PTK7: a cell polarity receptor with multiple facets. Cell Cycle 10, 1233–1236 [DOI] [PubMed] [Google Scholar]
- 18. Andreeva A., Lee J., Lohia M., Wu X., Macara I. G., Lu X. (2014) PTK7-Src Signaling at Epithelial Cell Contacts Mediates Spatial Organization of Actomyosin and Planar Cell Polarity. Dev. Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Golubkov V. S., Aleshin A. E., Strongin A. Y. (2011) Potential relation of aberrant proteolysis of human protein tyrosine kinase 7 (PTK7) chuzhoi by membrane type 1 matrix metalloproteinase (MT1-MMP) to congenital defects. J. Biol. Chem. 286, 20970–20976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Golubkov V. S., Chekanov A. V., Cieplak P., Aleshin A. E., Chernov A. V., Zhu W., Radichev I. A., Zhang D., Dong P. D., Strongin A. Y. (2010) The Wnt/planar cell polarity protein-tyrosine kinase-7 (PTK7) is a highly efficient proteolytic target of membrane type-1 matrix metalloproteinase: implications in cancer and embryogenesis. J. Biol. Chem. 285, 35740–35749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Golubkov V. S., Strongin A. Y. (2012) Insights into ectodomain shedding and processing of protein-tyrosine pseudokinase 7 (PTK7). J. Biol. Chem. 287, 42009–42018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Na H. W., Shin W. S., Ludwig A., Lee S. T. (2012) The cytosolic domain of protein-tyrosine kinase 7 (PTK7), generated from sequential cleavage by a disintegrin and metalloprotease 17 (ADAM17) and γ-secretase, enhances cell proliferation and migration in colon cancer cells. J. Biol. Chem. 287, 25001–25009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mossie K., Jallal B., Alves F., Sures I., Plowman G. D., Ullrich A. (1995) Colon carcinoma kinase-4 defines a new subclass of the receptor tyrosine kinase family. Oncogene 11, 2179–2184 [PubMed] [Google Scholar]
- 24. Saha S., Bardelli A., Buckhaults P., Velculescu V. E., Rago C., St Croix B., Romans K. E., Choti M. A., Lengauer C., Kinzler K. W., Vogelstein B. (2001) A phosphatase associated with metastasis of colorectal cancer. Science 294, 1343–1346 [DOI] [PubMed] [Google Scholar]
- 25. Gorringe K. L., Boussioutas A., Bowtell D. D. (2005) Novel regions of chromosomal amplification at 6p21, 5p13, and 12q14 in gastric cancer identified by array comparative genomic hybridization. Genes Chromosomes Cancer 42, 247–259 [DOI] [PubMed] [Google Scholar]
- 26. Müller-Tidow C., Schwäble J., Steffen B., Tidow N., Brandt B., Becker K., Schulze-Bahr E., Halfter H., Vogt U., Metzger R., Schneider P. M., Büchner T., Brandts C., Berdel W. E., Serve H. (2004) High-throughput analysis of genome-wide receptor tyrosine kinase expression in human cancers identifies potential novel drug targets. Clin. Cancer Res. 10, 1241–1249 [DOI] [PubMed] [Google Scholar]
- 27. Endoh H., Tomida S., Yatabe Y., Konishi H., Osada H., Tajima K., Kuwano H., Takahashi T., Mitsudomi T. (2004) Prognostic model of pulmonary adenocarcinoma by expression profiling of eight genes as determined by quantitative real-time reverse transcriptase polymerase chain reaction. J. Clin. Oncol. 22, 811–819 [DOI] [PubMed] [Google Scholar]
- 28. Easty D. J., Mitchell P. J., Patel K., Flørenes V. A., Spritz R. A., Bennett D. C. (1997) Loss of expression of receptor tyrosine kinase family genes PTK7 and SEK in metastatic melanoma. Int. J. Cancer 71, 1061–1065 [DOI] [PubMed] [Google Scholar]
- 29. Baudrier-Régnier A., Bodenant C., Proust F., Delangre T., Hemet J., Laquerrière A. (2000) An isochromosome 6p in a primary meningeal malignant melanoma. Cancer Genet. Cytogenet. 119, 80–82 [DOI] [PubMed] [Google Scholar]
- 30. Su Y. A., Yang J., Tao L., Nguyen H., He P. (2010) Undetectable and decreased expression of KIAA1949 (phostensin) encoded on chromosome 6p21.33 in human breast cancers revealed by transcriptome analysis. J. Cancer 1, 38–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Piao Z., Lee K. S., Kim H., Perucho M., Malkhosyan S. (2001) Identification of novel deletion regions on chromosome arms 2q and 6p in breast carcinomas by amplotype analysis. Genes Chromosomes Cancer 30, 113–122 [PubMed] [Google Scholar]
- 32. Chen R., Khatri P., Mazur P. K., Polin M., Zheng Y., Vaka D., Hoang C. D., Shrager J., Xu Y., Vicent S., Butte A. J., Sweet-Cordero E. A. (2014) A meta-analysis of lung cancer gene expression identifies PTK7 as a survival gene in lung adenocarcinoma. Cancer Res. 74, 2892–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fazilaty H., Mehdipour P. (2014) Genetics of breast cancer bone metastasis: a sequential multistep pattern. Clin. Exp. Metastasis 31, 595–612 [DOI] [PubMed] [Google Scholar]
- 34. Gärtner S., Gunesch A., Knyazeva T., Wolf P., Högel B., Eiermann W., Ullrich A., Knyazev P., Ataseven B. (2014) PTK 7 is a transforming gene and prognostic marker for breast cancer and nodal metastasis involvement. PLoS One 9, e84472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jin J., Ryu H. S., Lee K. B., Jang J. J. (2014) High expression of protein tyrosine kinase 7 significantly associates with invasiveness and poor prognosis in intrahepatic cholangiocarcinoma. PLoS One 9, e90247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paudyal A., Damrau C., Patterson V. L., Ermakov A., Formstone C., Lalanne Z., Wells S., Lu X., Norris D. P., Dean C. H., Henderson D. J., Murdoch J. N. (2010) The novel mouse mutant, chuzhoi, has disruption of Ptk7 protein and exhibits defects in neural tube, heart and lung development and abnormal planar cell polarity in the ear. BMC Dev. Biol. 10, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Golubkov V. S., Boyd S., Savinov A. Y., Chekanov A. V., Osterman A. L., Remacle A., Rozanov D. V., Doxsey S. J., Strongin A. Y. (2005) Membrane type-1 matrix metalloproteinase (MT1-MMP) exhibits an important intracellular cleavage function and causes chromosome instability. J. Biol. Chem. 280, 25079–25086 [DOI] [PubMed] [Google Scholar]
- 38. Kimura H., Hayashi K., Yamauchi K., Yamamoto N., Tsuchiya H., Tomita K., Kishimoto H., Bouvet M., Hoffman R. M. (2010) Real-time imaging of single cancer-cell dynamics of lung metastasis. J. Cell Biochem. 109, 58–64 [DOI] [PubMed] [Google Scholar]
- 39. Martin K. H., Hayes K. E., Walk E. L., Ammer A. G., Markwell S. M., Weed S. A. (2012) Quantitative measurement of invadopodia-mediated extracellular matrix proteolysis in single and multicellular contexts. JoVE, e4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zijlstra A., Lewis J., Degryse B., Stuhlmann H., Quigley J. P. (2008) The inhibition of tumor cell intravasation and subsequent metastasis via regulation of in vivo tumor cell motility by the tetraspanin CD151. Cancer Cell 13, 221–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lehti K., Valtanen H., Wickström S. A., Lohi J., Keski-Oja J. (2000) Regulation of membrane-type-1 matrix metalloproteinase activity by its cytoplasmic domain. J. Biol. Chem. 275, 15006–15013 [DOI] [PubMed] [Google Scholar]
- 42. Ridley A. J. (2011) Life at the leading edge. Cell 145, 1012–1022 [DOI] [PubMed] [Google Scholar]
- 43. Egeblad M., Werb Z. (2002) New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2, 161–174 [DOI] [PubMed] [Google Scholar]
- 44. Itoh Y., Seiki M. (2006) MT1-MMP: a potent modifier of pericellular microenvironment. J. Cell Physiol. 206, 1–8 [DOI] [PubMed] [Google Scholar]
- 45. Monteiro P., Rossé C., Castro-Castro A., Irondelle M., Lagoutte E., Paul-Gilloteaux P., Desnos C., Formstecher E., Darchen F., Perrais D., Gautreau A., Hertzog M., Chavrier P. (2013) Endosomal WASH and exocyst complexes control exocytosis of MT1-MMP at invadopodia. J. Cell Biol. 203, 1063–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Artym V. V., Zhang Y., Seillier-Moiseiwitsch F., Yamada K. M., Mueller S. C. (2006) Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function. Cancer Res. 66, 3034–3043 [DOI] [PubMed] [Google Scholar]
- 47. Yu X., Zech T., McDonald L., Gonzalez E. G., Li A., Macpherson I., Schwarz J. P., Spence H., Futó K., Timpson P., Nixon C., Ma Y., Anton I. M., Visegrády B., Insall R. H., Oien K., Blyth K., Norman J. C., Machesky L. M. (2012) N-WASP coordinates the delivery and F-actin-mediated capture of MT1-MMP at invasive pseudopods. J. Cell Biol. 199, 527–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Morrison C. J., Butler G. S., Rodríguez D., Overall C. M. (2009) Matrix metalloproteinase proteomics: substrates, targets, and therapy. Curr. Opin. Cell Biol. 21, 645–653 [DOI] [PubMed] [Google Scholar]
- 49. Rodríguez D., Morrison C. J., Overall C. M. (2010) Matrix metalloproteinases: What they do not do. New substrates and biological roles identified by murine models and proteomics. Biochim. Biophys. Acta 1803, 39–54 [DOI] [PubMed] [Google Scholar]
- 50. Hotary K., Li X. Y., Allen E., Stevens S. L., Weiss S. J. (2006) A cancer cell metalloprotease triad regulates the basement membrane transmigration program. Genes Dev. 20, 2673–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hotary K. B., Allen E. D., Brooks P. C., Datta N. S., Long M. W., Weiss S. J. (2003) Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell 114, 33–45 [DOI] [PubMed] [Google Scholar]
- 52. Hotary K., Allen E., Punturieri A., Yana I., Weiss S. J. (2000) Regulation of cell invasion and morphogenesis in a three-dimensional type I collagen matrix by membrane-type matrix metalloproteinases 1, 2, and 3. J. Cell Biol. 149, 1309–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sabeh F., Ota I., Holmbeck K., Birkedal-Hansen H., Soloway P., Balbin M., Lopez-Otin C., Shapiro S., Inada M., Krane S., Allen E., Chung D., Weiss S. J. (2004) Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J. Cell Biol. 167, 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Belkin A. M., Akimov S. S., Zaritskaya L. S., Ratnikov B. I., Deryugina E. I., Strongin A. Y. (2001) Matrix-dependent proteolysis of surface transglutaminase by membrane-type metalloproteinase regulates cancer cell adhesion and locomotion. J. Biol. Chem. 276, 18415–18422 [DOI] [PubMed] [Google Scholar]
- 55. Deryugina E. I., Bourdon M. A., Jungwirth K., Smith J. W., Strongin A. Y. (2000) Functional activation of integrin alpha V beta 3 in tumor cells expressing membrane-type 1 matrix metalloproteinase. Int. J. Cancer 86, 15–23 [DOI] [PubMed] [Google Scholar]
- 56. Kajita M., Itoh Y., Chiba T., Mori H., Okada A., Kinoh H., Seiki M. (2001) Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J. Cell Biol. 153, 893–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kim J. H., Kwon J., Lee H. W., Kang M. C., Yoon H. J., Lee S. T., Park J. H. (2014) Protein tyrosine kinase 7 plays a tumor suppressor role by inhibiting ERK and AKT phosphorylation in lung cancer. Oncol. Rep. 31, 2708–2712 [DOI] [PubMed] [Google Scholar]
- 58. Liu Y., Chen J., Sethi A., Li Q. K., Chen L., Collins B., Gillet L. C., Wollscheid B., Zhang H., Aebersold R. (2014) Glycoproteomic analysis of prostate cancer tissues by SWATH mass spectrometry discovers N-acylethanolamine acid amidase and protein tyrosine kinase 7 as signatures for tumor aggressiveness. Mol. Cell Proteomics 13, 1753–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shin W. S., Kwon J., Lee H. W., Kang M. C., Na H. W., Lee S. T., Park J. H. (2013) Oncogenic role of protein tyrosine kinase 7 in esophageal squamous cell carcinoma. Cancer Sci. 104, 1120–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.