

Background: GPCRs may be expressed in a monomer/dimer equilibrium at the plasma membrane.
Results: For the dimeric CRF1R, we found a constant monomer/dimer equilibrium not only at the plasma membrane but also in the ER.
Conclusion: The monomer/dimer equilibrium of the CRF1R is established already in the ER.
Significance: Our findings shed new light on the monomer/dimer equilibrium of GPCRs and on ER functions.
Keywords: Confocal Microscopy, Endoplasmic Reticulum (ER), Fluorescence Correlation Spectroscopy (FCS), G Protein-coupled Receptor (GPCR), Plasma Membrane, Corticotropin-releasing Factor Receptors, Fluorescence Cross-correlation Spectroscopy, Monomer/Oligomer Equilibrium, Receptor Oligomerization
Abstract
G protein-coupled receptors (GPCRs) represent the most important drug targets. Although the smallest functional unit of a GPCR is a monomer, it became clear in the past decades that the vast majority of the receptors form dimers. Only very recently, however, data were presented that some receptors may in fact be expressed as a mixture of monomers and dimers and that the interaction of the receptor protomers is dynamic. To date, equilibrium measurements were restricted to the plasma membrane due to experimental limitations. We have addressed the question as to where this equilibrium is established for the corticotropin-releasing factor receptor type 1. By developing a novel approach to analyze single molecule fluorescence cross-correlation spectroscopy data for intracellular membrane compartments, we show that the corticotropin-releasing factor receptor type 1 has a specific monomer/dimer equilibrium that is already established in the endoplasmic reticulum (ER). It remains constant at the plasma membrane even following receptor activation. Moreover, we demonstrate for seven additional GPCRs that they are expressed in specific but substantially different monomer/dimer ratios. Although it is well known that proteins may dimerize in the ER in principle, our data show that the ER is also able to establish the specific monomer/dimer ratios of GPCRs, which sheds new light on the functions of this compartment.
Introduction
The family of GPCRs3 forms the largest group of transmembrane receptors in the plasma membrane of mammalian cells (1). Because of the numerous drugs affecting GPCRs, there is a strong interest in understanding their function and signaling. Although it is known that the smallest functional unit of a (prototypical) GPCR is a monomer (2–8), it became clear that most of the receptors may form homo- or heterodimers (9–13) (see Footnote 4 for terminology concerning GPCR dimers and oligomers). In fact, an exclusive expression of a GPCR as a monomer seems to be rare and was experimentally shown only for a limited amount of GPCRs, such as the CRF2(a)R (14).
The functional significance of GPCR dimerization was addressed by many studies, and it was shown to play a role for different functions such as receptor trafficking (15, 16), ligand binding (17–19), and G protein coupling and selectivity (20–23).
The vast majority of the studies addressed GPCR dimerization only at a qualitative level, i.e. it was analyzed whether or not dimers are formed. Only very recently, it was examined whether dimeric GPCRs are expressed exclusively as dimers or as a mixture of monomers and dimers. In these studies, it was shown by smTIRFM that the M1 muscarinic receptor (24), the N-formyl peptide receptor (25), and the β1- and β2-adrenergic receptors (26) are expressed in the plasma membrane as such a mixture. Moreover, it was demonstrated that dimers can fall apart, i.e. that the interaction of the individual protomers is dynamic. The detected amount of dimers was different and ranged between 20% for the M1 muscarinic receptor (24) up to 60% for the β2-adrenergic receptor (26).
However, several questions addressing the monomer/dimer (M/D) equilibrium of GPCRs remain unclear. Most importantly, although it is known that GPCRs may dimerize in intracellular compartments such as the ER, it is not clear whether the M/D is also formed in these compartments or whether it is only present at the plasma membrane. It is also unclear whether the M/D is a general feature of GPCRs and whether it may undergo changes during the receptor's life cycle. Studies addressing the M/D of GPCRs so far mainly used smTIRFM (24–26). Although this technique has many advantages such as the possibility of defining the exact dimeric/oligomeric state of GPCRs and the dynamic interactions of the protomers at a single molecule level, it also has one major drawback, the measurements are limited to the plasma membrane, and consequently, the M/D of GPCRs in intracellular compartments cannot be studied.
To measure the M/D also intracellularly, we used fluorescence cross-correlation spectroscopy (FCCS). This single molecule-based method was applied successfully to monitor GPCR dimerization (14, 27, 28). As a model, we took a class B receptor, namely the CRF1R, which forms dimers (14, 29, 30) and is expressed mainly in the anterior pituitary where it plays a central role in the regulation of the hypothalamic-pituitary-adrenal stress axis in mammals (31). As a control, we took the CRF2(a)R, which forms exclusively monomers due to the presence of a unique domain at the N tail of the receptor, its pseudo signal peptide (14).
In this study, we show that the CRF1R is expressed in a specific M/D at the plasma membrane that is established early during receptor biogenesis and is kept constant throughout the receptor's life cycle. In addition, we demonstrate for seven additional GPCRs that they are expressed in specific but substantially different M/Ds at the plasma membrane.
EXPERIMENTAL PROCEDURES
Materials
The vector plasmids pECFP-N1, pEGFP-N1, pEYFP-N1, pmCherry-N1, and the ER marker plasmid pCFP-ER were obtained from Clontech. The latter plasmid encodes an N-terminal fusion of the ER targeting sequence of calreticulin and a C-terminal fusion of the ER retention sequence (KDEL) to CFP. The pPal7 expression vector was from the Profinity eXactTM cloning and expression kits (Bio-Rad). The transfection reagent LipofectamineTM 2000 was purchased from Invitrogen. The plasma membrane marker CellMaskTM Deep Red was from Molecular Probes (Darmstadt, Germany). Oligonucleotides were from Biotez (Berlin, Germany). Brefeldin A was purchased from Calbiochem. The polyclonal rabbit anti-GFP02 antibody (GFP/YFP antibody) was described (27). The ligand sauvagine was synthesized in our laboratory as described previously (32). Pitstop 2 was described (33) and kindly provided by Volker Haucke (FMP, Berlin, Germany). All other reagents were from Sigma.
DNA Manipulations
Standard DNA manipulations were carried out according to the handbook of Sambrook and Russel (34). The nucleotide sequences of the plasmid constructs were verified using the Sanger sequencing service from Source Bioscience (Berlin, Germany).
Receptor Constructs
Details of the cloning procedures are provided on request. The full-length rat CRF1R, rat CRF2(a)R, human V2R, human ETBR, human TSHR, human LHR, and human PAR1 were fused C-terminally with GFP or mCherry, thereby deleting the stop codons. The vector plasmids were pEGFP-N1 and pmCherry-N1. The resulting GFP-tagged constructs were CRF1R.GFP, CRF2(a)R.GFP, V2R.GFP, ETBR.GFP, TSHR.GFP, LHR.GFP, and PAR1.GFP; the corresponding mCherry-tagged constructs were CRF1R.mCherry, CRF2(a)R.mCherry, V2R.mCherry, ETBR.mCherry, TSHR.mCherry, LHR.mCherry, and PAR1.mCherry. The full-length CRF1R was also C-terminally tagged with CFP or YFP (vector plasmids pECFP-N1, pEYFP-N1) yielding constructs CRF1R.CFP and CRF1R.YFP respectively.
cAMP Accumulation Assay
Activation of CRF1R.GFP in transiently transfected HEK 293 cells was monitored by measuring sauvagine-mediated cAMP accumulation as described (cAMP-RIA) (35). The cAMP determination was performed using two different sauvagine concentrations (0.5 and 5 nm) in the presence and absence of pitstop 2 (30 μm, 20-min incubation prior to the addition of ligand).
Cell Lines, Cell Culture, and Transfection
Transiently transfected HEK 293 cells were used in this study. Cells were grown in DMEM containing 10% (v/v) fetal calf serum. Cells were cultured at 37 °C and 5% CO2. For microscopic analysis, DMEM without phenol red was used. Transfection of the cells with LipofectamineTM 2000 was carried out according to the supplier's recommendations 24 h after seeding the cells.
LSM, Localization of the CRF1R in the ER
Transiently transfected HEK 293 cells (3 × 105) co-expressing CRF1R.mCherry and the ER marker ER-CFP were grown on 30-mm glass coverslips (pretreated with 100 μg/ml poly-l-lysine) in 35-mm dishes. After 24 h of incubation, coverslips were transferred into a self-made chamber (details on request). Fluorescence signals were visualized in the absence and presence of brefeldin A (1 μg/ml, 1 h at 37 °C) using the laser scanning microscope (LSM) system LSM710-ConfoCor3 (Carl Zeiss Microscopy GmbH, Jena, Germany, 63×/1.3 oil objective). The mCherry fluorescence signals were detected on one channel (diode-pumped solid state laser, λexc = 561 nm, emission 565–640 nm bandpass filter) and the ER-CFP fluorescence signals on a second channel (argon laser, λexc = 458 nm, emission 460–540 nm bandpass filter). The overlay of the signals was computed. Images were analyzed using the ZEN 2010 software (Carl Zeiss Microscopy GmbH, Jena, Germany).
LSM, Inhibition of Ligand-induced CRF1R Internalization Using Pitstop 2
Transiently transfected HEK 293 cells (3 × 105) expressing CRF1R.GFP were grown on 30-mm glass coverslips (pretreated with 100 μg/ml poly-l-lysine) in 35-mm dishes. After 24 h of incubation, coverslips were transferred into a self-made chamber (details on request). Cells were pretreated with 30 μm pitstop 2 or DMSO as a control (dilution in DPBS) for 20 min. Thereafter, cells were stimulated with 100 nm sauvagine, and GFP signals were detected at room temperature using a LSM710-ConfoCor3 system (63×/1.3 oil objective, argon laser λexc = 488 nm, emission 505 nm long pass filter). Time series were taken (5 images: 1 every 5 min) to analyze internalization of CRF1R.GFP in live cells.
FCCS Measurements
Principles of FCCS and its use with LSM systems have been described previously (36–38). Transiently transfected HEK 293 cells co-expressing the GFP and mCherry-tagged receptor constructs were grown as described above. FCCS measurements were performed at room temperature on an LSM710-ConfoCor3 system enabling FCCS measurements at the basal plasma membrane without reflections from the coverslip. Using this setup, recordings were less noisy due to the limited flexibility of the basal plasma membrane in comparison with the apical side. GFP and mCherry fluorescence signals were recorded using a 40×/1.2 water objective (GFP: argon laser, λexc = 488 nm, 505–540 nm bandpass filter; mCherry: diode-pumped solid state laser, λexc = 561 nm, 580 nm long pass filter), a main beam splitter 488/561, and a dichroic mirror 565, respectively. Under these conditions, cross-talk between the GFP and mCherry signals contributed ∼8%. Membranes were located by z-scans. Intensity fluctuations were recorded for 4 s and 25 repetitions. Average auto-correlation and cross-correlation curves were derived from the fluctuations using the LSM710 software ZEN 2010 (Carl Zeiss Microscopy GmbH, Jena Germany). For average calculations, only convergent curves were used. The auto-correlation function is defined as shown in Equation 1,
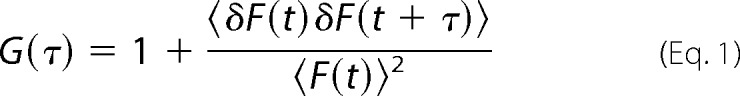 |
where 〈〉 indicates the average time-varying signal and the fluctuations around the mean intensity. Correlation curves were derived using a two-component model of free diffusion in two dimensions with triplet fraction and offset for membrane-associated proteins (Equation 2) using the ZEN 2010 software (39). A two-component model was used for the two-dimensional fits to obtain satisfactory fits. The first component was too fast to reflect membrane diffusion, and thus the diffusion time of the second component was considered to be significant (36). The analytical function of the model is described by Equation 2,
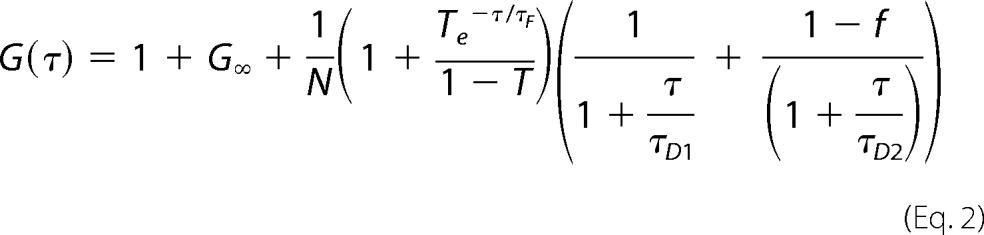 |
where G∞ is the offset from 1. N and T represent the total number of particles and the triplet fraction, respectively. τD, 1 and τD, 2 represent free diffusion times (the subscripts indicate the different molecule species). τF is the triplet time; f and 1 − f are the fractions of species 1 and 2, and τ is the correlation time.
In the case of the FCCS measurements in the ER, analyses are complicated by the fact that the axis of the laser beam may be aligned parallel to the ER membrane. Here, a three-dimensional fit with two components gave the best results. The quality of the fits was not improved by additional components.
Cross-correlation (CC) values are given as G(0)x/G(0)min, where G(0)x is the cross-correlation amplitude, and G(0)min is the lower autocorrelation amplitude.
FRET Experiments
To demonstrate that CRF1R dimerization is specific, the FRET signal of CRF1R.CFP and CRF1R.YFP was recorded, and changes by co-expressed nonfluorescent CRF1R or V2R were measured. The experimental setup of the FRET experiments was described previously (14).
smTIRFM Analyses
We used a custom-built TIRF system described previously (40). The 488-nm laser line (Coherent Sapphire 488 LP, 150 milliwatt) was reflected into the high numerical aperture objective (Nikon, Apo 100×/1.49) via a custom TIRF illumination system (40) using a dichroic mirror (BrightLine 405/488/561/635, Semrock). The resulting average laser intensity at the specimen was 8 microwatts/μm2. Emission was collected via a bandpass filter (BrightLine HC 525/45, Semrock) using an EMCCD camera (iXon-897E, 512 × 512 pixels of 16 μm size, Andor) at a 20-ms exposure and a gain of 120–300. Microscope and camera were controlled by a PC work station running open source software for acquisition (μManager, University of California at San Francisco) (41).
To analyze the derived data, we used the image analysis software GMimPro (42), which enables a single molecule tracking procedure. We first analyzed the intensity of single immobilized fluorophores (YFP). To this end, a 30-mm poly-l-lysine-coated coverslip was transferred into a self-made chamber (details on request), and a polyclonal GFP/YFP antibody (1:500) was attached to the coverslip by incubation for 10 min at room temperature. After three washing steps with DPBS, a 3 nm solution of purified YFP was added to the coverslip and incubated at room temperature for 10 min to allow binding to the antibody. After three washing steps with DPBS, the intensity of the immobilized YFP fluorophores was detected using the setting described above. The mean intensity of different fluorescent spots was recorded for purified YFP, and single-step photobleaching was observed (data not shown). Thereafter, transiently transfected HEK 293 cells (3 × 105) expressing CRF1R.YFP were grown on 30-mm glass coverslips (pretreated with 100 μg/ml poly-l-lysine) in 35-mm dishes. After 24 h of incubation, coverslips were transferred into a self-made chamber (details on request). Single molecule detection at the basal plasma membrane was performed, and the intensities of different fluorescent spots were analyzed. Only cells with a low protein expression were chosen (clearly visible single moving spots, data not shown). Discrete bleaching steps for individual tracked fluorescent spots could be observed. Areas containing no fluorescently labeled construct were used for background correction. The relatively broad distribution of spot intensity detected in live cells is due to a combination of factors already described (24): (i) uneven illumination of the cell; (ii) variability of the contact distance between the plasma membrane and the glass coverslip; and (iii) appearance of shot noise or camera noise as well as temporal intensity variation.
Statistics
Analyses were performed using the Student's t test (GraphPad t test calculator, GraphPad Software, Inc., La Jolla, CA); p values <0.0001 were considered to be significant.
RESULTS
Selection, Cloning, Expression, and Subcellular Location of GPCR Fusion Proteins
To analyze the M/D of various unrelated GPCRs, we took the following class A receptors as model proteins: the human V2R, human ETBR, human TSHR, human LHR, and the human PAR1. As class B receptors, we took the CRF1R and the CRF2(a)R. All these receptors were described to form dimers (14, 17, 29, 30, 43–45) with the exception of the CRF2(a)R, which is expressed exclusively as a monomer (14). For FCCS, the full-length receptors were C-terminally fused to GFP or mCherry. It was shown previously that C-terminally fused GFP (or one of its derivatives) does not impair transport of these receptors to the plasma membrane (17, 46–50). An influence of the mCherry tag, however, was not analyzed so far. Thus, we first studied expression of the mCherry-tagged receptors in the plasma membrane of transiently transfected HEK 293 cells. To this end, the receptor's mCherry signals were co-localized in live cells with the plasma membrane marker CellMaskTM Deep Red by confocal LSM. For all analyzed receptors, a substantial co-localization of the mCherry signals and the CellMaskTM Deep Red signals was observed, demonstrating that all constructs are readily expressed at the plasma membrane (data not shown).
Analyzed GPCRs Are Expressed in Specific but Substantially Different M/Ds at the Plasma Membrane
To analyze the M/Ds of the various GPCRs, we used FCCS. This method allows the detection of fluorescence signals in live cells at a single molecule level in both the plasma membrane and intracellular compartments. If two different diffusing fluorescent molecules are detected in a confocal volume, cross-correlation analyses with mathematical procedures can be performed (see under “Experimental Procedures”). A significant cross-correlation describes co-diffusion of the two fluorescent molecules and consequently their interaction. In contrast, a lack of cross-correlation in these experiments indicates independent diffusion and monomeric molecules. In this study, FCCS measurements were realized by recording intensity fluctuations of the GFP and mCherry-tagged GPCRs in transiently co-transfected HEK 293 cells. To determine the exact M/D out of the auto- and cross-correlation values in our experiments, we defined the following criteria.
1) Ideally, the amount of GFP- and mCherry-tagged receptors in the cells should be equal, which is not necessarily the case upon co-transfection. Therefore, we preselected cells expressing the GFP- and mCherry-tagged receptors in equivalent amounts. As a control for a 50:50 ratio of expression, we used a GFP-mCherry tandem fusion construct that necessarily leads to equal amounts of GFP and mCherry signals. The settings derived from this construct were used to preselect cells expressing matching amounts of the GFP- and mCherry-tagged receptors (Fig. 1A) and were not changed during the whole set of experiments.
FIGURE 1.

Prerequisites to determine the M/D of the analyzed GPCRs out of FCCS data in transiently transfected HEK 293 cells. A, ratio of the fluorescence intensities of the co-transfected GFP and mCherry-tagged GPCRs. Only cells with equal GFP and mCherry fluorescence intensities (as shown in the diagram) were selected for the FCCS measurements. The settings for the intensity detection were defined using a GFP-mCherry tandem fusion construct (see below). Columns represent mean values (± S.E.) of the ratio of the GFP and mCherry signals. n.s., not significant. B, representative auto- and cross-correlation data points and fitted curves of co-transfected separate GFP and mCherry proteins (left panel, GFP + mCherry) and a GFP-mCherry tandem fusion protein (right panel, GFP-mCherry). Auto-correlation data are depicted in red (mCherry) and green (GFP), CC data, are shown in black. Below the diagrams, the residuals of the relevant fittings are depicted (3D_1C, i.e. three dimensions, one component; autocorrelation data: GFP = green and mCherry = red). C, minimum CC value (GFP + mCherry, black square, monomers) and the maximum CC value (GFP-mCherry, gray square, dimers) are shown. To confirm a linear relation (black line), a transient co-transfection of the GFP-mCherry tandem fusion (dimer) and the photoconvertible protein mKikGR (monomer) was performed in HEK 293 cells. The fluorescence of mKikGR was stepwise photoconverted from green to red thereby reducing gradually the amount of detected monomers. CC values of GFP-mCherry in the presence of the green mKikGR signals (in different amounts) were determined. Using this approach, CC values for known ratios of monomers and GFP-mCherry pairs could be determined and fitted by linear regression. The diagram allows rescaling of the CC values obtained for the GFP and mCherry-tagged GPCRs. D, assumed statistical distribution of the interactions of GFP and mCherry (mCh)-tagged GPCRs.
2) To assess for the experimental dynamic range of our FCCS measurements, we determined the CC values of exclusive monomeric and exclusive dimeric molecules. The GFP-mCherry tandem construct (see above) defines an exclusive dimeric molecule. The maximal detectable CC value derived out of the auto- and cross-correlation curves of this construct (Fig. 1B, right panel) was 57% (Fig. 1C, gray square). Unfused co-transfected separate GFP and mCherry proteins were used to measure exclusive monomeric molecules. Here, the CC value was 8% (Fig. 1, B, left panel, and C, black square) reflecting the cross-talk of GFP into the mCherry channel. Fitting parameters and CC values of the vector constructs are given in Table 1.
TABLE 1.
FCCS fitting parameters (± S.E.) for unfused, co-expressed GFP and mCherry (GFP + mCherry), and the GFP-mCherry tandem fusion protein (GFP-mCherry) in live transiently transfected HEK 293 cells
Triplet fraction (TF (%)), triplet decay time (τD,F (μs)), decay time (τD (μs)), and minimum (GFP + mCherry) and maximum (GFP-mCherry) CC values (G(0)x/G(0)min) are listed. FCS curves were fitted assuming free diffusion in three dimensions and one component (3D_1C).
| Vector constructs | TF | τD,F | τD | N | G(0)x/G(0)min |
|---|---|---|---|---|---|
| % | μs | μs | % | ||
| GFP + mCherry | |||||
| GFP | 33 ± 2 | 16 ± 2 | 364 ± 6 | 220 | 8.3 ± 0.3 |
| mCherry | 44 ± 1 | 58 ± 2 | 443 ± 22 | ||
| GFP-mCherry | |||||
| GFP | 39 ± 2 | 20 ± 3 | 505 ± 11 | 152 | 56.10 ± 0.7 |
| mCherry | 42 ± 1 | 68 ± 2 | 726 ± 20 | ||
To verify the linear relation between minimal and maximal detectable CC values of monomeric and dimeric molecules, we used co-transfection of the GFP-mCherry tandem fusion (green-red pair) and the monomeric photoconvertible protein Kikume Green-Red (mKikGR). In the case of the GFP-mCherry tandem fusion, the same intensities are obtained in the green and red channel. The co-transfected mKikGR protein adds an additional fluorescence in the green channel, which can be gradually modified (reduced) by partial photoconversion using 405 nm irradiation (laser diode). Taking the known fluorescence quantum yields into account, the ratio of monomeric (green mKikGR) to dimeric (GFP-mCherry tandem fusion) molecules can be calculated. This methodology consequently allows the calculation of CC values in the case of defined changes of the M/D. Using this approach, the linear relation between minimal and maximal detectable CC values of monomeric and dimeric molecules could be confirmed (Fig. 1C, gray dots). The linear regression is given as shown in Equation 3,
Taking these calculations into account, the dynamic range of the CC values for the GFP- and mCherry-tagged receptor constructs (see above) can be rescaled.
3) We assumed a statistical distribution of interactions of the individual receptor molecules (Fig. 1D). In our measurements, however, GFP-tagged receptor dimers cannot be distinguished from monomeric receptors carrying GFP moieties. Thus, the rescaled CC values (see above) need further correction. The total number of detected green molecules (N) can be expressed as shown in Equation 4,
where GR is the number of receptor dimers containing GFP and mCherry (GFP.mCherry and mCherry.GFP); GG is the number of dimers carrying solely GFP (GFP.GFP); and G is the monomeric GFP-tagged receptors. For the number of receptor dimers containing GFP and mCherry, Equation 5 applies,
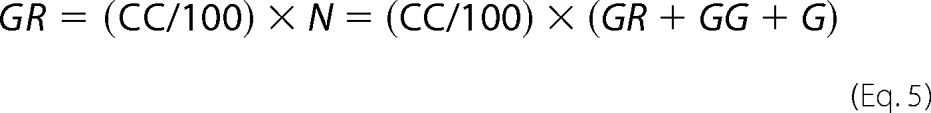 |
Because GFP.GFP receptor dimers are detected as monomers, GG can be expressed as GR/2 and consequently GR as shown in Equation 6,
or Equation 7,
The number of GFP-tagged receptor monomers is then given by Equation 8,
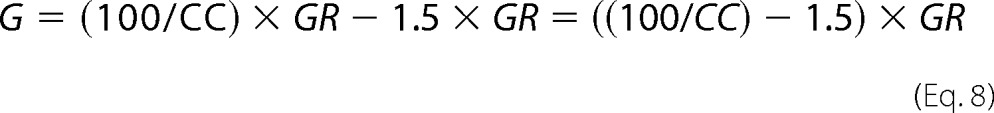 |
The M/D of the receptors (G/GR = M/D) can consequently be expressed as shown in Equation 9,
and the percentage of dimers as shown in Equation 10,
 |
4) As a single molecule technique, FCCS measurements require a very low expression of the analyzed proteins. The obvious advantage is that this expression rate resembles the situation of endogenous receptor expression. Under the settings described above, the count rates for all detected constructs were in the range of 50–200 kHz.
5) We preclude the possibility that the observed CC amplitudes at the plasma membrane are compromised by receptors that only co-diffuse within the same microdomain. Such a co-diffusion would lead to a strong increase in diffusion time, which was not observed in our experiments. The detected diffusion times were consistent with the literature (30), and the cross-correlation amplitudes derived from the FCCS experiments thus only reflect receptor interactions. In the case of the FCCS measurements at the ER membrane, however, we observed in some experiments an increase in diffusion time to a certain extent. Those curves were excluded from our analyses, because we were only interested in the mobile portion of the GPCRs studied. Moreover, we preclude the possibility that the detected interaction signals are affected by endogenous receptors because the GPCRs used in this study are not expressed in HEK 293 cells in significant amounts (51).
Taking the criteria outlined above into account, we performed FCCS using transiently transfected HEK 293 cells expressing the GFP- and mCherry-tagged GPCRs. Representative auto- and cross-correlation curves of individual GPCRs are shown in Fig. 2, and CC values and diffusion parameters are given in Table 2. No CC amplitudes were observed for the CRF2(a)R, confirming our previous results that this receptor is expressed exclusively as a monomer (14). In contrast, for all the other analyzed GPCRs, significant CC amplitudes were detected indicating their dimerization. We then rescaled the raw CC values (according to the criteria 2 and 3) and obtained the actual M/Ds for the analyzed GPCRs (Fig. 3). These data demonstrate that the analyzed GPCRs are expressed in specific but substantially different M/Ds in the plasma membrane of live cells. Most importantly, all analyzed receptors are expressed predominantly as monomers. The amount of dimers ranged from 7% for the TSHR up to 22% for the CRF1R.
FIGURE 2.

FCCS measurements using transiently co-transfected HEK 293 cells expressing the GFP and mCherry-tagged receptor constructs. The laser beam was focused at the plasma membrane. Representative auto- and cross-correlation data points and fitted curves for the analyzed receptor constructs are shown. Auto-correlation data are depicted in red (mCherry) and green (GFP), and CC data are shown in black. Curves were obtained by two-dimensional fits with two components (2D_2C), which gave the best results. Below each diagram, the residuals of this fitting are shown (autocorrelation data: GFP = green and mCherry = red). In addition, the residuals of suboptimal fits with two dimensions and one component (2D_1C) are depicted to demonstrate the necessity of 2D_2C fits.
TABLE 2.
FCCS fitting parameters (± S.E.) for the GFP- and mCherry-tagged GPCRs in the plasma membrane of transiently transfected HEK 293 cells
Decay times (τD, 1 (ms); τD, 2 (ms); fractions (τD, 1 (%); τD, 2 (%)), and CC values (G(0)x/G(0)min) are listed. The best fits were obtained assuming free diffusion in two dimensions and two components (2D_2C). Triplet fractions (TF (%)) ranged from 20 to 50% and triplet decay times (τD,F (μs)) from 20 to 200 μs.
| GPCRs | τD, 1 | τD, 2 | τD, 1 | τD, 2 | N | G(0)x/G(0)min |
|---|---|---|---|---|---|---|
| % | % | ms | ms | % | ||
| CRF1 | ||||||
| GFP | 40 ± 1 | 60 ± 1 | 0.43 ± 0.02 | 36 ± 1 | 108 | 17.2 ± 0.5 |
| mCherry | 56 ± 1 | 44 ± 1 | 0.15 ± 0.01 | 39 ± 2 | ||
| CRF2(a)R | ||||||
| GFP | 51 ± 1 | 49 ± 1 | 0.57 ± 0.03 | 37 ± 1 | 93 | 9.3 ± 0.3 |
| mCherry | 63 ± 1 | 37 ± 1 | 0.16 ± 0.01 | 38 ± 2 | ||
| TSHR | ||||||
| GFP | 33 ± 1 | 67 ± 1 | 0.33 ± 0.03 | 34 ± 1 | 98 | 11.4 ± 0.4 |
| mCherry | 58 ± 1 | 42 ± 1 | 0.11 ± 0.01 | 33 ± 1 | ||
| LHR | ||||||
| GFP | 53 ± 3 | 47 ± 3 | 0.29 ± 0.02 | 25 ± 2 | 42 | 16.3 ± 1.1 |
| mCherry | 60 ± 2 | 40 ± 2 | 0.13 ± 0.03 | 22 ± 3 | ||
| V2R | ||||||
| GFP | 41 ± 1 | 59 ± 1 | 0.46 ± 0.03 | 37 ± 2 | 117 | 14.6 ± 0.5 |
| mCherry | 60 ± 1 | 40 ± 1 | 0.13 ± 0.01 | 38 ± 2 | ||
| ETBR | ||||||
| GFP | 46 ± 2 | 54 ± 2 | 0,26 ± 0,02 | 23 ± 1 | 86 | 14.0 ± 0.6 |
| mCherry | 66 ± 1 | 34 ± 1 | 0.08 ± 0.01 | 25 ± 2 | ||
| PAR1 | ||||||
| GFP | 64 ± 2 | 36 ± 2 | 0.27 ± 0.01 | 28 ± 1 | 84 | 14.0 ± 0.5 |
| mCherry | 73 ± 1 | 27 ± 1 | 0.15 ± 0.01 | 23 ± 1 | ||
FIGURE 3.

M/D of the analyzed GPCRs in the plasma membrane of transiently transfected HEK 293 cells. The diagram shows the rescaled and distribution-corrected data of the FCCS experiments of Fig. 2.
CRF1R Does Not Form Higher Order Oligomers in the Plasma Membrane of Live Cells
To assess for the subcellular compartment in which the specific M/D of a GPCR is established and to analyze potential changes of this equilibrium during the receptor's life cycle, we took the CRF1R as an example. Whereas FCCS can be performed in intracellular compartments, it does not give detailed information about the oligomeric state of the receptor, i.e. whether the receptor is expressed as a dimer or as a higher order oligomer. To investigate the dimeric state of the CRF1R in detail and to validate the FCCS-derived data above, we performed smTIRFM for the YFP-tagged CRF1R (construct CRF1R.YFP) at the plasma membrane. We used a custom-built smTIRF microscope possessing a high numerical aperture objective lens, a laser power of 8 microwatts/μm2, and a frame rate of 20 ms (enabled by an EMCCD camera; see under “Experimental Procedures”).
Prior to the smTIRFM experiments using CRF1R.YFP, we determined the intensity of single purified YFP molecules immobilized on a coverslip by a poly-l-lysine-attached GFP/YFP antibody. A Gaussian distribution for the YFP intensities of all detected objects (fluorescent spots) was obtained (Fig. 4A, left panel). Fitting of this distribution gave a mean intensity value of 25 ± 6 a.u. for monomeric YFP. Using the above setup, we then performed single particle tracking of CRF1R.YFP in transiently transfected HEK 293 cells. The best fitting of the CRF1R.YFP intensities was obtained by a two-component Gaussian function (Fig. 4A, right panel). The first maximum represents the monomeric (25 ± 6 a.u.) and the second the dimeric (50 ± 11 a.u.) CRF1R.YFP molecules. No additional maxima representing higher order oligomers were detected. The single molecule intensity traces sometimes showed a stepwise decrease that either results from single YFP bleaching or dissociation of a dimer into monomers (Fig. 4B). The smTIRFM data demonstrate that the CRF1R forms dimers in the plasma membrane but no higher order oligomers. Moreover, the amount of dimers detected in the FCCS measurements (22%) is in good agreement with that determined by smTIRFM (29%).
FIGURE 4.

Detection of CRF1R.YFP dimers in the plasma membrane of transiently transfected HEK 293 cells using smTIRFM. A, left panel, normalized intensity distribution histogram of purified YFP immobilized by a monoclonal GFP/YFP antibody on a poly-l-lysine-coated coverslip. In four independent experiments, 2534 fluorescent YFP spots were detected. Best fitting is achieved by a Gaussian function with a mean value of 25 ± 6 a.u. Right panel, normalized intensity distribution histogram of CRF1R.YFP. In four independent experiments, 7594 fluorescent spots were analyzed (at least five cells in each experiment). Best fitting is achieved by a two gaussian function. The mean of the first component represents monomeric CRF1R.YFP (25 ± 6 a.u.; red curve), the mean of the second component represents dimeric CRF1R.YFP (50 ± 11 a.u.; black curve). B, intensity changes of individual tracks of CRF1R.YFP. Left panel, examples for intensity traces of moving fluorescent spots representing monomeric CRF1R.YFP (upper traces) in comparison with the traces resulting from areas containing no fluorescently labeled receptors (lower traces). Center panel, as above, but examples for intensity traces representing dimeric CRF1R.YFP are shown. Right panel, as above, but examples for intensity traces representing dimeric CRF1R.YFP are shown, which shift abruptly to the level of a single YFP fluorophore, either by photobleaching or by dimer dissociation. The red line indicates the mean value of the individual traces.
Dimerization of the CRF1R Is Specific
To validate our FCCS data, we also determined whether CRF1R dimerization is specific in transiently transfected HEK 293 cells using FRET experiments. To this end, the FRET signal of co-transfected CRF1R.CFP and CRF1R.YFP was recorded, and changes by co-expressed nonfluorescent CRF1R (positive control) or V2R (negative control) were measured (Fig. 5). Whereas the presence of the untagged CRF1R clearly reduced the FRET signal of CRF1R.CFP + CRF1R.YFP (ET, from 20.5 to 10.9%), no changes were observed in the presence of the untagged V2R. These results demonstrate that CRF1R dimerization is specific.
FIGURE 5.
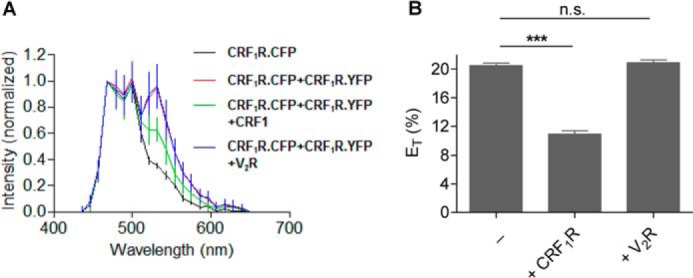
FRET experiments analyzing CRF1R dimerization specificity in transiently transfected HEK 293 cells. A, FRET spectra (± S.D.) were recorded for cells expressing CRF1R.CFP alone (black, n = 73) or the CRF1R.YFP/CRF1R.CFP FRET pair (red, n = 120). In addition, cells expressing CRF1R.YFP/CRF1R.CFP were co-transfected with the untagged CRF1R (green, n = 95) or V2R (blue, n = 140). B, energy transfer efficiencies (ET% ± S.D.) calculated out of A. ET values were derived as described previously (14). n.s., not significant; ***, highly significant.
The M/D of the CRF1R Is Established in the ER
To analyze the M/D of the CRF1R in the ER, we treated transiently transfected HEK 293 cells with the drug brefeldin A (1 μg/ml) to trap the receptors in this compartment. Under these conditions, a substantial increase in co-localization of the CRF1R.mCherry fluorescence signals and those of the ER marker ER-CFP was observed verifying the ER exit block (Fig. 6A). Next, FCCS was carried out using transiently transfected HEK 293 cells expressing CRF1R.GFP and CRF1R.mCherry. The laser beam was focused on the area of the rough ER immediately surrounding the nucleus (Fig. 6B, arrow). Auto- and cross-correlation curves were derived (Fig. 6C), and the CC values were rescaled and corrected as described above. The CC values obtained at the plasma membrane (Table 2) and the ER (Table 3) were not significantly different (17.2 ± 0.5% and 17.8 ± 0.7%, respectively). These data demonstrate that the M/D of the CRF1R is established already in the ER, i.e. early after receptor biosynthesis and folding. The presence of the same M/D in the ER and the plasma membrane suggests that the equilibrium also remains constant in the vesicular transport steps to the cell surface, although this question was not addressed directly.
FIGURE 6.

M/D of the CRF1R is established in the ER in transiently transfected HEK 293 cells. A, co-localization of CRF1R.mCherry and the ER marker ER-CFP following brefeldin A treatment in co-transfected cells. The mCherry signals (red, left panel) and the ER-CFP signals (green, center panel) were localized in live cells by confocal LSM and were computer-overlaid (right panel, co-localization is indicated by yellow). The scans show representative cells. Scale bar, 10 μm. Similar data were obtained in three independent experiments. B, left panel, representative brefeldin A-treated cell co-transfected with CRF1R.mCherry and CRF1R.GFP. The image was recorded by confocal LSM and shows an overlay of the CRF1R.mCherry (red) and CRF1R.GFP fluorescence signals (green). Co-localization is indicated by yellow. The arrow indicates the region of the rough ER at the interface of the nucleus where the FCCS measurements were conducted. Right panel, split green and red channels of the same cell. Scale bar, 10 μm. C, FCCS measurements in the ER using co-transfected cells expressing CRF1R.mCherry and CRF1R.GFP. The laser was focused at the ER membrane. Representative auto- and cross-correlation data points and fitted curves are shown. Auto-correlation data of the fluorophores are depicted in red (mCherry) and green (GFP), and CC data are shown in black. Below the diagram, the residuals of the relevant fitting is shown (3D_2C; i.e. three dimensions, two components; autocorrelation data: GFP = green, mCherry = red).
TABLE 3.
FCCS fitting parameters (± S.E.) for the GFP- and mCherry-tagged CRF1R and ETBR in the ER membrane of transiently transfected HEK 293 cells
Decay times (τD, 1 (ms); τD, 2 (ms); fractions (τD, 1 (%); τD, 2 (%)), and CC values (G(0)x/G(0)min) are listed. The best fits were obtained assuming free diffusion in three dimensions and two components (3D_2C). Triplet fractions (TF (%)) ranged from 20 to 50% and triplet decay times (τD,F (μs)) from 20 to 200 μs.
| GPCRs | τD, 1 | τD, 2 | τD, 1 | τD, 1 | N | G(0)x/G(0)min |
|---|---|---|---|---|---|---|
| % | % | ms | ms | % | ||
| CRF1R | ||||||
| GFP | 56 ± 1 | 44 ± 1 | 0.37 ± 0.02 | 34 ± 1 | 89 | 17.8 ± 0.7 |
| mCherry | 61 ± 1 | 39 ± 1 | 0.20 ± 0.04 | 28 ± 2 | ||
| ETBR | ||||||
| GFP | 34 ± 1 | 66 ± 1 | 0.62 ± 0.04 | 51 ± 2 | 77 | 14.6 ± 0.7 |
| mCherry | 62 ± 1 | 38 ± 1 | 0.13 ± 0.01 | 45 ± 3 | ||
To address the possibility that the constant M/D of the CRF1R at the plasma membrane and in the ER occurs at random, similar FCCS measurements were carried out using ETBR.GFP and ETBR.mCherry. The CC values obtained at the plasma membrane (Table 2) and in the ER (Table 3) were also not substantially different in the case of this receptor (14.0 ± 0.6% and 14.6 ± 0.7%, respectively). These results suggest that a constant M/D in the ER and at the plasma membrane may be a more general feature of GPCRs.
Receptor Activation Does Not Influence the M/D of the CRF1R
We next addressed the question whether the M/D of the CRF1R is changed upon ligand-induced receptor activation. However, these experiments are complicated by the fact that the CRF1R internalizes rapidly after its activation by a clathrin-dependent mechanism (52). To overcome this problem and to trap the activated receptors at the plasma membrane, we used the recently described substance pitstop 2, which inhibits the clathrin terminal domain and consequently blocks selectively clathrin-mediated endocytosis (33). A prerequisite for the use of pitstop 2 in these kinds of experiments is, however, that receptor activation is affected neither by the impaired internalization of the receptors nor by the substance itself. Pitstop 2 treatment (30 μm) indeed blocked sauvagine-induced (100 nm) internalization of CRF1R.GFP in transiently transfected HEK 293 cells (Fig. 7A). We next measured the influence of pitstop 2 (30 μm) on sauvagine-induced (0.5 nm or 5 nm) cAMP formation by CRF1R.GFP in transiently transfected HEK 293 cells using a cAMP RIA (Fig. 7B). No influence of the substance on second messenger formation could be detected in this experiment demonstrating that pitstop 2 can be used to trap activated receptors at the plasma membrane. Thereafter, FCCS was carried out at the plasma membrane of transiently transfected HEK 293 cells expressing CRF1R.GFP and CRF1R.mCherry as described above. To get reliable results, measurements were carried out using the same cell as follows: (i) without pitstop 2 treatment, (ii) after addition of pitstop 2, and (iii) after addition of the agonist sauvagine. We could not detect any significant change in mean CC values indicating no difference in the M/D of nonactivated and activated CRF1Rs (Table 4). Taken together, our results show that the M/D of the CRF1R remains constant not only following early receptor biogenesis in the ER but also following receptor activation.
FIGURE 7.

M/D of the CRF1R at the plasma membrane remains constant following agonist-induced receptor activation in transiently transfected HEK 293 cells. A, internalization assay demonstrating that pitstop 2 blocks internalization of CRF1R.GFP. Cells expressing the receptor construct were pretreated with pitstop 2 (30 μm). The agonist sauvagine was added in saturating concentrations (100 nm), and the fluorescence signals of CRF1R.GFP were recorded at times t = 0 and t = 30 min by confocal LSM. Cells treated with DMSO instead of pitstop 2 were used as a control. The arrows indicate vesicles containing internalized CRF1R.GFP in the case of the DMSO control. The scans are representative of three independent experiments. Scale bar, 10 μm. B, pitstop 2 treatment does not prevent second messenger formation mediated by CRF1R.GFP. Cells expressing the receptor construct were pretreated with pitstop 2 (30 μm) or DMSO (−) and were stimulated by sauvagine (0.5 or 5 nm). Formation of the second messenger cAMP was measured by a cAMP-RIA. Columns represent mean values of cAMP formation (± S.E.) of three independent experiments.
TABLE 4.
FCCS fitting parameters (± S.E.) of the GFP- and mCherry-tagged CRF1R in the plasma membrane of transiently transfected HEK 293 cells without and with pitstop2 (30 μm) and sauvagine (5 nM) treatment
Decay times (τD, 1 (ms); τD, 2 (ms); fractions (τD, 1 (%); τD, 2 (%)), and CC values (G(0)x/G(0)min) are listed for untreated, pitstop 2-treated, and sauvagine (Sauv.)-treated cells. Best fits were obtained assuming free diffusion in two dimensions and two components (2D_2C). Triplet fractions (TF (%)) ranged from 20 to 50% and triplet decay times (τD,F (μs)) from 20 to 200 μs.
| GPCRs | Pitstop2 | Sauv. | τD, 1 | τD, 2 | τD, 1 | τD, 2 | N | G(0)x/G(0)min |
|---|---|---|---|---|---|---|---|---|
| % | % | ms | ms | % | ||||
| CRF1R | ||||||||
| GFP | − | − | 47 ± 3 | 53 ± 3 | 0.41 ± 0.05 | 36 ± 3 | 54 | 16.3 ± 1.4 |
| mCherry | 53 ± 3 | 47 ± 3 | 0.10 ± 0.02 | 32 ± 3 | ||||
| CRF1R | ||||||||
| GFP | + | − | 47 ± 2 | 53 ± 2 | 0.31 ± 0.02 | 33 ± 2 | 54 | 17.6 ± 1.6 |
| mCherry | 58 ± 2 | 42 ± 2 | 0.25 ± 0.10 | 30 ± 2 | ||||
| CRF1R | ||||||||
| GFP | + | + | 52 ± 2 | 48 ± 2 | 0.30 ± 0.02 | 33 ± 2 | 54 | 16.6 ± 1.3 |
| mCherry | 54 ± 2 | 46 ± 2 | 0.12 ± 0.03 | 29 ± 2 | ||||
DISCUSSION
We show in this study for five class A GPCRs and for one class B GPCR, namely the CRF1R, that they are expressed in different but specific M/Ds at the plasma membrane. Expression in such an equilibrium thus seems to be a more general feature of dimeric GPCRs. This raises several questions which should be discussed as follows.
1) We have shown here that the M/D of the CRF1R is already established in the early secretory pathway, namely the ER. Although it seems to be clear that dimerization of GPCRs and other membrane proteins may take place in this compartment (9, 10), the finding that the M/D of the CRF1R does not differ between the ER and the plasma membrane is nevertheless surprising at first sight. Folding is facilitated by molecular chaperones. In case of the frequently N-glycosylated GPCRs, the lectin chaperones calnexin/calreticulin play an important role, and this was also demonstrated for the CRF1R (46). It is conceivable that chaperone-assisted folding engages initially monomeric rather than dimeric GPCRs, and thus a higher M/D may be expected in the ER. However, the CRF1R may fold very quickly thereby complicating the detection of these changes. Consistent with this view, only a very limited amount of high mannose, immature forms of the CRF1R was detected in previous immunoprecipitation studies (46). Thus, GPCRs with a slower folding dynamic should be considered in future studies, too. However, the fact that the M/D of the ETBR at the plasma membrane and in the ER was also not substantially different suggests that a constant M/D may also be a more general feature of GPCRs.
2) The observed amount of dimers in the equilibrium is surprisingly low, ranging here from 7% (TSHR) to 22% (CRF1R). An influence of the methodology on these values is unlikely because, at least in the case of the CRF1R, similar values were obtained by both FCCS and smTIRFM measurements. Our results are also in good agreement with those published for the M1 muscarinic receptor (24) and β1-adrenergic receptor (26) using smTIRFM. Both FCCS and smTIRFM analyses are single molecule techniques, and cells with low receptor expression were chosen for the experiments. Although this may converge to endogenous receptor expression, an influence of the receptor expression levels to the M/D must be taken into account. Assuming free two-dimensional diffusion and unhampered interactions, two-dimensional KD values can be calculated out of the receptor densities and the M/D values according to the law of mass order (Table 5) (25). It should be stressed, however, that diffusion and interactions may be hindered due to the presence of unknown protein factors/scaffolds, for example. The influence of protomer concentration must thus be addressed more rigorously in future studies by using expression titration analyses prior to the FCCS measurements, for example. Such experiments should at least provide an indication of whether a simple model of free diffusion/interaction applies or whether, for example, scaffolds are involved.
TABLE 5.
Receptor densities and calculated two-dimensional KD values for the association of the listed GPCRs in the plasma membrane of transiently transfected HEK 293 cells
Calculation of the two-dimensional KDvalues was performed as described in Ref. 25.
| GPCRs | Density | Two-dimensional KD |
|---|---|---|
| Receptor entities/μm2 | Receptor entities/μm2 | |
| CRF1R | 416 | 914 |
| CRF2(a)R | 430 | 428,712 |
| TSHR | 68 | 1967 |
| LHR | 130 | 434 |
| V2R | 320 | 1340 |
| ETBR | 170 | 915 |
| PAR1 | 78 | 420 |
3) Our result that the M/D of the CRF1R remains constant upon receptor activation is consistent with previous suggestions for this receptor using FRET experiments (29, 30). This result may be specific for the CRF1R. However, the recently published smTIRFM studies for the N-formyl peptide receptor (25) and the β1- and β2-adrenergic receptors (26) also showed that the M/Ds of these receptors are unaffected by receptor activation. A constant M/D may thus be a more general feature of GPCRs. It should be addressed whether this holds true, in particular by using GPCRs for which older studies using FRET suggested an influence of ligand binding on the dimeric state.
4) Another point is whether the presence of a specific M/D has any functional significance. In the case of the CRF1R, it is known that the dimeric state influences G protein selectivity. The dimeric CRF1R couples to Gs and at higher occupancy also to Gi, leading to a biphasic, bell-shaped concentration/response curve for the second messenger cAMP. CRF1R monomers, in contrast, couple exclusively to Gs (14, 47). It may thus be speculated that the specific M/D is involved in the fine-tuning of the signal transduction of this receptor. In the case of the CRF1R, the relatively high amount of dimers may prevent via Gi coupling an overshoot in the cAMP response at high occupancy.
So far, mainly the equilibrium of monomers and homodimers was studied in detail. However, many GPCRs are known to form heterodimers, too (10–12). In this case, the situation is more complex, because it is conceivable that monomers, homodimers, and heterodimers are expressed as a mixture. The ratio of homo- and heterodimers is not known, and it would be of particular interest to analyze this relation. In case of the CRF1R, for example, the individual receptor subpopulations resulting from the described heterodimerization with the vasopressin-1b-receptor (53) could be quantified.
Acknowledgments
We thank Jenny Eichorst for excellent technical assistance. Bettina Kahlich helped in cell culture and Jessica Tröger in purifying YFP. Gregory Mashanov (National Institute of Medical Research, London, UK) provided the open access GMimPro software.
For most GPCRs, it was not studied whether they form dimers or also higher order oligomers. In this paper, we thus use the term “dimer” for readability. We focused in this study on the CRF1R, and for this receptor, we could preclude the presence of higher order oligomers (see under “Results”). In the case of the other GPCRs studied, however, the presence of higher order oligomers is not excluded, and the term “dimer” should be read in this case as “dimer/oligomer.”
- GPCR
- G protein-coupled receptor
- CC
- cross-correlation
- CRF1R
- corticotropin-releasing factor receptor type 1
- CRF2(a)R
- corticotropin-releasing factor receptor type 2(a)
- DPBS
- Dulbecco's phosphate-buffered saline
- ER
- endoplasmic reticulum
- ETBR
- endothelin B receptor
- FCCS
- fluorescence cross-correlation spectroscopy
- GFP
- green fluorescent protein
- LSM
- laser scanning microscopy
- LHR
- luteinizing hormone receptor
- M/D
- monomer/dimer equilibrium
- PAR
- protease-activated receptor
- RIA
- radioimmunoassay
- smTIRFM
- single molecule total internal reflection fluorescence microscopy
- TSHR
- thyroid stimulating hormone receptor
- V2R
- vasopressin V2 receptor
- CC
- cross-correlation
- CFP
- cyan fluorescent protein.
REFERENCES
- 1. Pierce K. L., Premont R. T., Lefkowitz R. J. (2002) Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 3, 639–650 [DOI] [PubMed] [Google Scholar]
- 2. Ernst O. P., Gramse V., Kolbe M., Hofmann K. P., Heck M. (2007) Monomeric G protein-coupled receptor rhodopsin in solution activates its G protein transducin at the diffusion limit. Proc. Natl. Acad. Sci. U.S.A. 104, 10859–10864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Whorton M. R., Bokoch M. P., Rasmussen S. G., Huang B., Zare R. N., Kobilka B., Sunahara R. K. (2007) A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc. Natl. Acad. Sci. U.S.A. 104, 7682–7687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Whorton M. R., Jastrzebska B., Park P. S., Fotiadis D., Engel A., Palczewski K., Sunahara R. K. (2008) Efficient coupling of transducin to monomeric rhodopsin in a phospholipid bilayer. J. Biol. Chem. 283, 4387–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kuszak A. J., Pitchiaya S., Anand J. P., Mosberg H. I., Walter N. G., Sunahara R. K. (2009) Purification and functional reconstitution of monomeric μ-opioid receptors: allosteric modulation of agonist binding by Gi2. J. Biol. Chem. 284, 26732–26741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tsukamoto H., Sinha A., DeWitt M., Farrens D. L. (2010) Monomeric rhodopsin is the minimal functional unit required for arrestin binding. J. Mol. Biol. 399, 501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bayburt T. H., Vishnivetskiy S. A., McLean M. A., Morizumi T., Huang C. C., Tesmer J. J., Ernst O. P., Sligar S. G., Gurevich V. V. (2011) Monomeric rhodopsin is sufficient for normal rhodopsin kinase (GRK1) phosphorylation and arrestin-1 binding. J. Biol. Chem. 286, 1420–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Terrillon S., Bouvier M. (2004) Roles of G-protein-coupled receptor dimerization. EMBO Rep. 5, 30–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bulenger S., Marullo S., Bouvier M. (2005) Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol. Sci. 26, 131–137 [DOI] [PubMed] [Google Scholar]
- 11. Milligan G., Canals M., Pediani J. D., Ellis J., Lopez-Gimenez J. F. (2006) The role of GPCR dimerisation/oligomerisation in receptor signalling. Ernst Schering Found. Symp. Proc. 2, 145–161 [DOI] [PubMed] [Google Scholar]
- 12. Smith N. J., Milligan G. (2010) Allostery at G protein-coupled receptor homo- and heteromers: uncharted pharmacological landscapes. Pharmacol. Rev. 62, 701–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lohse M. J. (2010) Dimerization in GPCR mobility and signaling. Curr. Opin. Pharmacol. 10, 53–58 [DOI] [PubMed] [Google Scholar]
- 14. Teichmann A., Rutz C., Kreuchwig A., Krause G., Wiesner B., Schülein R. (2012) The pseudo signal peptide of the corticotropin-releasing factor receptor type 2A prevents receptor oligomerization. J. Biol. Chem. 287, 27265–27274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pin J. P., Comps-Agrar L., Maurel D., Monnier C., Rives M. L., Trinquet E., Kniazeff J., Rondard P., Prézeau L. (2009) G-protein-coupled receptor oligomers: two or more for what? Lessons from mGlu and GABA B receptors. J. Physiol. 587, 5337–5344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Terrillon S., Barberis C., Bouvier M. (2004) Heterodimerisation of V1a and V2 vasopressin receptors determines the interaction with β-arrestin and their trafficking patterns. Proc. Natl. Acad. Sci. U.S.A. 101, 1548–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Urizar E., Montanelli L., Loy T., Bonomi M., Swillens S., Gales C., Bouvier M., Smits G., Vassart G., Costagliola S. (2005) Glycoprotein hormone receptors: link between receptor homodimerization and negative cooperativity. EMBO J. 24, 1954–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. El-Asmar L., Springael J. Y., Ballet S., Andrieu E. U., Vassart G., Parmentier M. (2005) Evidence for negative binding cooperativity within CCR5-CCR2b heterodimers. Mol. Pharmacol. 67, 460–469 [DOI] [PubMed] [Google Scholar]
- 19. Mesnier D., Banères J. L. (2004) Cooperative conformational changes in a G-protein-coupled receptor dimer, the leukotriene B4 receptor BLT1. J. Biol. Chem. 279, 49664–49670 [DOI] [PubMed] [Google Scholar]
- 20. George S. R., Fan T., Xie Z., Tse R., Tam V., Varghese G., O'Dowd B. F. (2000) Oligomerization of μ- and δ-opioid receptors: Generation of novel functional properties. J. Biol. Chem. 275, 26128–26135 [DOI] [PubMed] [Google Scholar]
- 21. Charles A. C., Mostovskaya N., Asas K., Evans C. J., Dankovich M. L., Hales T. G. (2003) Coexpression of δ-opioid receptors with μ receptors in GH3 cells changes the functional response to μ agonists from inhibitory to excitatory. Mol. Pharmacol. 63, 89–95 [DOI] [PubMed] [Google Scholar]
- 22. Mellado M., Rodríguez-Frade J. M., Vila-Coro A. J., Fernández S., Martín de Ana A., Jones D. R., Torán J. L., Martínez-A C. (2001) Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. 20, 2497–2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Allen M. D., Neumann S., Gershengorn M. C. (2011) Occupancy of both sites on the thyrotropin (TSH) receptor dimer is necessary for phosphoinositide signaling. FASEB J. 25, 3687–3694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hern J. A., Baig A. H., Mashanov G. I., Birdsall B., Corrie J. E., Lazareno S., Molloy J. E., Birdsall N. J. (2010) Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proc. Natl. Acad. Sci. U.S.A. 107, 2693–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kasai R. S., Suzuki K. G., Prossnitz E. R., Koyama-Honda I., Nakada C., Fujiwara T. K., Kusumi A. (2011) Full characterization of GPCR monomer-dimer dynamic equilibrium by single molecule imaging. J. Cell Biol. 192, 463–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Calebiro D., Rieken F., Wagner J., Sungkaworn T., Zabel U., Borzi A., Cocucci E., Zürn A., Lohse M. J. (2013) Single-molecule analysis of fluorescently labeled G-protein-coupled receptors reveals complexes with distinct dynamics and organization. Proc. Natl. Acad. Sci. U.S.A. 110, 743–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schmidt A., Wiesner B., Weisshart K., Schulz K., Furkert J., Lamprecht B., Rosenthal W., Schülein R. (2009) Use of Kaede fusions to visualize recycling of G protein-coupled receptors. Traffic 10, 2–15 [DOI] [PubMed] [Google Scholar]
- 28. Schmidt V., Baum K., Lao A., Rateitschak K., Schmitz Y., Teichmann A., Wiesner B., Petersen C. M., Nykjaer A., Wolf J., Wolkenhauer O., Willnow T. E. (2012) Quantitative modelling of amyloidogenic processing and its influence by SORLA in Alzheimer's disease. EMBO J. 31, 187–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kraetke O., Wiesner B., Eichhorst J., Furkert J., Bienert M., Beyermann M. (2005) Dimerization of corticotropin-releasing factor receptor type 1 is not coupled to ligand binding. J. Recept. Signal Transduct. Res. 25, 251–276 [DOI] [PubMed] [Google Scholar]
- 30. Milan-Lobo L., Gsandtner I., Gaubitzer E., Rünzler D., Buchmayer F., Köhler G., Bonci A., Freissmuth M., Sitte H. H. (2009) Subtype-specific differences in corticotropin-releasing factor receptor complexes detected by fluorescence spectroscopy. Mol. Pharmacol. 76, 1196–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Denver R. J. (2009) Structural and functional evolution of vertebrate neuroendocrine stress systems. Ann. N.Y. Acad. Sci. 1163, 1–16 [DOI] [PubMed] [Google Scholar]
- 32. Beyermann M., Fechner K., Furkert J., Krause E., Bienert M. (1996) A single-point slight alteration set as a tool for structure-activity relationship studies of ovine corticotropin releasing factor. J. Med. Chem. 39, 3324–3330 [DOI] [PubMed] [Google Scholar]
- 33. von Kleist L., Stahlschmidt W., Bulut H., Gromova K., Puchkov D., Robertson M. J., MacGregor K. A., Tomilin N., Tomlin N., Pechstein A., Chau N., Chircop M., Sakoff J., von Kries J. P., Saenger W., Kräusslich H. G., Shupliakov O., Robinson P. J., McCluskey A., Haucke V. (2011) Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell 146, 471–484 [DOI] [PubMed] [Google Scholar]
- 34. Sambrook J., Russel D. W. (2001) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 35. Grantcharova E., Furkert J., Reusch H. P., Krell H. W., Papsdorf G., Beyermann M., Schulein R., Rosenthal W., Oksche A. (2002) The extracellular N terminus of the endothelin B (ETB) receptor is cleaved by a metalloprotease in an agonist-dependent process. J. Biol. Chem. 277, 43933–43941 [DOI] [PubMed] [Google Scholar]
- 36. Elson E. L. (2001) Fluorescence correlation spectroscopy measures molecular transport in cells. Traffic 2, 789–796 [DOI] [PubMed] [Google Scholar]
- 37. Rigler R., Mets U., Widengren J., Kask P. (1993) Fluorescence correlation spectroscopy with high count rate and low background: analysis of translational diffusion. Eur. Biophys. J. 22, 169–175 [Google Scholar]
- 38. Haustein E., Schwille P. (2003) Ultrasensitive investigations of biological systems by fluorescence correlation spectroscopy. Methods 29, 153–166 [DOI] [PubMed] [Google Scholar]
- 39. Schwille P., Korlach J., Webb W. W. (1999) Fluorescence correlation spectroscopy with single-molecule sensitivity on cell and model membranes. Cytometry 36, 176–182 [DOI] [PubMed] [Google Scholar]
- 40. Lampe A., Haucke V., Sigrist S. J., Heilemann M., Schmoranzer J. (2012) Multi-colour direct STORM with red emitting carbocyanines. Biol. Cell 104, 229–237 [DOI] [PubMed] [Google Scholar]
- 41. Edelstein A., Amodaj N., Hoover K., Vale R., Stuurman N. (2010) Computer control of microscopes using μManager. Curr. Protoc. Mol. Biol. 10.1002/0471142727.mb1420s92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mashanov G. I., Molloy J. E. (2007) Automatic detection of single fluorophores in live cells. Biophys. J. 92, 2199–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cottet M., Albizu L., Perkovska S., Jean-Alphonse F., Rahmeh R., Orcel H., Méjean C., Granier S., Mendre C., Mouillac B., Durroux T. (2010) Past, present and future of vasopressin and oxytocin receptor oligomers, prototypical GPCR models to study dimerization processes. Curr. Opin. Pharmacol. 10, 59–66 [DOI] [PubMed] [Google Scholar]
- 44. Gregan B., Schaefer M., Rosenthal W., Oksche A. (2004) Fluorescence resonance energy transfer analysis reveals the existence of endothelin-A and endothelin-B receptor homodimers. J. Cardiovasc. Pharmacol. 44, S30–S33 [DOI] [PubMed] [Google Scholar]
- 45. McLaughlin J. N., Patterson M. M., Malik A. B. (2007) Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc. Natl. Acad. Sci. U.S.A. 104, 5662–5667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rutz C., Renner A., Alken M., Schulz K., Beyermann M., Wiesner B., Rosenthal W., Schülein R. (2006) The corticotropin-releasing factor receptor type 2a contains an N-terminal pseudo signal peptide. J. Biol. Chem. 281, 24910–24921 [DOI] [PubMed] [Google Scholar]
- 47. Schulz K., Rutz C., Westendorf C., Ridelis I., Vogelbein S., Furkert J., Schmidt A., Wiesner B., Schülein R. (2010) The pseudo signal peptide of the corticotropin-releasing factor receptor type 2a decreases receptor expression and prevents Gi-mediated inhibition of adenylyl cyclase activity. J. Biol. Chem. 285, 32878–32887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zampatis D. E., Rutz C., Furkert J., Schmidt A., Wüstenhagen D., Kubick S., Tsopanoglou N. E., Schülein R. (2012) The protease-activated receptor 1 possesses a functional and cleavable signal peptide which is necessary for receptor expression. FEBS Lett. 586, 2351–2359 [DOI] [PubMed] [Google Scholar]
- 49. Westendorf C., Schmidt A., Coin I., Furkert J., Ridelis I., Zampatis D., Rutz C., Wiesner B., Rosenthal W., Beyermann M., Schülein R. (2011) Inhibition of biosynthesis of human endothelin B receptor by the cyclodepsipeptide cotransin. J. Biol. Chem. 286, 35588–35600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thielen A., Oueslati M., Hermosilla R., Krause G., Oksche A., Rosenthal W., Schülein R. (2005) The hydrophobic amino acid residues in the membrane-proximal C tail of the G protein-coupled vasopressin V2 receptor are necessary for transport-competent receptor folding. FEBS Lett. 579, 5227–5235 [DOI] [PubMed] [Google Scholar]
- 51. Atwood B. K., Lopez J., Wager-Miller J., Mackie K., Straiker A. (2011) Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genomics 12, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Perry S. J., Junger S., Kohout T. A., Hoare S. R., Struthers R. S., Grigoriadis D. E., Maki R. A. (2005) Distinct conformations of the corticotropin releasing factor type 1 receptor adopted following agonist and antagonist binding are differentially regulated. J. Biol. Chem. 280, 11560–11568 [DOI] [PubMed] [Google Scholar]
- 53. Murat B., Devost D., Andrés M., Mion J., Boulay V., Corbani M., Zingg H. H., Guillon G. (2012) V1b and CRHR1 receptor heterodimerization mediates synergistic biological actions of vasopressin and CRH. Mol. Endocrinol. 26, 502–520 [DOI] [PMC free article] [PubMed] [Google Scholar]